

Abstract
Rheb (Ras homolog enriched in brain) is a component of the phosphatidylinositol 3-kinase (PI3K)–target of rapamycin (TOR) signaling pathway, functioning as a positive regulator of TOR. Constitutively active mutants of Rheb induce oncogenic transformation in cell culture. The transformed cells are larger and contain more protein than their normal counterparts. They show constitutive phosphorylation of the ribosomal protein S6 kinase (S6K) and the eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), two downstream targets of TOR. The TOR-specific inhibitor rapamycin strongly interferes with transformation induced by constitutively active Rheb, suggesting that TOR activity is essential for the oncogenic effects of mutant Rheb. Rheb-induced transformation is also dependent on a C-terminal farnesylation signal that mediates localization to a cellular membrane. An engineered N-terminal myristylation signal can substitute for the farnesylation. Immunofluorescence localizes wild type (wt) and mutant Rheb to vesicular structures in the cytoplasm, overlapping with the endoplasmic reticulum (ER).
Keywords: Rheb, PI 3-kinase, Akt, TOR, S6-kinase, 4E-binding protein, GTP-binding
Introduction
Rheb is a member of the Ras superfamily of small G-proteins. It was isolated in a screen for genes that are induced in neurons by synaptic activity (Yamagata et al., 1994). Subsequent studies revealed that Rheb is expressed in various tissues, but most abundantly in skeletal and cardiac muscle, testes, and ovary (Gromov et al., 1995). The Rheb gene is highly conserved in eukaryotes from yeast to mammals (Urano et al., 2001). Rheb shares high homology with human Rap2 (Ras-related protein-2), yeast RAS1, and human H-Ras (37.7%, 43.3%, and 34% identity, respectively) (Yamagata et al., 1994). It contains five G-boxes that are involved in the recognition and hydrolysis of GTP (Urano et al., 2000). Biochemical and cell biological evidence suggests that Rheb has a low intrinsic GTPase activity and occurs preferentially in an activated, GTP-bound state (Clark et al., 1997; Im et al., 2002). The tuberous sclerosis complex (TSC), a tumor suppressor formed by the TSC1 (hamartin) and TSC2 (tuberin) proteins, functions as a GTPase-activating protein (GAP) and hence as a negative regulator of Rheb (Castro et al., 2003; Garami et al., 2003; Inoki et al., 2003; Tee et al., 2003; Zhang et al., 2003). Drosophila TCTP (translationally controlled tumor protein) has recently been identified as the putative GEF (guanine nucleotide exchange factor) for Rheb in that species (Hsu et al., 2007).
In yeast, disruption of Rheb causes increased uptake of arginine, whereas hyperactivated Rheb, such as that in TSC1/2 mutant cells, leads to decreased uptake of arginine (Urano et al., 2000; van Slegtenhorst et al., 2004). In the Drosophila cell line S2 and in S. pombe, knockdown of Rheb results in cell cycle arrest in the G0/G1 phase, whereas overexpression of Rheb leads to a significant increase of S-phase cells (Mach et al., 2000; Patel et al., 2003). Complementary screens for loss- and gain-of-function mutations affecting cell size in Drosophila identified Rheb as an essential regulator of cell size; overexpression of Rheb results in larger cells (Patel et al., 2003; Saucedo et al., 2003; Stocker et al., 2003).
Genetic and biochemical studies have placed Rheb upstream of TOR and downstream of Akt (v-akt murine thymoma viral oncogene homolog) and TSC1/2 in the PI3K/Akt/TOR/S6K signaling pathway (Castro et al., 2003; Garami et al., 2003; Inoki et al., 2003; Saucedo et al., 2003; Stocker et al., 2003; Tee et al., 2003; Zhang et al., 2003). In the canonical PI3K/Akt/TOR signaling model, Class IA PI3Ks convert PIP2 (phosphatidylinositol 4,5-bisphosphate) to PIP3 (phosphatidylinositol 3,4,5-trisphosphate) upon growth factor stimulation. PIP3 recruits the serine/threonine kinases PDK1 (phosphoinositide-dependent kinase 1) and Akt to the membrane through their pleckstrin homology domains. Akt is then activated by phosphorylation at T308 by PDK1 and at S473 by PDK2 (Alessi et al., 1997a; Alessi et al., 1997b; Sarbassov et al., 2004; Sarbassov et al., 2005; Stephens et al., 1998; Stokoe et al., 1997). Activated Akt is the predominant and essential mediator for the regulation of both growth and proliferation by PI3K. Akt phosphorylates a broad spectrum of protein substrates that affect cell growth, survival, and metabolism (Alessi and Cohen, 1998; Aoki and Vogt, 2004; Chan et al., 1999; Datta et al., 1999; Downward, 1998; Lawlor and Alessi, 2001). Akt phosphorylates TSC2 at the residues S939, S1130, and T1462, thus inhibiting the GAP activity of the TSC complex and enhancing the charge of Rheb with GTP (Castro et al., 2003; Dan et al., 2002; Garami et al., 2003; Inoki et al., 2003; Inoki et al., 2002; Manning et al., 2002; Tee et al., 2003; Zhang et al., 2003). Activation of TOR by Rheb-GTP leads to phosphorylation and activation of ribosomal S6 kinase (S6K), and phosphorylation and inactivation of eukaryotic translation initiation factor eIF-4E binding protein 1 (4E-BP1) (Brown et al., 1994; Sabatini et al., 1994; Sabers et al., 1995). Through its modification of S6K and 4E-BP1 activities, TOR controls the translation of numerous proteins that regulate cell growth.
Rheb binds to the TOR complex directly (Long et al., 2005a), and recent observations suggest that it activates TOR by relieving an inhibitory interaction between TOR and FKBP38 (FK506-binding protein 38) (Bai et al., 2007). Withdrawal of amino acids inhibits the binding of Rheb to TOR, although it does not change the level of GTP bound to Rheb. Overexpressed Rheb can restore the TOR-dependent phosphorylation of S6K when amino acids are withdrawn (Long et al., 2005b). In vertebrate cells Rheb with a high GTP:GDP ratio appears more effective in activating TOR than Rheb with lower GTP:GDP ratio, yet Rheb-GDP interacts with TOR more strongly than Rheb-GTP (Long et al., 2005a). In fission yeast the opposite result has been obtained: constitutively active Rheb mutants interact more strongly with TOR than wild type (wt) (Urano et al., 2005). Rheb likely has other functions in addition to the control of TOR, as suggested by rapamycin-resistant activities of Rheb (Saito et al., 2005).
In some mammalian tumors and tumor cell lines, the level of Rheb transcripts is elevated (Basso et al., 2005; Gromov et al., 1995), but overexpression of either wt or of a constitutively active Rheb mutant does not cause morphological or growth transformation of NIH 3T3 cells (Clark et al., 1997). Here we show that a constitutively active Rheb has oncogenic potential and induces transformation of chicken embryonic fibroblasts (CEF) via activated TOR. The transforming activity requires localization of Rheb to a cellular membrane.
Results
Rheb mutants that constitutively bind GTP induce oncogenic transformation in cell culture
Ras, a close relative of Rheb, is mutationally activated by a glutamine to leucine substitution at position 61 (Q61L). The mutated Ras is insensitive to the GTPase activating protein, is constitutively charged with GTP and permanently active. Rheb has a conserved glutamine at position 64 in a homologous region of the protein. The Rheb Q64L mutant behaves similar to its Ras counterpart; it is nearly 90% bound to GTP (Inoki et al., 2003; Li et al., 2004; Long et al., 2005a). The activity of the Rheb mutant, measured indirectly by the kinase level of its downstream target TOR, is substantially elevated (Li et al., 2004; Long et al., 2005a). In addition to the Q64L mutant, several novel hyperactive mutants of Rheb were recently identified (Urano et al., 2005; Yan et al., 2006). The mutants identified in S. pombe carry single amino acid changes (V17G, S21G, K120R, N153T) that confer resistance to canavanine, a toxic arginine analogue. These mutations lower the affinity of the Rheb protein to GDP, thus increasing the ratio of Rheb-GTP to Rheb-GDP in the cell. We introduced the gain-of-function mutations singly into the sequence of the human wt Rheb, generating Rheb V17G, S21G, Q64L, K120R and N153T. The mutant constructs were fused to a FLAG tag at the N terminus, cloned into the avian retroviral expression vector RCAS and transfected into CEF. RCAS constructs produce infectious retrovirus upon transfection. This virus progeny was used to express the mutant Rheb proteins and study their properties. Western blot analysis confirmed RCAS-mediated expression of wt Rheb, Q64L (Fig. 1a), and N153T (data not shown). The S21G mutant was expressed at a very low level, and V17G and K120R could not be detected in the infected cell cultures (data not shown). The mutants Q64L and N153T induced morphological changes characteristic of oncogenic transformation in monolayer cultures and conferred upon the infected cells the ability to produce colonies in nutrient agar (Fig. 1b and 1c). In contrast, CEF expressing wt Rheb showed only barely detectable changes in cell morphology and generated only few small colonies in soft agar (Fig. 1b and 1c). These results demonstrate the strong oncogenic potential of Q64L and N153T. We have chosen Q64L for further detailed investigation.
Fig. 1.

(a) Protein expression of wt and mutant Rheb in CEF. CEF transfected with recombinant RCAS encoding wt Rheb or Q64L were lysed 10 days post transfection. The lysates were separated on a 4-20% gradient SDS/PAGE gel and transferred to a polyvinylidene difluoride membrane. The membrane was probed with anti-FLAG and anti-tubulin antibody. (b) Focus formation induced by mutant Rheb. CEF were infected with the RCAS vector or recombinant RCAS viruses expressing wt Rheb, Q64L or N153T at different dilutions as indicated. The cultures were overlaid with nutrient agar and stained with crystal violet on day 14. (c) Anchorage-independent colony formation induced by mutant Rheb. CEF were transfected with the RCAS vector or recombinant RCAS viruses expressing wt Rheb, Q64L or N153T, then seeded in nutrient agar 12 days post transfection and photographed on day 10 after seeding.
The morphological changes in CEF expressing Q64L include increased size and vacuolization (Fig. 2a). The average volume of the mutant-expressing cells as determined by a CASY Cell Counter (Scharfe System GmbH) is about 25% above that of cells expressing wt Rheb or vector alone (Fig. 2b). This result was confirmed by flow cytometry forward scatter analysis of fixed cells and by measurements with the Beckman-Coulter Vi-CELL XR cell viability analyzer. CEF transformed by Q64L also showed a 30% greater protein content than vector controls (Fig. 2c). Although Q64L-transformed cells have the ability of multilayered and anchorage-independent growth, showing altered morphology and increased protein content, their rate of propagation and cell cycle profile do not significantly differ from those of wt Rheb or vector-only expressing cells (data not shown).
Fig. 2.
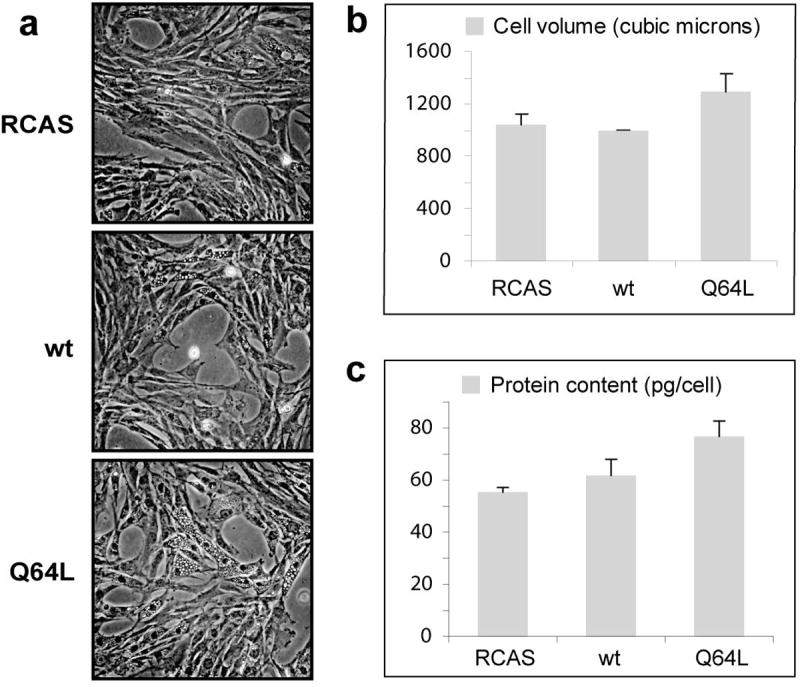
Q64L-transformed CEF are larger, have increased vacuolization, and contain more protein than their normal counterparts. (a) Micrographs of CEF transfected with vector, wt Rheb, or Q64L (phase-contrast optics, X10 objective lens). (b) Cell volume of CEF transfected with constructs as indicated. (c) Protein content (pg/cell) of CEF transfected with constructs as indicated.
Cellular transformation induced by Rheb requires TOR activity
Rheb binds to TOR and activates TOR in a GTP-dependent manner (Long et al., 2005a). In order to determine whether the oncogenic transformation induced by Q64L is TOR-dependent, we tested the effect of rapamycin, a specific inhibitor of TOR, on the transformation process. At a concentration of 10 ng/ml, rapamycin effectively interfered with the formation of transformed cell foci as well as with anchorage-independent growth induced by Q64L (Fig. 3). This concentration of rapamycin had no effect on the transforming efficiency of the Jun oncoprotein tested in the same cells (Fig. 3b). The experiments with rapamycin were complemented by determinations on TOR targets. Transient expression of Rheb in Drosophila S2 cells and HEK293 cells increases the phosphorylation of the ribosomal protein S6 kinase and of the eukaryotic initiation factor 4E binding protein 4E-BP1, both targets of TOR (Castro et al., 2003; Garami et al., 2003; Inoki et al., 2003; Saucedo et al., 2003; Tee et al., 2003). In serum-starved CEF, uninfected or infected with vector alone, phosphorylation of S6K was low, but insulin induced a strong phosphorylation on threonine 389 within 15 min. In contrast, CEF transformed by Q64L showed a high level of phosphorylation on threonine 389, even under conditions of serum deprivation, suggesting that S6K is constitutively activated in the Q64L-transformed cells (Fig. 4). Similarly, phosphorylation of 4E-BP1 was constitutively increased in Q64L-transformed cells but not in control cells. These results suggest that oncogenic transformation by Q64L depends on TOR activity and is correlated with constitutive activation of TOR signaling.
Fig. 3.
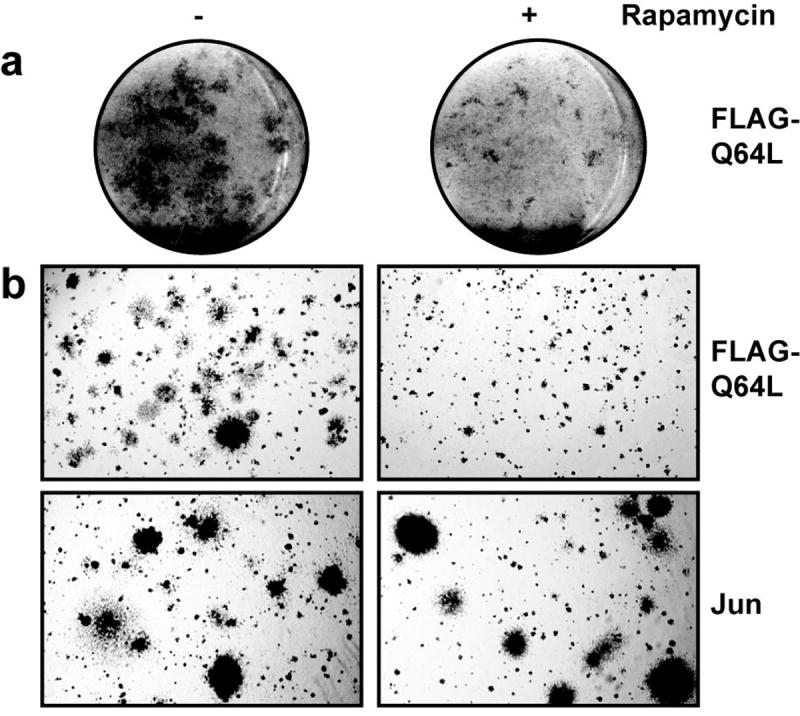
Rapamycin strongly interferes with transformation induced by Q64L. (a) CEF were infected with Q64L, overlaid with nutrient agar containing 10 ng/ml rapamycin (right column) or solvent only (left column). After 14 days, the cultures were fixed and stained. (b) CEF were infected with the RCAS viruses expressing Q64L or the oncoprotein Jun, then seeded in nutrient agar supplemented with 10 ng/ml rapamycin (right column) or solvent only (left column) 12 days post infection and photographed on day 10 after seeding.
Fig. 4.
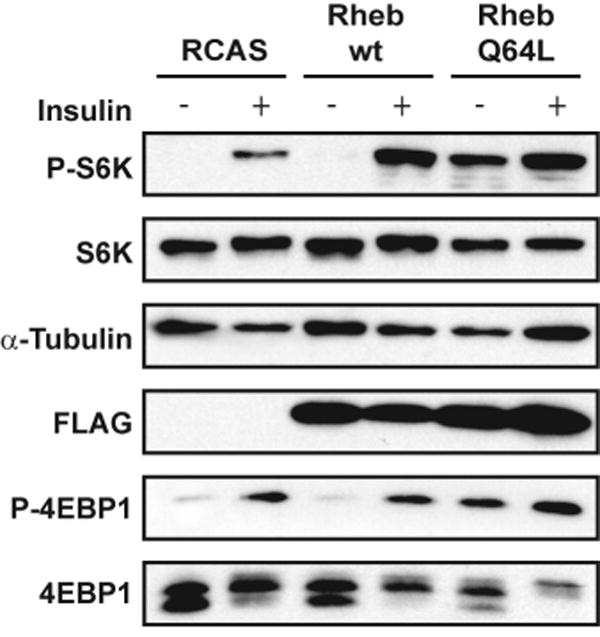
Constitutive activation of TOR signaling in Q64L-transformed CEF. CEF transfected with RCAS vector, wt Rheb or Q64L were serum starved for 40 h and subsequently stimulated with insulin for 15 min. Cells were then lysed, and the proteins were separated on a 4-20% gradient SDS/PAGE gel. The transferred blot was probed with antibodies directed against total proteins or phosphorylated proteins as indicated.
FOXO-1 and YB-1 interfere with the oncogenic transformation induced by Rheb
The winged helix transcription factor FOXO-1 (forkhead box protein class-O 1) is a growth-attenuating and pro-apoptotic protein. Phosphorylation of FOXO-1 by Akt induces exclusion of FOXO-1 from the nucleus, thus preventing its transcriptional activity and mediating its proteolytic degradation (Aoki et al., 2004; Biggs et al., 1999; Cahill et al., 2001). The Y box-binding protein 1 (YB-1) is a regulator of transcription and translation. It is transcriptionally downregulated in Akt-transformed CEF, and ectopic expression of YB-1 induces a strong cellular resistance to transformation by constitutively active forms of PI3K and Akt (Bader et al., 2003). Because FOXO-1 and YB-1 proteins are growth-inhibitory, we explored their potential for interference with Rheb-induced transformation. As shown in Fig. 5, both FOXO-1 and YB-1 dramatically reduced the anchorage-independent growth induced by Q64L. In contrast, the dominant negative Akt (dnAkt, T308A/S473A), which interferes with PI3K-induced oncogenic transformation (Aoki et al., 1998) but functions upstream of Rheb, does not inhibit Q64L-induced agar colony formation. These results are in accord with the canonical PI3K/Akt signaling pathway, which places Akt above and YB-1 below Rheb. The interference by FOXO-1 suggests that oncogenic activity of Rheb also requires signaling molecules that are regulated by FOXO-1.
Fig. 5.

Effect of FOXO-1, YB-1 and dnAkt on Q64L-induced transformation. CEF were transfected with the RCAS(A) vector or recombinant RCAS(A) viruses expressing FOXO-1 or YB-1 or dnAkt. After three passages, they were seeded and challenged with Q64L expressed by RCAS(B). Equal amounts of cells were seeded in nutrient agar 10 days after challenge infection. Agar colonies were counted and photographed on day 10 after seeding. The result of one representative experiment is shown.
Western blot analysis demonstrated that phosphorylation of Akt is downregulated in Q64L-transformed CEF (data not shown). This result agrees with previous findings and probably reflects a negative feedback regulation of Akt activity by TOR (Briaud et al., 2005; Hers and Tavare, 2005; Shah and Hunter, 2006; Shah et al., 2004; Wan et al., 2007). Akt phosphorylates and thereby negatively regulates FOXO-1. Lower Akt activity in Q64L-transformed cells translates into less phosphorylation of and hence higher activity of FOXO-1 (data not shown). However, that activity may be insufficient to significantly affect the overexpressed Rheb mutant.
Cellular transformation by Rheb requires a functional membrane address
Like other Ras family members, Rheb contains a C-terminal farnesylation motif, CAAX (where ‘A’ represents an aliphatic amino acid). Farnesylation of Rheb is essential for cell cycle progression in S. pombe (Yang et al., 2001), and is also required for arginine uptake in S. cerevisiae (Urano et al., 2000). In mammalian cells, loss of the farnesylation motif markedly reduces the ability of Rheb to induce TOR-dependent phosphorylation of S6K (Castro et al., 2003; Tee et al., 2003). We determined the role of farnesylation in the transformation process induced by Q64L by constructing several mutants with a blocked or defective farnesylation signal, i.e., Q64L-FLAG, FLAG-C181S, C181S-FLAG, and Q64L/C181S-FLAG. We also generated mutants, myr-Q64L-FLAG and myr-Q64L/C181S-FLAG, with an inactive farnesylation motif but an added myristylation signal to test whether farnesylation-negative mutants can be rescued by providing an alternative membrane address. All mutants in which the farnesylation signal was either inactivated or blocked by a C-terminally fused FLAG tag failed to induce oncogenic transformation as measured by focus formation on monolayer culture and by anchorage-independent growth in nutrient agar (Fig. 6). Addition of a myristylation signal at the N-terminus of these mutants restored transforming activity (Fig. 6). The farnesylation-negative mutants also failed to induce the growth-factor independent phosphorylation of S6K and of 4E-BP1; myristylation rescued this signaling activity (Fig. 7). These observations suggest that oncogenic transformation by Q64L requires membrane localization. This requirement is met by the C-terminal farnesylation signal of Rheb, but myristylation will serve the same function.
Fig. 6.

Cellular transformation induced by myristylated mutant Rheb. In focus formation assay (round photographs), CEF were infected with RCAS viruses expressing FLAG-Q64L, Q64L-FLAG, FLAG-C181S, Q64L/C181S-FLAG, myr-Q64L-FLAG, or myr-Q64L/C181S-FLAG. The cultures were overlaid with nutrient agar and stained with crystal violet on day 14. In anchorage-independent colony formation assay (rectangular photographs), CEF were transfected with recombinant RCAS expressing FLAG-Q64L, Q64L-FLAG, FLAG-C181S, Q64L/C181S-FLAG, myr-Q64L-FLAG, or myr-Q64L/C181S-FLAG. The cells were seeded in nutrient agar 12 days post transfection and photographed on day 10 after seeding.
Fig. 7.
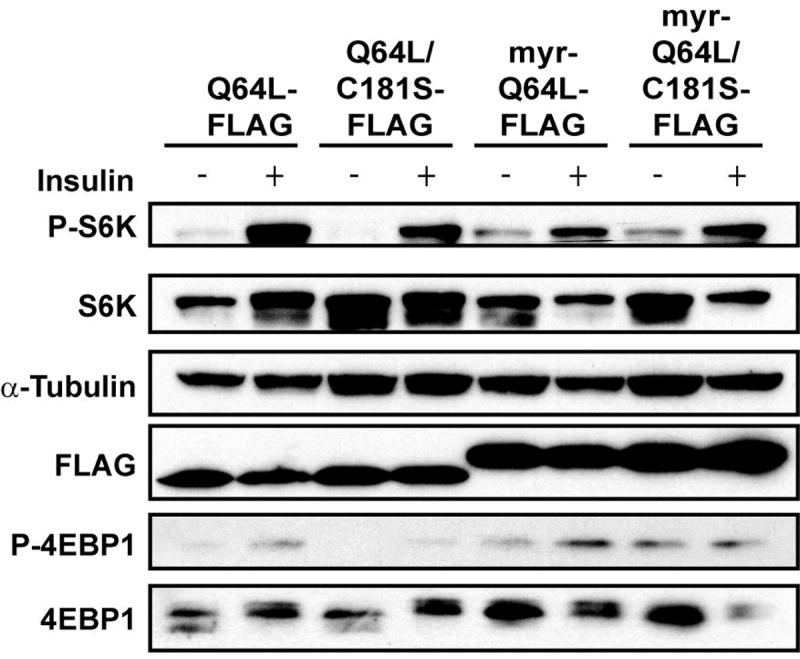
Constitutive activation of TOR signaling by myristylated mutant Rheb. CEF transfected with Q64L-FLAG, Q64L/C181S-FLAG, myr-Q64L-FLAG, or myr-Q64L/C181S-FLAG were serum starved for 40 h and subsequently stimulated with insulin for 15 min. Cells were then lysed, and the proteins were separated on a 4-20% gradient SDS/PAGE gel. The transferred blot was probed with antibodies directed against total proteins or phosphorylated proteins as indicated.
Observations with immunofluorescence directed to the FLAG tag showed that wt Rheb and Q64L are located in the cytoplasm, associated with vesicular structures (Fig. 8a). This cellular distribution is in agreement with recently published results (Buerger et al., 2006; Heo et al., 2006; Takahashi et al., 2005). Additionally, CEF expressing Q64L are characterized by pronounced cytoplasmic vacuolization, which has also been seen with wt Rheb in mammalian cells (Saito et al., 2005). In contrast, the farnesylation-negative mutants Q64L-FLAG, FLAG-C181S, C181S-FLAG, and Q64L/C181S-FLAG showed identical patterns of localization: partially in the nucleus and partially in the cytoplasm (Fig. 8a, FLAG-C181S is shown as representative for all these mutants). So far, no nuclear localization signal has been identified in Rheb. Addition of a myristylation signal at the N-terminus of these mutants, i.e., myr-Q64L-FLAG and myr-Q64L/C181S-FLAG, restored the cytoplasmic distribution and the association with vesicular structures. The constructs carrying a myristylation signal, myr-Q64L-FLAG and myr-Q64L/C181S-FLAG, both induced the formation of conspicuous filopodia on the cell surface (Fig. 8b). Using antibodies raised against ER-specific protein ERp72, we found that wt Rheb, Q64L, myr-Q64L-FLAG and myr-Q64L/C181S-FLAG partially co-localized with the ER but extended beyond the ER network. In contrast, FLAG-C181S showed much reduced association with ER (Fig. 8a). Assays of protein binding to large multilamellar vesicles (LMVs) were conducted for wt Rheb, Q64L, C181S, and myr-Q64L/C181S. Compared to wt and Q64L, C181S showed much lower binding to phospholipids, whereas myr-Q64L/C181S showed much higher affinity for phospholipids than wt or Q64L (Fig. 8c).
Fig. 8.
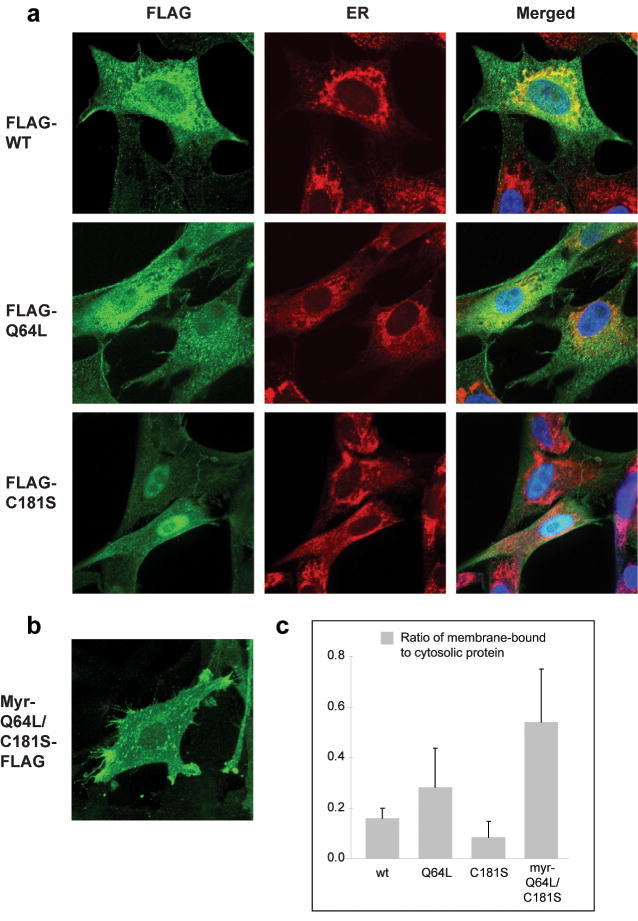
Subcellular localization of Rheb. CEF expressing either (a) wt Rheb, Q64L, C181S (all with an N-terminal FLAG tag), or (b) myr-Q64L/C181S-FLAG were fixed in 3.7% formaldehyde and treated with mouse anti-FLAG and FITC-conjugated secondary antibody, or together with rabbit anti-ERp72 (Calbiochem) and Texas red-conjugated goat anti-rabbit antibody (Santa Cruz Biotechnology). Nuclei were stained with 4′ 6-Diamidino-2-phenylindole (DAPI). Pictures were taken with a Bio-Rad Radiance 2100 Rainbow confocal laser scanning microscope, attached to a Nikon TE2000-U microscope with infinity corrected optics. (c) Assays of protein binding to large multilamellar vesicles (LMVs). A mixture of brain phospholipids (10 mg/ml) was incubated with a protein sample from CEF transfected with wt Rheb, Q64L, C181S, and myr-Q64L/C181S, respectively. The lipid-protein complex was spun down (designated as M for membrane-bound), whereas the cytosolic proteins in the supernatant (designated as C for cytosolic) was precipitated by TCA. Sample M and C were separated on a 4-20% Tris-Glycine SDS polyacrylamide gel. Blots were probed with anti-FLAG antibody and the intensity of the bands was quantified by the ImageJ software (NIH). The figure shows the ratios of membrane-bound to cytosolic proteins.
Discussion
Constitutively active, GTP-bound Rheb has oncogenic potential; it transforms primary avian cells in culture and imparts on these cells the ability of anchorage–independent growth. This growth-promoting activity of Rheb-GTP is of particular significance in view of the fact that Rheb is frequently overexpressed in various human cancer cells derived from carcinomas of the lung, colon, and ovary (Basso et al., 2005; Gromov et al., 1995). Rheb is also overexpressed in brain tumors (Holland, personal communication). The gain of function in Rheb may contribute to the malignant phenotype of the tumor cells. The Rheb-induced transformation in avian cell cultures is correlated with an increase in cell size and protein content, but it does not result in accelerated cell replication.
The oncogenic activity of Q64L is probably not due to increased expression. At 10 days post transfection, there is no significant difference of protein levels between wt and Q64L, yet transformation is evident. However after prolonged culture, Q64L is often expressed at higher levels than wt. This is likely due to an increase of protein synthesis in Q64L-transformed CEF. The GTPase activity of Q64L is sensitive to the GAP activity of TSC when both proteins are cotransfected (Li et al., 2004). However, in the experiments described here, only Q64L is overexpressed by the RCAS vector, whereas TSC is endogenous. Under these conditions, the higher levels of Q64L will likely overcome endogenous TSC-GAP activity. Overexpression of wt Rheb might also overcome endogenous TSC-GAP activity, but its GTP/GDP charge ratio is not high enough to trigger constitutive activation of TOR.
The growth promoting activity of Rheb is well documented in Drosophila, where wt Rheb enhances cell replication and induces an increase of cell size and where reduced or deleted Rheb function leads to cell cycle arrest (Patel et al., 2003; Saucedo et al., 2003; Stocker et al., 2003). However, in mammalian NIH 3T3 cells, expression of constitutively active Rheb has not resulted in an altered phenotype (Clark et al., 1997). Mammalian cells are refractory to the action of most single oncogenes; in contrast, avian cells are highly sensitive indicators of the oncogenic potential of single agents. They can be readily transformed by single oncoproteins, for example, Myc, Jun, Qin, P3K or Akt, all of which are either inactive or extremely inefficient as single oncogenic agents in mammalian cell systems (Aoki et al., 1998; Bister et al., 1977; Chang et al., 1997; Himly et al., 1998).
Rheb is an essential component of the Akt-TOR signaling chain. The transforming activity of Rheb is highly sensitive to the TOR inhibitor rapamycin and therefore is TOR-dependent. Accordingly, Rheb-induced transformation is correlated with enhanced phosphorylation of the TOR targets S6K and 4E-BP1. Yet not all activities of Rheb are mediated by TOR. Rheb interacts with the B-Raf kinase in a rapamycin-resistant manner and inhibits its function, resulting in an interference with H-Ras (G12V)-induced transformation of NIH 3T3 cells (Clark et al., 1997; Im et al., 2002; Karbowniczek et al., 2004; Karbowniczek et al., 2006; Yee and Worley, 1997). The pronounced vacuolization observed in Rheb-expressing cells by Saito and coworkers and by us appears to reflect another TOR-independent function of Rheb (Saito et al., 2005). Exposure of Rheb-transformed cells to rapamycin for extended periods (6 to 24 hrs) has no detectable effect on the extent of vacuolization but effectively shuts down TOR activity within 2 hrs as judged by phosphorylation of S6K and 4E-BP1 (data not shown).
We have identified two effective antagonists of the growth-promoting activity of Rheb, YB-1 and FOXO-1. Both interfere with Rheb-induced oncogenic transformation. YB-1 is transcriptionally downregulated in Akt-transformed cells, and re-expression of YB-1 induces a strong cellular resistance to Akt signaling and to Akt-dependent transformation (Bader et al., 2003). This effect of YB-1 is probably due to an inhibition of cap-dependent protein synthesis (Bader and Vogt, 2005; Bader and Vogt, 2007). The inhibition appears to be selective for certain growth-stimulating proteins generated from messages with complex 5′ untranslated structures that require an abundance of eIF4E (eukaryotic initiation factor 4E) for effective translation. YB-1 acts downstream of TOR, and its inhibitory action of Rheb-induced transformation is in accord with the position of Rheb in the Akt signaling pathway. The inhibitory effect of FOXO-1 on Rheb-induced transformation is likely the result of FOXO-1-induced enhancement in the transcription of GADD45 (growth arrest- and DNA damage-inducible protein 45) and of p27Kip, two negative regulators of the cell cycle (Cunningham et al., 2004; Furukawa-Hibi et al., 2002; Medema et al., 2000; Stahl et al., 2002). The antagonistic effect of YB-1 on Rheb is specific for the Akt-TOR signaling pathway; that of FOXO-1 will likely extend to a broad spectrum of oncoproteins, representing different signaling pathways. As expected, dominant-negative Akt does not interfere with Rheb-induced transformation. Akt is situated upstream of Rheb, and constitutively active Rheb functions independently of Akt. In Rheb-transformed cells, phosphorylation of serine 473 and threonine 308 of Akt is significantly reduced (data not shown). Whether this reduction translates into attenuated Akt activity is not known.
Oncogenic transformation by Rheb requires a functional farnesylation signal. Expression of constructs with a mutated or blocked farnesylation signal has no detectable effect on the cellular phenotype. However, such constructs can be rescued with the addition of an N-terminal myristylation signal. The functional equivalence of myristylation and farnesylation suggests the relevant effect of these post-translational modifications of Rheb is membrane localization. It is not known whether attachment to any cellular lipid membrane satisfies this requirement or whether a functionally specialized membrane has to be addressed. Location of Rheb by immunofluorescence suggests an at least partial overlap with the ER. In our experiments Rheb is overexpressed, and it is therefore not surprising that its distribution exceeds the areas defined by the ER-specific marker. The mandatory association of Rheb with cellular membranes is likely to extend to TOR. Rheb physically interacts with TOR and therefore could recruit TOR to a membrane. The subcellular localization of TOR is likely signal-dependent and cells observed under different conditions yield different results. Although a nuclear localization has been reported for TOR (Zhang et al., 2002), TOR has also been found in the cytoplasm and might shuttle between cytoplasm and nucleus (Kim and Chen, 2000; Sabatini et al., 1999). Drenan et al. demonstrate a predominantly perinuclear distribution of TOR and show a close association with the ER and the Golgi apparatus (Drenan et al., 2004). Our data then suggest that the intracellular membranes composed of the ER and the Golgi network might offer a platform for Rheb and TOR to interact in signal transduction.
In summary, our studies reveal an oncogenic potential of Rheb that is both dependent on TOR activity and on binding to a cellular membrane. The overexpression of Rheb in human gliomas suggests that it may be a therapeutically valuable target.
Materials and methods
Plasmid construction
Human Rheb (hRheb) gene (AF493921) was obtained from Openbiosystems (Huntsville, AL). Kozak sequence and FLAG tag were introduced into hRheb by PCR using primers 5′- AGCTCCGCGGGCCACCATGGACTACAAGGATGACGATGACAAGCCGCAGTCCAAGTCCCGGAAGA-3′, and 5′- AGCTTCTAGATCACATCACCGAGCATGAAG-3′. PCR-amplified Rheb was subsequently cloned into pBSFI vector (Aoki et al., 1998), then the SfiI DNA fragment was cloned into the avian retrovirus vector RCAS(B). Sfi (Ahmed et al., 1997; Hughes et al., 1987). For C-terminally FLAG-tagged Rheb, primers 5′- ACTGGCGGCCGCGCCACCATGCCGCAGTCCAAGTCCCG-3′ and 5′- TGCCACGCGTTCACTTGTCATCGTCATCCTTGTAGTCCATCACCGAGCATGAAGACT-3′ were used. The mutant constructs containing a V17G, S21G, Q64L, K120R, N153T substitution were generated by using the Quick Change site-directed mutagenesis kit (Stratagene, La Jolla, CA) and the following primers: 5′-V17G (5′- CTGGGCTACCGGTCTGGGGGGAAATCCTCATTG -3′) and 3′-V17G (5′- CAATGAGGATTTCCCCCCAGACCGGTAGCCCAG-3′); 5′-S21G (5′- GGTCTGTGGGGAAATCCGGATTGACGATTCAATTTG-3′) and 3′-S21G (5′- CAAATTGAATCGTCAATCCGGATTTCCCCACAGACC-3′); 5′-Q64L (5′- GTAGACACAGCCGGGCTAGATGAATATTCTATC-3′) and 3′-Q64L (5′- GATAGAATATTCATCTAGCCCGGCTGTGTCTAC-3′); 5′-K120R (5′- GTTGGTTGGGAATAGGAAAGACCTGCATATG-3′) and 3′-K120R (5′- CATATGCAGGTCTTTCCTATTCCCAACCAAC-3′); 5′-N153T (5′- CTTCTGCTAAAGAAACTCAGACTGCTGTGG-3′) and 3′-N153T (5′- CCACAGCAGTCTGAGTTTCTTTAGCAGAAG-3′). For myr-Rheb, myristylation signal was added at the 5′-end and FLAG was added at the 3′-end using primers 5′- ACTGGCGGCCGCGCCACCATGGGGAGCAGCAAGAGCAAGCCCAAGGACCCCAGCCAGCGCCCGCAGTCCAAGTCCCGGAA-3′, and 5′- TGCCACGCGTTCACTTGTCATCGTCATCCTTGTAGTCCATCACCGAGCATGAAGACT-3′. Farnesylation mutant C181S was constructed using primers 5′-FLAG-C181S (5′- ACTGGCGGCCGCGCCACCATGGACTACAAGGATGACGATGACAAGCCGCAGTCCAAGTCCCGGAA-3′) and 3′-FLAG-C181S (5′- TGCCACGCGTTCACATCACCGACGATGAAGACTTGCCTTGTGAAG-3′).
Cell culture and transformation assays
Primary cultures of CEF were prepared from White Leghorn embryos obtained from Charles River Breeding Laboratories (Preston, CT). For focus assays with infectious retroviral vectors, DNA was transfected into CEF by using the dimethyl sulfoxide-polybrene method (Bos et al., 1990; Chang et al., 1997; Duff and Vogt, 1969). CEF were infected with culturing medium containing the RCAS vector or recombinant RCAS viruses. The cultures were overlaid with nutrient agar and stained with crystal violet on day 14. Rapamycin (Calbiochem, La Jolla, CA) was added directly to the nutrient agar overlay of the infected cells. Controls received DMSO vehicle instead of rapamycin. Infected cells were fed every other day with nutrient agar for 14 days and stained with crystal violet. The ability of cells to grow in soft nutrient agar suspension was tested according to published techniques (Cohen et al., 2001; Himly et al., 1998; Sonderegger and Vogt, 2003). Colonies were counted after 14 days of incubation. Pictures were taken using a Zeiss Axiovert 35 microscope and a Canon 300D digital camera. Mean cell size was determined by a CASY Cell Counter (Scharfe System GmbH) with live cells, or by flow cytometry forward scatter analysis of fixed cells, or by a Beckman-Coulter Vi-CELL XR cell viability analyzer. FACS analysis was following standard procedures.
Serum starvation and insulin stimulation
For serum starvation, CEF were cultured in Ham's F-10 medium (Sigma, St. Louis, MO) containing 0.5% FCS and 0.1% chicken serum for 40 h. Then the cells were stimulated with 10 nM insulin (Sigma) for 15 min. For rapamycin treatment, rapamycin (10 ng/ml) was added to the culture 2 h before the addition of insulin.
Western blot analysis
CEF were grown to confluence, rinsed with PBS, and lysed in protein lysis buffer containing 1% Nonidet P-40, 10% glycerol, 20 mM Tris (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 1 mM PMSF, 50 mM NaF, 1 mM DTT, 50 mM β-glycerophosphate, 1 mM Na3VO4, and 1× Cømplete protease inhibitor mixture (Roche, Pleasanton, CA). Lysates containing 30-40 μg of protein were separated on a 4-20% Tris-Glycine SDS polyacrylamide gel (Invitrogen, Carlsbad, CA), and transferred to an Immobilon P membrane (Millipore, Billerica, MA). The membrane was blocked with 5% nonfat dry milk in 0.1% TBS-T (0.1% Tween-20 in 1× Tris-buffered saline) for 1 h at room temperature and incubated overnight at 4°C with primary antibodies (5% BSA in 0.1% TBS-T for polyclonal antibody, and 5% nonfat dry milk in 0.1% TBS-T for monoclonal antibody). After rinsing with 0.1% TBS-T, the blots were incubated with secondary antibodies in 5% nonfat dry milk/0.1% TBS-T for 1 h at room temperature. The reactive bands were visualized by chemiluminescence (Pierce, Rockford, IL). Anti-Akt, anti-phospho-Akt (Ser-473, and Thr-308), anti-S6K, anti-phospho-S6K (Thr-389), anti-4E-BP1, anti-phospho-4E-BP1 (Ser-65), anti-FOXO-1, anti-phospho-FOXO-1 (Ser-256) antibodies were purchased from Cell Signaling Technology (Beverly, MA), anti-tubulin antibody from ICN (Costa Mesa, CA), anti-FLAG antibody from Sigma.
Protein binding to large multilamellar vesicles (LMVs)
Assays of protein binding to LMVs (large multilamellar vesicles) follow the procedure described in a previous publication (Davletov et al., 1998). A mixture of brain phospholipids (10 mg/ml, Avanti Polar Lipids Inc., Alabaster, AL) was vortexed to form LMVs. A 100 μg protein sample prepared with passive lysis buffer (Promega) was mixed with 320 μg lipids and diluted into 500 μl HEPES/NaCl buffer (50 mM HEPES, 100 mM NaCl, pH 7.2). After 15 min incubation at room temperature, the lipids were spun down with associated proteins at 10,700 rpm for 10 min. The pellet, designated as M for membrane-bound, was washed once with the HEPES/NaCl buffer and dissolved in protein sample buffer, whereas the supernatant was transferred to another tube and an equal volume of 40% TCA (trichloroacetic acid) was added and further incubated on ice for 15 min. This portion of cytosolic proteins, designated as C for cytosolic, was spun down and washed with 500 μl HEPES/NaCl buffer, then dissolved in protein sample buffer. Sample M and C were separated on a 4-20% Tris-Glycine SDS polyacrylamide gel. Blots were probed with anti-FLAG antibody and the intensity of the bands was quantified by the ImageJ software (NIH). The ratios of membrane-bound to cytosolic proteins are shown in Fig. 8c.
Immunofluorescence
Cells grown overnight on glass coverslips were washed once with phosphate-buffered saline (PBS), fixed with 3.7% formaldehyde for 30 min, and permeabilized with PBS containing 0.1% Triton X-100 (Sigma) for 30 min. After a wash with PBS, the coverslips were incubated with 10% goat serum (Sigma) for 1 h at room temperature in a humidified container followed by incubation with mouse monoclonal anti-FLAG antibody (Sigma) for another hour. Coverslips were washed with PBS three times and incubated with fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin G (Sigma) for 1 h. During the last 5 min, 4,6-diamidino-2-phenylindole (DAPI) was added. The coverslips were again washed three times with PBS and mounted on glass slides using Slowfade mounting medium (Molecular Probes, Eugene, OR). Primary and secondary antibodies were used at a final dilution of 1:100, and the final concentration of DAPI was 2 ng/μl. Anti-ERp72 and anti-GRASp65 antibodies were purchased from Calbiochem. Pictures were taken with confocal laser scanning microscopy.
Acknowledgments
The authors thank Lynn Ueno, Dr. Marco Gymnopoulos, and Dr. William B. Kiosses (Core Microscopy, The Scripps Research Institute) for expert technical assistance. Work of the authors is supported by grants from the National Cancer Institute. This manuscript is number 19286 of The Scripps Research Institute.
Abbreviations
- 4E-BP1
eukaryotic initiation factor 4E-binding protein 1
- Akt
v-akt murine thymoma viral oncogene homolog
- CEF
chicken embryonic fibroblast
- eIF4E
eukaryotic initiation factor 4E
- ER
endoplasmic reticulum
- FKBP38
FK506-binding protein 38
- FOXO-1
forkhead box protein class-O 1
- GADD45
growth arrest- and DNA damage-inducible protein 45
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- PI3K
phosphatidylinositol 3-kinase
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- Rap2
Ras-related protein-2
- Ras
rat sarcoma
- Rheb
Ras-homolog enriched in brain
- S6K
ribosomal protein S6 kinase
- TCTP
translationally controlled tumor protein
- TOR
target of rapamycin
- TSC
tuberous sclerosis complex
- YB-1
Y box-binding protein 1
References
- Ahmed NN, Grimes HL, Bellacosa A, Chan TO, Tsichlis PN. Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc Natl Acad Sci U S A. 1997;94:3627–32. doi: 10.1073/pnas.94.8.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. Curr Opin Genet Dev. 1998;8:55–62. doi: 10.1016/s0959-437x(98)80062-2. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol. 1997a;7:776–89. doi: 10.1016/s0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997b;7:261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Aoki M, Batista O, Bellacosa A, Tsichlis P, Vogt PK. The akt kinase: molecular determinants of oncogenicity. Proc Natl Acad Sci U S A. 1998;95:14950–5. doi: 10.1073/pnas.95.25.14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki M, Jiang H, Vogt PK. Proteasomal degradation of the FoxO1 transcriptional regulator in cells transformed by the P3k and Akt oncoproteins. Proc Natl Acad Sci U S A. 2004;101:13613–7. doi: 10.1073/pnas.0405454101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki M, Vogt PK. Retroviral oncogenes and TOR. Curr Top Microbiol Immunol. 2004;279:321–38. doi: 10.1007/978-3-642-18930-2_19. [DOI] [PubMed] [Google Scholar]
- Bader AG, Felts KA, Jiang N, Chang HW, Vogt PK. Y box-binding protein 1 induces resistance to oncogenic transformation by the phosphatidylinositol 3-kinase pathway. Proc Natl Acad Sci U S A. 2003;100:12384–9. doi: 10.1073/pnas.2135336100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader AG, Vogt PK. Inhibition of protein synthesis by Y box-binding protein 1 blocks oncogenic cell transformation. Mol Cell Biol. 2005;25:2095–106. doi: 10.1128/MCB.25.6.2095-2106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader AG, Vogt PK. Phosphorylation by Akt disables the anti-oncogenic activity of YB-1. Oncogene. 2007 doi: 10.1038/sj.onc.1210719. [DOI] [PubMed] [Google Scholar]
- Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, et al. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977–80. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- Basso AD, Mirza A, Liu G, Long BJ, Bishop WR, Kirschmeier P. The farnesyl transferase inhibitor (FTI) SCH66336 (lonafarnib) inhibits Rheb farnesylation and mTOR signaling. Role in FTI enhancement of taxane and tamoxifen anti-tumor activity. J Biol Chem. 2005;280:31101–8. doi: 10.1074/jbc.M503763200. [DOI] [PubMed] [Google Scholar]
- Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–6. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bister K, Hayman MJ, Vogt PK. Defectiveness of avian myelocytomatosis virus MC29: isolation of long-term nonproducer cultures and analysis of virus-specific polypeptide synthesis. Virology. 1977;82:431–48. doi: 10.1016/0042-6822(77)90017-4. [DOI] [PubMed] [Google Scholar]
- Bos TJ, Monteclaro FS, Mitsunobu F, Ball AR, Jr, Chang CH, Nishimura T, et al. Efficient transformation of chicken embryo fibroblasts by c-Jun requires structural modification in coding and noncoding sequences. Genes Dev. 1990;4:1677–87. doi: 10.1101/gad.4.10.1677. [DOI] [PubMed] [Google Scholar]
- Briaud I, Dickson LM, Lingohr MK, McCuaig JF, Lawrence JC, Rhodes CJ. Insulin receptor substrate-2 proteasomal degradation mediated by a mammalian target of rapamycin (mTOR)-induced negative feedback downregulates protein kinase B-mediated signaling pathway in beta-cells. J Biol Chem. 2005;280:2282–93. doi: 10.1074/jbc.M412179200. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–8. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Buerger C, DeVries B, Stambolic V. Localization of Rheb to the endomembrane is critical for its signaling function. Biochem Biophys Res Commun. 2006;344:869–80. doi: 10.1016/j.bbrc.2006.03.220. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Tzivion G, Nasrin N, Ogg S, Dore J, Ruvkun G, et al. Phosphatidylinositol 3-kinase signaling inhibits DAF-16 DNA binding and function via 14-3-3-dependent and 14-3-3-independent pathways. J Biol Chem. 2001;276:13402–10. doi: 10.1074/jbc.M010042200. [DOI] [PubMed] [Google Scholar]
- Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem. 2003;278:32493–6. doi: 10.1074/jbc.C300226200. [DOI] [PubMed] [Google Scholar]
- Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- Chang HW, Aoki M, Fruman D, Auger KR, Bellacosa A, Tsichlis PN, et al. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 1997;276:1848–50. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- Clark GJ, Kinch MS, Rogers-Graham K, Sebti SM, Hamilton AD, Der CJ. The Ras-related protein Rheb is farnesylated and antagonizes Ras signaling and transformation. J Biol Chem. 1997;272:10608–15. doi: 10.1074/jbc.272.16.10608. [DOI] [PubMed] [Google Scholar]
- Cohen SB, Waha A, Gelman IH, Vogt PK. Expression of a downregulated target, SSeCKS, reverses v-Jun-induced transformation of 10T1/2 murine fibroblasts. Oncogene. 2001;20:141–6. doi: 10.1038/sj.onc.1204077. [DOI] [PubMed] [Google Scholar]
- Cunningham MA, Zhu Q, Hammond JM. FoxO1a can alter cell cycle progression by regulating the nuclear localization of p27kip in granulosa cells. Mol Endocrinol. 2004;18:1756–67. doi: 10.1210/me.2004-0071. [DOI] [PubMed] [Google Scholar]
- Dan HC, Sun M, Yang L, Feldman RI, Sui XM, Ou CC, et al. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J Biol Chem. 2002;277:35364–70. doi: 10.1074/jbc.M205838200. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–27. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Davletov B, Perisic O, Williams RL. Calcium-dependent membrane penetration is a hallmark of the C2 domain of cytosolic phospholipase A2 whereas the C2A domain of synaptotagmin binds membranes electrostatically. J Biol Chem. 1998;273:19093–6. doi: 10.1074/jbc.273.30.19093. [DOI] [PubMed] [Google Scholar]
- Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10:262–7. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- Drenan RM, Liu X, Bertram PG, Zheng XF. FKBP12-rapamycin-associated protein or mammalian target of rapamycin (FRAP/mTOR) localization in the endoplasmic reticulum and the Golgi apparatus. J Biol Chem. 2004;279:772–8. doi: 10.1074/jbc.M305912200. [DOI] [PubMed] [Google Scholar]
- Duff RG, Vogt PK. Characteristics of two new avian tumor virus subgroups. Virology. 1969;39:18–30. doi: 10.1016/0042-6822(69)90344-4. [DOI] [PubMed] [Google Scholar]
- Furukawa-Hibi Y, Yoshida-Araki K, Ohta T, Ikeda K, Motoyama N. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J Biol Chem. 2002;277:26729–32. doi: 10.1074/jbc.C200256200. [DOI] [PubMed] [Google Scholar]
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–66. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- Gromov PS, Madsen P, Tomerup N, Celis JE. A novel approach for expression cloning of small GTPases: identification, tissue distribution and chromosome mapping of the human homolog of rheb. FEBS Lett. 1995;377:221–6. doi: 10.1016/0014-5793(95)01349-0. [DOI] [PubMed] [Google Scholar]
- Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, et al. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314:1458–61. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hers I, Tavare JM. Mechanism of feedback regulation of insulin receptor substrate-1 phosphorylation in primary adipocytes. Biochem J. 2005;388:713–20. doi: 10.1042/BJ20041531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himly M, Foster DN, Bottoli I, Iacovoni JS, Vogt PK. The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology. 1998;248:295–304. doi: 10.1006/viro.1998.9290. [DOI] [PubMed] [Google Scholar]
- Hsu YC, Chern JJ, Cai Y, Liu M, Choi KW. Drosophila TCTP is essential for growth and proliferation through regulation of dRheb GTPase. Nature. 2007;445:785–8. doi: 10.1038/nature05528. [DOI] [PubMed] [Google Scholar]
- Hughes SH, Greenhouse JJ, Petropoulos CJ, Sutrave P. Adaptor plasmids simplify the insertion of foreign DNA into helper-independent retroviral vectors. J Virol. 1987;61:3004–12. doi: 10.1128/jvi.61.10.3004-3012.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im E, von Lintig FC, Chen J, Zhuang S, Qui W, Chowdhury S, et al. Rheb is in a high activation state and inhibits B-Raf kinase in mammalian cells. Oncogene. 2002;21:6356–65. doi: 10.1038/sj.onc.1205792. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Karbowniczek M, Cash T, Cheung M, Robertson GP, Astrinidis A, Henske EP. Regulation of B-Raf kinase activity by tuberin and Rheb is mammalian target of rapamycin (mTOR)-independent. J Biol Chem. 2004;279:29930–7. doi: 10.1074/jbc.M402591200. [DOI] [PubMed] [Google Scholar]
- Karbowniczek M, Robertson GP, Henske EP. Rheb inhibits C-raf activity and B-raf/C-raf heterodimerization. J Biol Chem. 2006;281:25447–56. doi: 10.1074/jbc.M605273200. [DOI] [PubMed] [Google Scholar]
- Kim JE, Chen J. Cytoplasmic-nuclear shuttling of FKBP12-rapamycin-associated protein is involved in rapamycin-sensitive signaling and translation initiation. Proc Natl Acad Sci U S A. 2000;97:14340–5. doi: 10.1073/pnas.011511898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903–10. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- Li Y, Inoki K, Guan KL. Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol Cell Biol. 2004;24:7965–75. doi: 10.1128/MCB.24.18.7965-7975.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005a;15:702–13. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- Long X, Ortiz-Vega S, Lin Y, Avruch J. Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J Biol Chem. 2005b;280:23433–6. doi: 10.1074/jbc.C500169200. [DOI] [PubMed] [Google Scholar]
- Mach KE, Furge KA, Albright CF. Loss of Rhb1, a Rheb-related GTPase in fission yeast, causes growth arrest with a terminal phenotype similar to that caused by nitrogen starvation. Genetics. 2000;155:611–22. doi: 10.1093/genetics/155.2.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–62. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–7. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Patel PH, Thapar N, Guo L, Martinez M, Maris J, Gau CL, et al. Drosophila Rheb GTPase is required for cell cycle progression and cell growth. J Cell Sci. 2003;116:3601–10. doi: 10.1242/jcs.00661. [DOI] [PubMed] [Google Scholar]
- Sabatini DM, Barrow RK, Blackshaw S, Burnett PE, Lai MM, Field ME, et al. Interaction of RAFT1 with gephyrin required for rapamycin-sensitive signaling. Science. 1999;284:1161–4. doi: 10.1126/science.284.5417.1161. [DOI] [PubMed] [Google Scholar]
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270:815–22. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- Saito K, Araki Y, Kontani K, Nishina H, Katada T. Novel role of the small GTPase Rheb: its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J Biochem (Tokyo) 2005;137:423–30. doi: 10.1093/jb/mvi046. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol. 2003;5:566–71. doi: 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- Shah OJ, Hunter T. Turnover of the active fraction of IRS1 involves raptor-mTOR- and S6K1-dependent serine phosphorylation in cell culture models of tuberous sclerosis. Mol Cell Biol. 2006;26:6425–34. doi: 10.1128/MCB.01254-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–6. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Sonderegger CK, Vogt PK. Binding of the corepressor TLE1 to Qin enhances Qin-mediated transformation of chicken embryo fibroblasts. Oncogene. 2003;22:1749–57. doi: 10.1038/sj.onc.1206308. [DOI] [PubMed] [Google Scholar]
- Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, et al. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–31. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–4. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, et al. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003;5:559–65. doi: 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF, et al. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–70. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Nakagawa M, Young SG, Yamanaka S. Differential membrane localization of ERas and Rheb, two Ras-related proteins involved in the phosphatidylinositol 3-kinase/mTOR pathway. J Biol Chem. 2005;280:32768–74. doi: 10.1074/jbc.M506280200. [DOI] [PubMed] [Google Scholar]
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–68. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- Urano J, Comiso MJ, Guo L, Aspuria PJ, Deniskin R, Tabancay AP, Jr, et al. Identification of novel single amino acid changes that result in hyperactivation of the unique GTPase, Rheb, in fission yeast. Mol Microbiol. 2005;58:1074–86. doi: 10.1111/j.1365-2958.2005.04877.x. [DOI] [PubMed] [Google Scholar]
- Urano J, Ellis C, Clark GJ, Tamanoi F. Characterization of Rheb functions using yeast and mammalian systems. Methods Enzymol. 2001;333:217–31. doi: 10.1016/s0076-6879(01)33058-6. [DOI] [PubMed] [Google Scholar]
- Urano J, Tabancay AP, Yang W, Tamanoi F. The Saccharomyces cerevisiae Rheb G-protein is involved in regulating canavanine resistance and arginine uptake. J Biol Chem. 2000;275:11198–206. doi: 10.1074/jbc.275.15.11198. [DOI] [PubMed] [Google Scholar]
- van Slegtenhorst M, Carr E, Stoyanova R, Kruger WD, Henske EP. Tsc1+ and tsc2+ regulate arginine uptake and metabolism in Schizosaccharomyces pombe. J Biol Chem. 2004;279:12706–13. doi: 10.1074/jbc.M313874200. [DOI] [PubMed] [Google Scholar]
- Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–40. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Sanders LK, Kaufmann WE, Yee W, Barnes CA, Nathans D, et al. rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J Biol Chem. 1994;269:16333–9. [PubMed] [Google Scholar]
- Yan L, Findlay GM, Jones R, Procter J, Cao Y, Lamb RF. Hyperactivation of mammalian target of rapamycin (mTOR) signaling by a gain-of-function mutant of the Rheb GTPase. J Biol Chem. 2006;281:19793–7. doi: 10.1074/jbc.C600028200. [DOI] [PubMed] [Google Scholar]
- Yang W, Tabancay AP, Jr, Urano J, Tamanoi F. Failure to farnesylate Rheb protein contributes to the enrichment of G0/G1 phase cells in the Schizosaccharomyces pombe farnesyltransferase mutant. Mol Microbiol. 2001;41:1339–47. doi: 10.1046/j.1365-2958.2001.02599.x. [DOI] [PubMed] [Google Scholar]
- Yee WM, Worley PF. Rheb interacts with Raf-1 kinase and may function to integrate growth factor- and protein kinase A-dependent signals. Mol Cell Biol. 1997;17:921–33. doi: 10.1128/mcb.17.2.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shu L, Hosoi H, Murti KG, Houghton PJ. Predominant nuclear localization of mammalian target of rapamycin in normal and malignant cells in culture. J Biol Chem. 2002;277:28127–34. doi: 10.1074/jbc.M202625200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5:578–81. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]