

Abstract
Organic anion transporting polypeptide (OATP) 1B1 and 1B3 are widely acknowledged as important and rate-limiting to the hepatic uptake of many drugs in clinical use. Accordingly, to better understand the in vivo relevance of OATP1B transporters, targeted disruption of murine Slco1b2 gene was carried out. Interestingly, Slco1b2−/− mice were fertile, developed normally, and did not exhibit any overt phenotypic abnormalities. We confirmed the loss of Oatp1b2 expression in liver using real-time PCR, Western Blot analysis and immunohistochemistry. Oatp1a4 and Oatp2b1, but not Oatp1a1 expression was greater in female Slco1b2−/− mice, but expression of other non-oatp transporters did not significantly differ between wildtype and Slco1b2−/− males. Total bilirubin level was elevated by 2-fold in the Slco1b2−/− mice despite that liver enzymes ALT and AST were normal. Pharmacological characterization was carried out using two prototypical substrates of human OATP1B1 and 1B3, rifampin and pravastatin. After a single intravenous dose of rifampin (1mg/kg), a 1.7-fold increase in plasma AUC was observed while the liver-to-plasma ratio was reduced by 5-fold, and nearly 8-fold when assessed at steady -state conditions after 24 hours of continuous subcutaneous (SC) infusion in Slco1b2−/− mice. Similarly, continuous SC-infusion at low dose rate (8 μg/hr) or high dose rate (32 μg/hr) pravastatin resulted in a 4-fold lower liver-plasma ratio in the in Slco1b2−/− mice. This is the first report of altered drug disposition profile in the Slco1b2 knockout mice and suggests the utility of this model for understanding the in vivo role of hepatic OATP transporters in drug disposition.
INTRODUCTION
Carrier-mediated, membrane transport is now widely appreciated as a critical determinant of drug uptake, distribution and elimination (Ho and Kim, 2005). The organic anion transporting polypeptide (OATP) transporters belong to a large family of membrane proteins that mediate the sodium-independent cellular uptake of a variety of amphipathic compounds, including hormones, bile acids, eicosanoids, environmental toxins, and many drugs in clinical use today (Tirona and Kim, 2007). Members of this family are expressed in a variety of organs of relevance to drug disposition or response, such as liver, kidney, brain and intestine (Marzolini et al., 2004). One subset of the OATP transporters that has received significant attention in relation to hepatic drug uptake is the human OATP1B subfamily. There are two members, OATP1B1 (SLC21A6, OATP-C, OATP2, LST-1) and OATP1B3 (SLC21A8, OATP-8, LST-2) in humans, which share 80% sequence homology (Hagenbuch and Meier, 2004). Both transporters have been described to be highly expressed in human liver (Abe et al., 2001;Konig et al., 2000;Abe et al., 1999;Ho et al., 2006) and are localized to the basolateral membrane, facilitating the hepatocellular uptake of endo- and xenobiotic substrates prior to their metabolism and efflux from the liver (Shimizu et al., 2005). In rodents, there is only one member of the Oatp1b subfamily. This transporter termed oatp1b2 (Slco1b2, Lst-1, oatp4,), has been identified in rat and mouse and is thought to be the closest ortholog of both human OATP1B1 and 1B3 (Cattori et al., 2000;Choudhuri et al., 2000;Ogura et al., 2000).
Although a number of in vitro and cell-based model systems for the study of OATP transporters exit, as yet a murine Slco knockout mouse model has not been reported. The lack of such a model has prevented the delineation of the overall contribution of a given Oatp transporter to the organ-specific elimination of drugs shown to be substrates of multiple Oatp transporters in vitro.
Since the mouse genome contains only one Slco1b subfamily member, we hypothesized that the targeted disruption of this transporter should allow for a better delineation and extrapolation of the in vivo relevance of human OATP1B1 and OATP1B3 to the hepatic uptake of substrate drugs. Accordingly, we now report on the generation of a Slco1b2 knockout mouse model. In addition to assessing the relative impact of this transporter to alterations of liver function or phenotype, we wanted to evaluate the utility of this model in terms of clarifying the relevance of Oatp1b2 in the hepatic elimination of prototypical substrates of human OATP1B1 and OATP1B3, such as rifampin and pravastatin. Indeed studies from a number of laboratories including ours would suggest that OATP1B1 is a major transporter for rifampin (Tirona et al., 2003). Similarly, 3-hydroxy-3-methylgutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) have also been shown to be highly dependent on OATP1B1 and OATP1B3 to exert their effects on intracellular hepatic HMG-CoA reductase and thereby reduce cardiovascular events associated with hypercholesterolemia and atherosclerosis (Schachter, 2005). Pravastatin has been widely studied as a substrate for OATP transporters including OATP1B1 and recent studies have linked single nucleotide polymorphisms (SNPs) in SLCO1B1 as a major predictor of pravastatin pharmacokinetics (Nakai et al., 2001;Ho et al., 2007).
We now provide data that suggest loss of Oatp1b2 (Slco1b2) does not result in a major phenotype in terms of viability or liver function, but the ability to eliminate drugs such as pravastatin and rifampin is significantly compromised. Taken together, our findings suggest that the Slco1b2−/− mouse model may prove to be a useful in vivo model for assessing the contribution of OATP1B transporters to the hepatic uptake of drugs.
MATERIALS AND METHODS
Materials
Rifampin, diclofenac, dimethylsulfoxide, and pravastatin were obtained from Sigma (St. Louis, MO). Atorvastatin was obtained from Pfizer Global Research and Development (Ann Arbor, MI). Ammonium formate and all other chemical reagents were obtained from Sigma (St. Louis, MO). Isoflurane (IsoFlo) was obtained from Abbott laboratories (Abbott Park, IL). All other compounds used were reagents grade.
Targeting of Slco1b2 in mouse ES cells
The targeting vector was designed using the Slco1b2 genomic locus form the Celera mouse genomic sequence database and the mRNA sequence (NM_178235). As shown in figure 1 homologous recombination resulted in the deletion of a 6772 bp fragment containing exon 10 to 12 of the mouse Slco1b2 genomic sequence and insertion of a neomycin resistance cassette. The 5′- and the 3′- regions of homology were amplified from DBA1/lacJ mouse genomic DNA using the Expand High Fidelity PCR system (Roche, Laval, QC, Canada). The following primer pairs (5′-OAC-6141F, 5′-ggatccgagttgtcttttctgagtgttagg-3′ and 5′-OAC-9873R, 5′-ggatccatgacagttgtctgctcattgc-3′; and 3′-′OAC-17027F, 5′-ctcgagatgtttggctgagagcatctgc-3′ and 3′-OAC-21988R, 5′-gcggccgctcatcttcatagcaaccctttcc-3′ were used to amplify a 4139 bp and 4962 bp fragment of the genomic DNA, respectively. The resulting PCR fragments were cloned into the pCR 2.1-TOPO (Invitrogen, Carlsbad, CA). After sequence verification, the homologous fragments were cloned into the backbone of the targeting vector plasmid that contained both a neomycin (neo) and a thymidine kinase (tk) expression cassette (see figure 1). Subsequently the linearized Slco1b2 targeting vector was electroporated into DBA1/lacJ ES cells. Those cells were cultured in the presence of G418 (positive selection due to neomycin cassette) and gancyclovir (negative selection due to thymidine kinase cassette) for selecting the correctly targeted ES cells.
Figure 1.
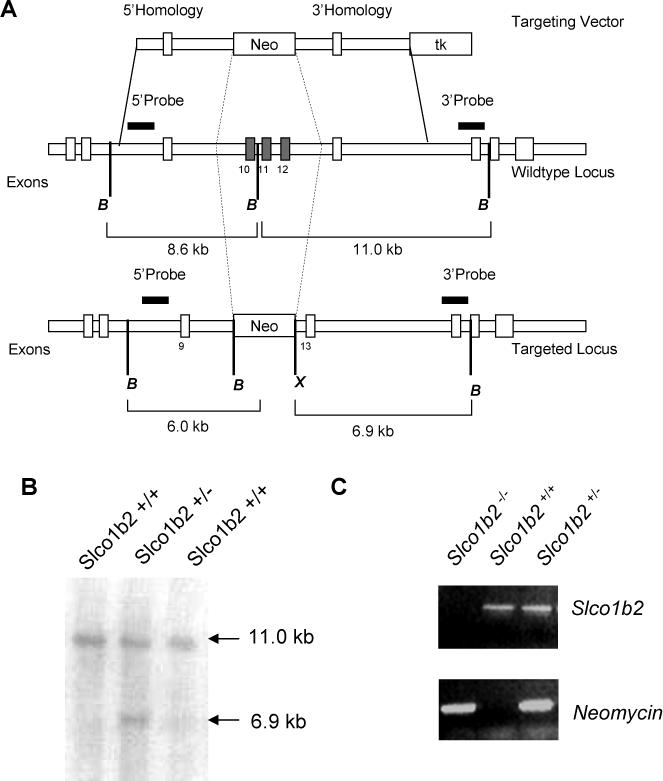
The Slco1b2-knockout mouse was generated by excision of exon 10 to 12 of the Slco1b2 gene locus by standard targeting methods. The targeting vector consisted of 5′ and 3′ sequence homology arms flanking the neomycin selection cassette (A). For negative selection with gancyclovir, a thymidine kinase cassette was included in the targeting vector (A). Digest of the wildtype locus with BamHI results in a 11.0 kb fragment detected by Southern blotting using a 3′ probe for detection whereas successful targeting with deletion of exon 10 to 12 resulted in a 6.9 kb fragment after digest with BamHI and XhoI (B). For genotyping of the resulting mice, PCRs detecting a fragment of the Slco1b2 gene locus or neomycin were performed (C).
Southern Blot Screening of ES Cells for Slco1b2 Homologous Recombination
Southern Blot analysis was performed to screen for successful integration of the targeting vector that resulted in the deletion of exon 10 to exon 12 and insertion of the neomycin cassette. Briefly, genomic DNA was isolated from ES cells after electroporation with the Slco1b2 targeting vector and selection with G418/gancyclovir. DNA was digested with BamHI and XhoI. After electrophoretic separation on a 0.7% agarose gel (BioProducts, Rockland, Maine) the DNA was transferred to Hybond N+ nylon membrane (Amersham Pharmacia Biotech, Buckinghamshire, UK). For detection of the genomic DNA fragments two probes were used to screen for homologous recombination between the Slco1b2 targeting vector and endogenous locus. The 3′-Southern Probe was a 717 bp-fragment (OAC-23209F, 5′-cacttggaagcaaacattagtcc-3′ and OAC-23926R, 5′-taatccttgcaggaaagatacc-3′) that recognized an 11.0 kb endogenous Slco1b2 BamHI fragment and also a 6.9 kb BamHI / XhoI fragment in a targeted clone. Clones demonstrating targeting with the 3′ probe were then confirmed by Southern analysis using a second probe spanning 524 bp of the endogenous locus (OAC-5660F, 5′-tatctgtcgctgtaccagtacc-3′ and OAC-6184R, 5′-gatggttcactggagtactctgc-3′).
ES cell microinjection and identification of homozygote Slco1b2 knockout mice
Slco1b2 targeted ES cells (15−20 cells) were microinjected into blastocyst stage (3.5 day) C57BL/6 embryos (Charles River Laboratories, Wilmington, MA).. Surviving embryos were implanted into the uteri of day 2.5 pseudopregnant CD-1 (Charles River Laboratories) females. Chimeric males were identified from the offspring of these injections by the presence of mixed coat color, black (C57BL/6 host derived) and brown (Slco1b2 ES cell derived). Chimeric males were bred to wild type DBA1/LacJ females to identify Slco1b2 ES cell derived germline offspring. DNA was isolated from tail biopsies of the germline offspring and genotyped by PCR (Neo-833F, 5′-gcaggatctcctgtcatctcacc-3′ and Neo-1023R, 5′-gatgctcttcgtccagatcatcc-3′) for the presence (heterozygous, +/−) or absence (wild type, +/+) of the neomycin cassette. The Neo-833F and Neo-1023R oligonucleotide set amplifies a 190 bp fragment. Slco1b2 heterozygous males and females were bred together to produce homozygous (−/−) knockouts. Offspring were genotyped by PCR using 2 oligonucleotide sets, one being the Neo set and the other a set specific for part of the Slco1b2 knockout region. The Slco1b2 oligonucleotide set (OAC-15409F, 5′-tggacaatgtgcaatgggagc-3′ and OAC-15906R, 5′-gaaagagctgattagagatacg-3′) amplifies a 497 bp fragment contained within the knockout region. Hence, by genotyping using these 2 oligonucleotide sets, a knockout (−/−) animal would be negative for the Slco1b2 PCR while positive for the Neo PCR. A heterozygous (+/−) mouse would be positive for both PCR sets. And a wild type (+/+) mouse would be positive for the Slco1b2 PCR while negative for the neo PCR.
Animals
Mice were housed in an AAALAC–accredited (Association for Assessment and Accreditation of Laboratory Animal Care International) facility and handled according to Pfizer Global Research guidelines complying with the U.S. Public Health Service Policy for the Care and Use of Laboratory Animals. Slco1b2 knockout and wildtype (DBA1/lacJ) mice were obtained from Pfizer Groton with breeding colony housing and expansion provided by Charles River Laboratories. All mice used in this report were between 9 and 14 weeks of age. Animals were kept in a temperature-controlled environment with a 12-h light/dark cycle and received a standard diet and water ad libitum.
Clinical biochemistry
Serum separator tubes containing mouse blood collected at necropsy were allowed to stand at room temperature until clotted (approximately 30 minutes). Samples were then centrifuged for at least 8 minutes at <3500 rpm (2851 g) at 2−8°C to separate serum from the blood cells, minimizing changes in the electrolytes and preventing hemolysis of the samples. Samples were analyzed using the Bayer Advia 2400 Chemistry System (Tenley Town, NY). Advia 2400 clinical chemistry assay methodology is based on the change in color from the reaction of the analyte in the sample fluid with method-specific reagents, and electrolytes are measured by a potentiometric procedure that uses ion-selective electrodes (ISE). Clinical laboratory data was collected using Cerner version 8, and treatment comparisons were performed by pairwise comparison within one-factor analysis of variance (ANOVA).
Quantitative real-time PCR in mouse liver
Liver tissue was harvested from wildtype and Slco1b2−/− mice, snap frozen and stored at −80°C until isolation of total RNA. After mechanical homogenization of the tissue, RNA was isolated and purified using the Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA). following the manufacturer’s instructions. Subsequently, the amount and integrity of the isolated RNA was determined using an Agilent Bioanalyzer and the Agilent RNA 6000 Series II Nano Lab on Chip assay (Agilent, Santa Clara, CA). RNA was stored at −80°C.
Real-time PCR
TaqMan® Reverse Transcription Reagents supplied by Applied Biosystems (Foster City, CA,) were used for reverse transcription of total RNA. 2 μg of total RNA were transcribed in a 50 μl reaction containing 1× TaqMan RT-buffer, 5.5 mM MgCl2, 500 μM dNTPs, 2.5 μM random hexamers, 0.4 U/μl RNase inhibitor, and 1.25 U/μl Multiscribe Reverse Transcriptase. The reaction was performed under following thermocycler conditions: 25°C for 10 min, 48°C for 30 min and 95°C for 5 min. The resulting cDNA was used for quantitative real-time PCR.
Expression of Slco1b2 (Oatp1b2), Slco1a1 (Oatp1a1) , Slco1a4 (Oatp1a4), and 18S rRNA were determined using the pre-developed TaqMan© assays Mm00451513_m1, Mm00649796_m, Mm00453136_m1 and Hs99999901_s1, respectively. Expression of other transporters was determined using SYBR green assays. Primers are summarized in table 1. For all assays the recommended TaqMan cycler conditions were used.
Table 1.
Primers used for quantification of hepatic transporter expression in mice.
| Gene |
Primer forward and reverse |
Reference |
|---|---|---|
| Abcc2 | 5′-CTGAGTGCTTGGACCAGTGA-3′ | - |
| |
5′-CAAAGTCTGGGGGAGTGTGT-3′ |
|
| Abcc3 | 5′-CGCTCTCAGCTCACCATCAT-3′ | - |
| |
5′-GGTCATCCGTCTCCAAGTCA-3′ |
|
| Abcc4 | 5′-TTAGATGGCCTCTGGTTCT-3′ | (Wagner et al., 2003) |
| |
5′-GCCCACAATTCCAACCTTT-3′ |
|
| Abcg2 | 5′-AATGGAGCACCTCAACCTG-3′ | (Han and Sugiyama, 2006) |
| |
5′-CCCATCACAACGTCATCTTG-3′ |
|
| Ostα | 5′-GTCTCAAGTGATGAACTGCCA-3′ | (Zollner et al., 2006) |
| |
5′-TTGAGTGCTGAGTCCAGGTC-3′ |
|
| Ostß | 5′-GTATTTTCGTGCAGAAGATGCG-3′ | (Zollner et al., 2006) |
| |
5′-TTTCTGTTTGCCAGGATGCTC-3′ |
|
| Abcb11 | 5′-GTTCAGTTCCTCCGTTCAAA-3′ | |
| |
5′-AAGCTGCACTGTCTTTTCAC-3′ |
|
| Slc10a1 | 5′-CACCATGGAGTTCAGCAAGA-3′ | (Wagner et al., 2005) |
| |
5′-AGCACTGAGGGGCATGATAC-3′ |
|
| Abcb1b | 5′-TGCTTATGGATCCCAGAGTGAC-3′ | |
| |
5′-TTGGTGAGGATCTCTCCGGCT-3′ |
|
| Slc22a7 | 5′-CAGCTCCGACACGTCTCACT-3′ | |
| 5′-TGTCTGGTACACGGTCAGCC-3′ |
Immunohistochemistry
Paraffin-embedded liver sections were deparaffinized in two changes of xylene (5min each) followed by stepwise rehydration in decreasing ethanol and two washes in ddH2O. Heat-induced epitope retrieval was performed by boiling the slides in citrate buffer (pH 6.0). After several rinses in ice-cold PBS the sections were incubated with 5% fetal bovine serum (FBS) diluted in phosphate buffered saline (PBS) for 1.5 hours, followed by an overnight incubation at 4°C with the primary antibody diluted 1:50 in 5% FBS-PBS. The primary antibody against oatp1b2 is a novel custom antibody raised in rabbits immunized against the following epitope specific for murine oatp1b2 KNPVTNPTTQEKQAPAN (Invitrogen). After several washes in PBS the slides were incubated with the secondary anti-rabbit antibody provided in the Vectastain Kit (Vector Laboratories, Burlington, ON, Canada), which was then visualized using AEC substrate provided by Vector Laboratories.
Crude membrane preparation from mouse liver tissue
Mouse liver tissue was homogenized in 5 mM Tris-HCl, pH 7.5 containing protease inhibitors (Sigma Aldrich) using a Potter Elvejhem homogenizer. After 2 × 20 strokes on ice, the homogenate was incubated on ice under continuous stirring. Subsequently, the homogenate was centrifuged at 9 000 × g for 20 min at 4°C. The supernatant was transferred to an ultracentrifuge tube and centrifuged for 45 min at 100,000 × g at 4°C. The resulting pellet was dissolved in 5 mM Tris-HCl supplemented with protease inhibitors. Protein content was determined by BCA method (Pierce, Rockford, IL).
Western Blot analysis
After separating the crude membrane fraction by SDS-PAGE using 4−12% gradient acrylamide gels (NuPAGE, Invitrogen), the separated proteins were electrotransferred to a nitrocellulose membrane using a tank blotting system. The transfer of protein was assessed by Ponceau S staining (Sigma-Aldrich). Subsequently, the membranes were incubated in 5% low fat milk powder in TBS-T (tris-buffered saline, 0.1% Tween-20) for 2 hours. Afterwards, the membrane was incubated with the anti-mouse Oatp1b2 antibody at a dilution of 1:2000 or anti-calnexin at a dilution of 1:4000. After overnight incubation at 4°C, several washing steps with TBS-T and an additional incubation for 1.5 h with 5% FBS in TBS-T the blot was incubated with an HRP-labeled anti-rabbit secondary antibody (BioRad, Hercules, CA) diluted 1:2000 for 2 h. Before visualizing the immobilized secondary antibody using ECL Plus Reagent (GE Healthcare) the blot was washed in several changes of TBS-T. The chemiluminescent signal was detected using the KODAK ImageStation 4000MM (Mandel, Guelph, ON, Canada).
Rifampin and pravastatin pharmacokinetics in Slco1b2−/− and wildtype mice
Male Slco1b2 knockout and wildtype mice were obtained from Pfizer Groton (bred in-house by Genetic Technology). Mice received an intravenous dose of rifampin (1 mg/kg in 0.5% DMSO in water) through the tail vein. At predetermined time points, mice were anesthetized with isoflurane, and blood samples were obtained by cardiac puncture and transferred to EDTA-containing tubes. Plasma was retrieved after centrifugation of blood. Liver tissues were also collected from animals and all samples were stored at −80°C until analyzed by LC-MS/MS.
Rifampin and pravastatin continuous subcutaneous infusion in Slco1b2−/−
Alzet mini-osmotic pumps (Model 201D) were purchased from Durect Corporation (Mountain View, CA; U.S.A). Male wildtype and Slco1b2 knockout mice (n=4/genotype) had the mini pump containing dosing solution implanted dorsally (rate of drug release 8 μl/hr). For rifampin determination, blood samples were collected by cardiac puncture 24 hours post pump insertion, samples were centrifuged (3,000 × g) to obtain plasma and livers were collected for storage at −80°C until analyzed by liquid chromatography, tandem mass spectrometry (LC-MS/MS). For pravastatin analysis, blood samples were obtained by cardiac puncture and transferred to tubes containing NaF 0.72 mg and potasium oxalate 0.58 mg. Plasma was obtained by centrifugation. Subsequently, 10% 6 M Ammonium Acetate, pH 4.5 was added to all pravastatin plasma samples. Liver tissues were reconstituted and homogenized in 6 M Ammonium Acetate (pH 4.5). All samples were stored at −80°C until analyzed by LC-MS/MS.
Quantitation of pravastatin and rifampin levels
LC-MS/MS analysis was carried out using a high-performance liquid chromatography system consisting of a LC-10AD Shimadzu pump with CTC PAL autosampler interfaced to a TSQ-Quantum Ultra Finnigan mass spectrometer. Rifampin and internal standard (diclofenac) were separated on an Atlantis dc18 5μm column (2.1 × 50 mm). The mobile phase consisted of solvent A (5 mM ammonium formate with 0.1% formic acid) and solvent B (acetonitrile). The gradient was as follows: solvent B was held at 10% for 0.3 minutes, ramped from 10% to 90% over 1.7 minutes, ramped from 90% to 95% over 0.8 minutes, and then immediately brought back down to 10% for re-equilibration. Total run time was 3.5 minutes with a flow rate of 0.30 mL/min. The mass spectrometer was operated in positive ion ESI for the detection of rifampin and diclofenac. Multiple reaction monitoring analysis was performed with the following transitions: m/z 823 → 791 (rifampin) and m/z 296 → 215 (diclofenac). Standard samples in the appropriate drug-free matrix were prepared yielding a concentration range from 9.75−10,000 ng/ml. Pravastatin, and the internal standard (atorvastatin) were separated on a Hypersil Gold column (2.1 × 50mm, Thermo-Fisher) by gradient elution. The mobile phase consisted of solvent A (5 mM ammonium formate in water) and solvent B (acetonitrile). The gradient was as follows: solvent B was held at 25% for 0.3 min., linearly ramped from 25% to 95% over 1.8 min., held at 95% for 0.7 min. and then immediately brought back to 25% for re-equilibration. Total run time was 3 minutes with a flow rate of 0.275 mL/min. The mass spectrometer was operated in negative ion ESI mode for the detection of pravastatin and atorvastatin. Multiple reaction monitoring was performed with the transitions m/z 423 → 101 for pravastatin and m/z 557 → 278 for atorvastatin. All raw data was processed using Analyst Software ver. 1.4.1 (Applied Biosystems/ MDS Sciex Inc., Mississauga, ON, Canada). Calibration standards were prepared from the high calibration standard by serial dilution to the following levels: 1000, 500, 125, 31.5, and 3.9 ng/mL in mouse plasma. Pravastatin standard curve samples were prepared in appropriate drug-free matrix yielding a concentration range from 3.9−2000 ng/mL.
Pharmacokinetic calculations and statistical analysis
Rifampin clearance (CL) after IV bolus was calculated as Dose/AUC, where AUC is the area under the plasma concentration-time profile from t=0 to ∞. Volume of distribution at steady-state (VSS) was calculated as (AUMC × Dose)/AUC2, where AUMC is the area under the moment curve. The Student’s t-test was used to evaluate statistical significance between Slco1b2−/− and wild-type mice in gene expression and in pharmacokinetic studies.
RESULTS
Targeted deletion and characterization of Slco1b2−/− mice
The Slco1b2−/− mice were generated by deleting a 6772 bp fragment spanning exon 10 to exon 12 in the genomic DNA locus (figure 1A) with replacement with a neomycin resistance gene cassette through homologous recombination in mouse embryonic stem cells derived from DBA1/LacJ mice. DNA obtained from ES cells resistant to neomycin and gancyclovir was used for Southern blot analysis using two different probes detecting a 6.9 kb -fragment compared to 11.0 kb fragments present in wildtype DNA (figure 1B). Two targeted ES cell clones were identified from 189 clones screened. Karyotype analysis was done on these clones and both were shown to have normal 40XY karyotypes. Genotyping was carried out performing PCRs specific for Slco1b2 and neomycin identifying homozygote and heterozygote offspring (figure 1C). Mice homozygous for the disrupted allele, designated Slco1b2−/− were born normally and appeared indistinguishable from their wild-type counterparts. No differences were found between litter size and growth rates for the Slco1b2−/− animals as compared with wild-type littermate controls. The resulting Slco1b2−/− mice displayed no obvious phenotype, and livers appear macroscopically and histologically normal.
Comparison of oatp1b2 expression in wildtype and Slco1b2−/− mice
We utilized a novel custom synthesized anti-mouse Oatp1b2-antibody to conduct Western blot analysis and noted the lack of Oatp1b2 protein (∼65 kDa) in the liver homogenate of Slco1b2−/− mice (figure 2A). Immunohistochemical staining of paraffin sections using the same antibody demonstrated clear expression of Oatp1b2 on the basal membrane facing the perisinusoidal space of hepatocytes in the wildtype but not in Slco1b2−/− mice (figures 2B and 2C). Moreover, quantitative real-time PCR analysis revealed that Slco1b2 mRNA expression was markedly lower in the Slco1b2−/−livers relative to the wildtype mice livers suggesting that transcript lacking the deleted exons are unstable or rapidly degraded. Note that detection of low level Slco1b2 transcript in the Slco1b2 knockout mice is not unexpected since the primers and probe used for detection encompass exons 3 and 4. Interestingly, wildtype female mice appeared to express higher mRNA levels of the transporter compared to wildtype males (mean relative expression of Slco1b2 ± SD; wildtype female 1.47±0.33; wildtype male 1.01±0.16, p<0.05).
Figure 2.
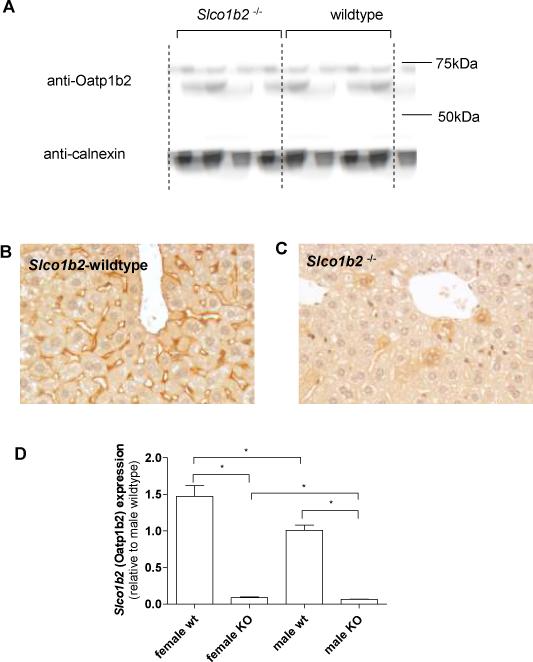
Detection of Oatp1b2 by Western Blot analysis revealed the absence of Oatp1b2 expression in Slco1b2−/− mice (A). Immunohistochemical staining of Oatp1b2 (red) in mouse liver showed expression of the transporter in the basal membrane of hepatocytes (B). In animals with targeted disruption of Slco1b2, expression of the transporter was not detectible (C). mRNA expression was assessed by real-time RT-PCR and showed a significant reduction of Slco1b2 in knock-out animals (D).
Serum biochemical analysis
Serum biochemical analyses did not reveal any significant changes in markers of liver function such as ALT or AST. Moreover no changes were observed in the serum levels of triglycerides, chloride, sodium, potassium, creatinine or urea nitrogen. However, a modest elevation of total bilirubin levels was detected in Slco1b2−/− mice (table 2). Further analysis revealed that most of the plasma bilirubin was conjugated to suggest Oatp1b2 may be involved in hepatic reuptake of conjugated bilirubin from circulation.
Table 2.
Serum biochemistry comparing wildtype and Slco1b2−/− animals.
| Parameter | wildtype animals | Slco1b2−/− animals |
|---|---|---|
| Triglycerides (mg/dL) | 119.00 ± 7.02 | 110.67 ± 9.87 |
| Aspartate Aminotransferase (U/L) | 129.00 ± 78.72 | 123.00 ± 38.66 |
| Alanine Aminotransferase (U/L) | 42.75 ± 5.62 | 66.5 ± 29.38 |
| Total Bilirubin (mg/dL) | 0.10 ± 0.00 | 0.23 ± 0.05 * |
| Chloride (mmol/L) | 111.75 ± 1.71 | 112.00 ± 2.16 |
| Sodium (mmol/L) | 149.00 ± 0.00 | 148.75 ± 0.96 |
| Potassium (mmol/L) | 5.50 ± 0.18 | 5.28 ± 0.22 |
| Creatinine (mg/dL) | 0.2 ± 0.0 | 0.2 ± 0.0 |
| Urea Nitrogen (mg/dL) | 21.50 ± 1.73 | 20.25 ± 0.50 |
Expression of other hepatic oatp transporters
To determine if compensatory changes in expression of other hepatic Oatp transporters occurred in the Slco1b2−/− mice, real-time PCR was performed to quantitate expression of Slco1a1, (Oatp1a1, Oatp1), Slco1a4 (Oatp1a4, Oatp2) and Slco2b1 (Oatp2b1, Oatp-b). Slco1al mRNA did not differ between wildtype and Slco1b2−/− animals (mean Slco1a1 expression relative to wildtype male ± SD; female wildtype 0.64 ± 0.26; female Slco1b2−/− 0.68 ± 0.1; male wildtype 1.02 ± 0.22; male Slco1b2−/− 1.22±0.32, n=5) (figure 3A). However, Slco1a4 (figure 3B; mean relative Slco1a4 expression ± SD; female wildtype 5.198 ± 1.417, female Slco1b2−/− 11.31 ± 5.24, p < 0.05, n=5) and oatp2b1 (figure 3C; mean relative Slco2b1 expression ± SD; female wildtype 1.97 ± 0.5; female Slco1b2−/− 3.21 ± 1.30; p<0.05, n=5) levels were significantly greater only in the female Slco1b2−/−. No differences in hepatic Slco1a4 expression were detected in male wildtype versus Slco1b2−/− mice (mean relative Slco1a4 expression ± SD; male wildtype 1.04 ± 0.32, male Slco1b2−/− male 1.49 ± 0.58, n=5) (figure 3B). Again, no difference in expression was noted for Slco2b1 expression in male wildtype versus Slco1b2−/− mice (mean relative Slco2b1 expression ± SD; male wildtype 1.01 ± 0.19; male Slco1b2−/− 1.33 ± 0.47, n=5). Expression of other hepatic transporters was measured including efflux transporters Abcc2, Abcc3, Abcc4 and Abcg2, Abcb1b and Abcb11, and the uptake transporters Slc10a1, Ostα, Ostβ and Slc22a7. Although modest gender-dependent differences were noted to be statistically different for Abbc4, Ostβ and Slc22a7, for the most part, no major differences were noted in the basal expressed levels of other uptake and efflux transporters (table 3).
Figure 3.
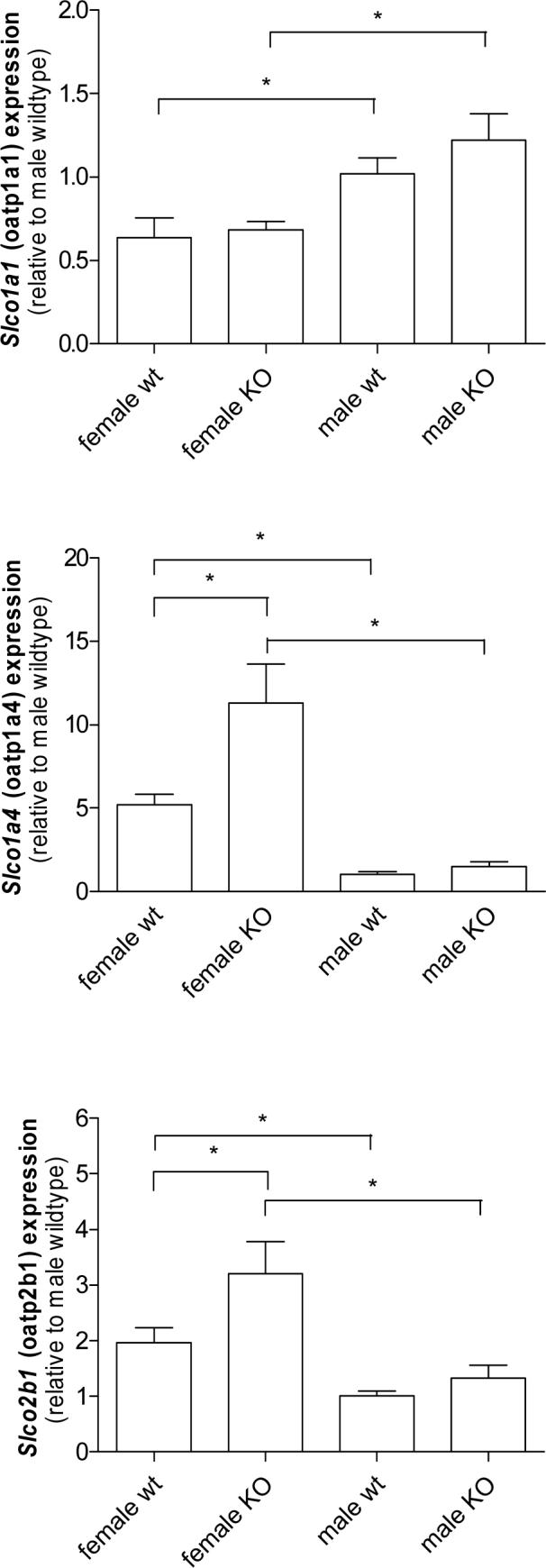
Hepatic mRNA expression of Slco1a1 (Oatp1a1) , Slco1a4 (Oatp1a4) and Slco2b1 (Oatp2b1) as determined by real-time PCR comparing wildtype and Slco1b2−/− animals. * p <0.05.
Table 3.
Expression of hepatic drug transporters in mouse liver comparing wildtype and Slco1b2-knockout animals.
| Wildtype | Slco1b2−/− | |||
|---|---|---|---|---|
| Female | male | female | male | |
| Abcc2 | 1.36 ± 0.56 | 1.01 ± 0.14 | 1.71 ± 0.58 | 0.90 ± 0.45 |
| Abcc3 | 1.99 ± 0.60 | 1.08 ± 0.36 | 2.83 ± 1.19 | 1.53 ± 0.35 |
| Abcc4 | 1.67 ± 0.29 | 1.03 ± 0.25 | 3.03 ± 0.47 (*) | 1.51 ± 0.63 |
| Abcg2 | 0.32 ± 0.06 | 1.01 ± 0.15 | 0.47 ± 0.14 | 1.11 ± 0.22 |
| Ostα | 1.05 ± 0.42 | 1.10 ± 0.47 | 1.94 ± 3.47 | 2.89 ± 3.47 |
| Ostβ | 3.65 ± 1.04 | 1.03 ± 0.29 | 3.61 ± 2.24 | 0.62 ± 0.18 (*) |
| Abcb11 | 0.71 ± 0.19 | 1.05 ± 0.33 | 0.98 ± 0.18 (*) | 0.79 ± 0.37 |
| Slc10a1 | 3.04 ± 1.51 | 1.20 ± 0.62 | 4.18 ± 3.93 | 1.87 ± 2.00 |
| Abcb1b | 0.41 ± 0.09 | 1.03 ± 0.26 | 0.75 ± 0.35 | 1.94 ± 0.35 |
| Slc22a7 | 1.76 ± 0.71 | 1.01 ± 0.12 | 1.16 ± 0.42 (*) | 0.75 ± 0.08 |
Data are presented as mean expression relative to wildtype male animals ± SD
p<0.05
Rifampin pharmacokinetics and liver/plasma ratio in Slco1b2−/− and wildtype mice
To assess the in vivo relevance of Oatp1b2, rifampin disposition was compared between male Slco1b2−/−and wildtype animals. First, we examined rifampin pharmacokinetics after a single IV dose of 1 mg/kg. As shown in figure 4A and 4B, there were significantly higher plasma rifampin levels in mice lacking Oatp1b2 expression. The plasma AUC in knockout mice was 1.7-fold greater than in wildtype mice (mean rifampin plasma AUC ± SD; wildtype animals 19586 ± 5971 ng*hr/mL, Slco1b2−/− animals 31931± 4949 ng*hr/mL, p<0.05). However, and consistent with Oatp1b2 function as a hepatic rifampin uptake transporter, mice lacking Oatp1b2 had a 2.5-fold reduction in liver rifampin exposure (mean rifampin area under the liver concentration-time curve (AUC) ± SD; wildtype animals 206434 ± 140712 ng*hr/g, Slco1b2−/− animals 86781 ± 58433 ng*hr/g, p=0.055) was noted. Liver-to-plasma concentration ratios were also determined for both genotypes and revealed a significant difference between wildtype and Slco1b2−/− animals (table 4). The plasma clearance (CL) of rifampin in Slco1b2−/− mice was 43% lower than wildtype mice (mean CL ± SD; wildtype mice 56 ± 20 mL/hr/kg, Slco1b2−/− mice 32 ± 5 mL/hr/kg, p<0.05) indicating that reduced uptake into liver had marked effects on drug elimination. Furthermore, the rifampin volume of distribution at steady-state (VSS) in Slco1b2−/− mice was 67% smaller than in wildtype mice (mean VSS ± SD; wildtype mice 374 ± 244 mL/kg, Slco1b2−/−mice 124 ± 18 mL/kg, p=0.054) suggesting that the liver is a major organ for rifampin distribution and that Oatp1b2 plays a significant role.
Figure 4.

Rifampin concentration in plasma (○) and liver (Δ) of wildtype (A) and Slco1b2−/− (B) mice (n=5−6/time point) after a single intravenous dose of rifampin (1mg/kg). Steady-state rifampin concentration in liver (C) and plasma (D) in animals treated with continuous subcutaneous infusion of rifampin at a rate of 8 μg/hr over 24 hrs using Alzet Mini-osmotic pumps (n=4). Panel E shows the calculated liver to plasma ratio comparing wildtype and Slco1b2−/− animals. Data are expressed as mean ± SD.
Table 4.
Liver-to-plasma ratio comparing wildtype and Slco1b2−/− animals following single intravenous dose of rifampin (1mg/kg)
| Time | wildtype | Slco1b2−/− |
|---|---|---|
| 5 min | 2.0 ± 1.2 | 0.6 ± 0.3 (*) |
| 15 min | 5.8 ± 2.9 | 1.6 ± 1.0 (*) |
| 30 min | 11.9 ± 6.7 | 2.4 ± 2.4 (*) |
| 1 h | 18.0 ± 16.6 | 3.0 ± 2.2 |
| 4 h | 4.7 ± 1.3 | 3.0 ± 0.4 (*) |
Data are expressed as mean liver-to-plasma ratio (mean ± SD)
p<0.05
To further understand the role of Oatp1b2 activity with respect to rifampin clearance we carried out additional studies of rifampin concentrations in plasma and liver tissues at steady-state conditions. After 24 hours of continuous subcutaneous infusion of rifampin at a rate of 8 μg/hr, the measured liver concentration of rifampin was significanlty decreased in Slco1b2−/−animals (mean liver concentration ± SD, wildtype animals 9440 ± 1904 ng/g; Slco1b2−/− animals 2348 ± 196 ng/g, p < 0.05) (figure 4C). This result was coupled with significantly higher steady-state plasma concentrations in knockout mice (mean rifampin steady-state plasma concentration ± SD, wildtype animals 249 ± 70 ng/ml; Slco1b2−/− animals 480 ± 113 ng/g, p < 0.05) (figure 4D). Our data strongly suggest that Oatp1b2 is a major contributor of hepatic rifampin uptake and clearly shows the relevance of this transporter in altering liver-to-plasma ratio of this drug (mean plasma-to-liver ratio ± SD, wildtype animals 39.0 ± 6.5; Slco1b2−/− animals 5.2 ± 2.1, p < 0.05) (figure 4 E).
Pravastatin pharmacokinetics and liver/plasma ratio in Slco1b2−/− and wildtype mice
Given the widely accepted notion that pravastatin is an excellent probe substrate of OATP transporters, we extended our investigations to assess the disposition profile of this drug. The male mice were given constant infusion of low (8 μg/hr) and high (32 μg/hr ) dose rate pravastatin. As shown in figure 5A when administered the low dose rate subcutaneous infusion, steady-state plasma concentrations of pravastatin were 1.8-fold higher in knockout compared to wild-type mice (12 ± 2.2 ng/mL versus 6.5 ± 1.3 ng/mL; p<0.05). Conversely, hepatic concentrations were 1.8-fold higher in wildtype compared to knockout mice (172 ± 17 ng/g versus 95 ± 6 ng/g p<0.05) (figure 5B) resulting in significant differences in the plasma-to-liver ratio of pravastatin (liver-to-plasma ratio ± SD; wildtype animals 26.9 ± 3.4; Slco1b2−/− animals 8.6 ± 1.0, p<0.05, figure 5C). Similar results were obtained after treating the animals with the high rate infusion. As shown in figure 5D, 5E and 5F, knockout animals consistently exhibited elevated pravastatin plasma levels (mean plasma level ± SD, wildtype 114.7 ± 14.0 ng/mL; Slco1b2−/− 209.9 ± 14.2 ng/mL, p< 0.05) and correspondingly lower liver concentrations in the Slco1b2−/− knockout mice (mean liver level ± SD wildtype 647.0 ± 282.7 ng/g; Slco1b2−/− 343.8 ± 70.8 ng/g, p = 0.053).
Figure 5.

Steady-state concentrations of pravastatin comparing wildtype (○) and Slco1b2−/− (•) animals. The concentration of pravastatin was determined in plasma (A & D) and liver (B & E) following 24 hours subcutaneous infusion of low dose rate (8 μg/hr) (n=4) or high dose rate (32 μg/hr) (n=5) pravastatin using Alzet Mini-osmotic pumps. The liver-to-plasma ratios are shown in panels C & F.
DISCUSSION
In the past decade, molecular cloning and functional characterization of drug transporters have significantly changed our view of the mechanisms underlying drug absorption, distribution, and elimination. It is clear that in every tissue compartment, expression of an array of transporters that mediate the cellular uptake and efflux of endo- and xenobiotics is required to maintain target tissue or organ homeostasis. Indeed in organs that are critical to the drug disposition process such as liver and intestine, the predominant expression of certain types or classes of transporters with broad substrate specificity facilitates the intestinal absorption and efficient hepatic extraction of drugs for subsequent metabolism and biliary elimination.
The clinical relevance of a number of efflux transporters such as MDR1 (P-gp) and various MRP (ABCC) transporters has been clarified via the creation and study of efflux transporter gene knockout mouse models. However, for uptake transporters, only recently have there been the reports of such models (Jonker et al., 2001;Eraly et al., 2006;Sweet et al., 2002). In terms hepatic drug uptake transporters, members of the OATP superfamily are believed to be the major determinants governing the hepatic extraction, drug-drug interactions, and in some cases, drug/toxin-induced liver injury. However, a mouse Oatp knockout model has yet to be reported. Among the OATP transporters expressed in human liver, OATP1B subfamily members OATP1B1 and OATP1B3, have been shown to be by far the most relevant to drug disposition due to their broad substrate specificity, expressed levels, and available genotype to phenotype correlations (Tirona and Kim, 2007). Therefore in addition to in vitro models for studying the activity of individual transporters, creation and pharmacological assessment of a murine Slco1b2 knockout model has the potential to be a valuable preclinical model for delineating the in vivo role and relative contribution of hepatic OATP1B transporters to drug substrates identified using in vitro systems.
Our data strongly support a major role for Oatp1b2 in the hepatic uptake of drugs as shown by 3 to 8-fold lower liver-to-plasma ratio of prototypical substrate drugs pravastatin and rifampin in knockout compared to wildtype mice (figure 5C and 4C).
We also note that there does not appear to be a major compensatory upregulation of other Oatps known to be expressed in rodent liver, such as Oatp1a1, Oatp1a4, and Oatp2b1, although gender related differences were noted (figure 3 A-C). This may suggest Oatp1b2, in addition to transporting xenobiotic substrates, may transport hormone or hormone metabolites that are essential in female mice. Expression of other hepatic transporters for the most part was not affected (table 3). While the mice exhibited a mild elevation in basal total bilirubin level, we did not observe any jaundice at any stage post-partum to suggest Oatp1b2 is essential for hepatic bilirubin clearance. Moreover, most of the measured bilirubin appeared to be conjugated bilirubin suggesting that the mild elevation in bilirubin may be due to loss of Oatp1b2-mediated hepatic reuptake of conjugated bilirubin (table 2). Therefore, it seems likely other transporters expressed in liver are capable of efficient unconjugated bilirubin removal and Oatp1b2 alone does not seem to significantly alter unconjugated bilirubin clearance. Taken together, our findings of normal liver function, histology, and lack of any other overt phenotype, Oatp1b2, and likely human OATP1B1 and 1B3 play a clinically relevant function in terms of xenobiotic response and clearance.
What is also important to recognize is the fact that extensive studies of Oatp1a1 and Oatp1a4 have suggested both of these transporters are also highly expressed in liver and both possess broad substrate specificity. In fact, it has been shown that rat Oatp1a1 is capable of transporting pravastatin (Hsiang et al., 1999), and rifampin interacts with rat Oatp1a4 (Slco1a4) (Vavricka et al., 2002). Yet in our Slco1b2−/− mice, profound differences in the liver-to-plasma ratios for both drugs were noted (figure 4 and 5). Pravastatin liver-to-plasma ratio during infusion is greater during low infusion rate compared to high infusion rate (Fig 5C, F), due likely to saturation of uptake transport. For rifampin, our data show time-dependent changes in the liver-to-plasma rifampin concentration ratio after I.V. bolus. Initial liver-to-plasma ratios up to 1 hour are more reflective of hepatic uptake processes than elimination or efflux. After 4 hours, distribution equilibrium was observed as is evidenced by approximate parallel declines in log plasma and liver rifampin concentrations over time. Indeed, liver-to-plasma ratios are up to 6-fold higher in Slco1b2−/− mice than wild-type mice during the initial distribution phase, indicating that hepatic rifampin uptake is impaired. During infusion of rifampin at steady-state, the liver-to-plasma ratio of rifampin is approximately 8-fold greater in Slco1b2−/− mice than wild-type mice. At this steady-state, hepatic drug levels will be controlled by the interplay of uptake, metabolism and efflux processes. Since the liver-to-plasma ratio differences between knockout and wildtype mice at steady-state are close in magnitude to that during the initial distribution phase after I.V bolus, this again suggests that Oatp1b2-mediated uptake plays a rate-limiting role in hepatic rifampin clearance.
Overall, our current findings would support the notion that members of the OATP1B subfamily are major contributors to hepatocellular uptake and therefore efficacy of most HMG-CoA reductase inhibitors in clinical use today. Accordingly, reliance on only in vitro studies to identify drug substrates of oatp transporters are likely to prove detrimental or inadequate in terms of being able to accurately predict which oatp transporter(s) significantly affect drug disposition in vivo.
Note that while we recognize that a large list of drug substrates for oatp/OATP transporters currently exist (Tirona and Kim, 2007), the key focus of this study was to utilize prototypical substrates of human OATP1B1 and 1B3 to see if an alteration in the disposition of such compounds are observed in the mouse model of Oatp1b2 deficiency. As noted earlier, our findings would support the utility of this mouse model for predicting the in vivo contribution of human OATP1B1 and 1B3. Note that in rodent liver, although Oatp1a1 and Oatp1a4 are highly expressed, direct human orthologes of such oatps do not exist, nor is human OATP1A2 found in hepatocytes. Therefore subtracting out the role of Oatp1a1 and Oatp1a4 may prove to be difficult when the liver-to-plasma ratio of OATP1B1 and 1B3 substrate drugs do not change in the Slco1b2−/− mice. It is likely that additional murine Slco genes will need to be deleted and relevant human SLCO genes introduced to more fully replicate human hepatic drug uptake capabilities. Nevertheless as shown by our data using pravastatin and rifampin, important new insights to the in vivo relevance of Oatp1b2 can already be obtained through a systematic evaluation of the pharmacological response using this novel oatp transporter knockout model.
In conclusion, to our knowledge this is a first report that describes and details functional characterization of a murine oatp transporter knockout model. We focused on Oatp1b2 because in humans, members of the OATP1B subfamily have turned out to be key determinants governing the hepatic uptake of many drugs in clinical use including the statin class of lipid lowering drugs. Accordingly, data presented in this study show the utility of the Slco1b2−/− mouse model in terms of in vivo pharmacological and functional assessment of hepatic OATP1B-mediated hepatic drug uptake and may therefore be a useful tool for understanding the contribution of hepatic uptake transporters in drug disposition.
ACKNOWLEDGMENTS
We would like to thank Dr. Ute I. Schwarz (Division of Clinical Pharmacology, University of Western Ontario) for helpful discussions.
Abbreviations
- ABC
ATP-Binding Cassette
- AUC
Area under the time-concentration curve
- SLC
solute carrier
- ALT
Alanine Aminotransferase
- AST
Aspartate Aminotransferase
REFERENCES
- Abe T, Kakyo M, Tokui T, Nakagomi R, Nishio T, Nakai D, Nomura H, Unno M, Suzuki M, Naitoh T, Matsuno S, Yawo H. Identification of a Novel Gene Family Encoding Human Liver-Specific Organic Anion Transporter LST-1. J Biol Chem. 1999;274:17159–17163. doi: 10.1074/jbc.274.24.17159. [DOI] [PubMed] [Google Scholar]
- Abe T, Unno M, Onogawa T, Tokui T, Kondo TN, Nakagomi R, Adachi H, Fujiwara K, Okabe M, Suzuki T, Nunoki K, Sato E, Kakyo M, Nishio T, Sugita J, Asano N, Tanemoto M, Seki M, Date F, Ono K, Kondo Y, Shiiba K, Suzuki M, Ohtani H, Shimosegawa T, Iinuma K, Nagura H, Ito S, Matsuno S. LST-2, a Human Liver-Specific Organic Anion Transporter, Determines Methotrexate Sensitivity in Gastrointestinal Cancers. Gastroenterology. 2001;120:1689–1699. doi: 10.1053/gast.2001.24804. [DOI] [PubMed] [Google Scholar]
- Cattori V, Hagenbuch B, Hagenbuch N, Stieger B, Ha R, Winterhalter KE, Meier PJ. Identification of Organic Anion Transporting Polypeptide 4 (Oatp4) As a Major Full-Length Isoform of the Liver-Specific Transporter-1 (Rlst-1) in Rat Liver. FEBS Lett. 2000;474:242–245. doi: 10.1016/s0014-5793(00)01596-9. [DOI] [PubMed] [Google Scholar]
- Choudhuri S, Ogura K, Klaassen CD. Cloning of the Full-Length Coding Sequence of Rat Liver-Specific Organic Anion Transporter-1 (Rlst-1) and a Splice Variant and Partial Characterization of the Rat Lst-1 Gene. Biochem Biophys Res Commun. 2000;274:79–86. doi: 10.1006/bbrc.2000.3105. [DOI] [PubMed] [Google Scholar]
- Eraly SA, Vallon V, Vaughn DA, Gangoiti JA, Richter K, Nagle M, Monte JC, Rieg T, Truong DM, Long JM, Barshop BA, Kaler G, Nigam SK. Decreased Renal Organic Anion Secretion and Plasma Accumulation of Endogenous Organic Anions in OAT1 Knock-Out Mice. J Biol Chem. 2006;281:5072–5083. doi: 10.1074/jbc.M508050200. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B, Meier PJ. Organic Anion Transporting Polypeptides of the OATP/ SLC21 Family: Phylogenetic Classification As OATP/ SLCO Superfamily, New Nomenclature and Molecular/Functional Properties. Pflugers Arch. 2004;447:653–665. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- Han Y, Sugiyama Y. Expression and Regulation of Breast Cancer Resistance Protein and Multidrug Resistance Associated Protein 2 in BALB/c Mice. Biol Pharm Bull. 2006;29:1032–1035. doi: 10.1248/bpb.29.1032. [DOI] [PubMed] [Google Scholar]
- Ho RH, Choi L, Lee W, Mayo G, Schwarz UI, Tirona RG, Bailey DG, Michael SC, Kim RB. Effect of Drug Transporter Genotypes on Pravastatin Disposition in European- and African-American Participants. Pharmacogenet Genomics. 2007;17:647–656. doi: 10.1097/FPC.0b013e3280ef698f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho RH, Kim RB. Transporters and Drug Therapy: Implications for Drug Disposition and Disease. Clin Pharmacol Ther. 2005;78:260–277. doi: 10.1016/j.clpt.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Ho RH, Tirona RG, Leake BF, Glaeser H, Lee W, Lemke CJ, Wang Y, Kim RB. Drug and Bile Acid Transporters in Rosuvastatin Hepatic Uptake: Function, Expression, and Pharmacogenetics. Gastroenterology. 2006;130:1793–1806. doi: 10.1053/j.gastro.2006.02.034. [DOI] [PubMed] [Google Scholar]
- Hsiang B, Zhu Y, Wang Z, Wu Y, Sasseville V, Yang WP, Kirchgessner TG. A Novel Human Hepatic Organic Anion Transporting Polypeptide (OATP2). Identification of a Liver-Specific Human Organic Anion Transporting Polypeptide and Identification of Rat and Human Hydroxymethylglutaryl-CoA Reductase Inhibitor Transporters. J Biol Chem. 1999;274:37161–37168. doi: 10.1074/jbc.274.52.37161. [DOI] [PubMed] [Google Scholar]
- Jonker JW, Wagenaar E, Mol CA, Buitelaar M, Koepsell H, Smit JW, Schinkel AH. Reduced Hepatic Uptake and Intestinal Excretion of Organic Cations in Mice With a Targeted Disruption of the Organic Cation Transporter 1 (Oct1 [Slc22a1]) Gene. Mol Cell Biol. 2001;21:5471–5477. doi: 10.1128/MCB.21.16.5471-5477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J, Cui Y, Nies AT, Keppler D. A Novel Human Organic Anion Transporting Polypeptide Localized to the Basolateral Hepatocyte Membrane. Am J Physiol Gastrointest Liver Physiol. 2000;278:G156–G164. doi: 10.1152/ajpgi.2000.278.1.G156. [DOI] [PubMed] [Google Scholar]
- Marzolini C, Tirona RG, Kim RB. Pharmacogenomics of the OATP and OAT Families. Pharmacogenomics. 2004;5:273–282. doi: 10.1517/phgs.5.3.273.29831. [DOI] [PubMed] [Google Scholar]
- Nakai D, Nakagomi R, Furuta Y, Tokui T, Abe T, Ikeda T, Nishimura K. Human Liver-Specific Organic Anion Transporter, LST-1, Mediates Uptake of Pravastatin by Human Hepatocytes. J Pharmacol Exp Ther. 2001;297:861–867. [PubMed] [Google Scholar]
- Ogura K, Choudhuri S, Klaassen CD. Full-Length CDNA Cloning and Genomic Organization of the Mouse Liver-Specific Organic Anion Transporter-1 (Lst-1). Biochem Biophys Res Commun. 2000;272:563–570. doi: 10.1006/bbrc.2000.2830. [DOI] [PubMed] [Google Scholar]
- Schachter M. Chemical, Pharmacokinetic and Pharmacodynamic Properties of Statins: an Update. Fundam Clin Pharmacol. 2005;19:117–125. doi: 10.1111/j.1472-8206.2004.00299.x. [DOI] [PubMed] [Google Scholar]
- Shimizu M, Fuse K, Okudaira K, Nishigaki R, Maeda K, Kusuhara H, Sugiyama Y. Contribution of OATP (Organic Anion-Transporting Polypeptide) Family Transporters to the Hepatic Uptake of Fexofenadine in Humans. Drug Metab Dispos. 2005;33:1477–1481. doi: 10.1124/dmd.105.004622. [DOI] [PubMed] [Google Scholar]
- Sweet DH, Miller DS, Pritchard JB, Fujiwara Y, Beier DR, Nigam SK. Impaired Organic Anion Transport in Kidney and Choroid Plexus of Organic Anion Transporter 3 (Oat3 (Slc22a8)) Knockout Mice. J Biol Chem. 2002;277:26934–26943. doi: 10.1074/jbc.M203803200. [DOI] [PubMed] [Google Scholar]
- Tirona RG, Kim RB. In: Organic Anion Transporting Polypeptides (OATPs), in Drug Transporters. You G, Morris ME, editors. Wiley-VCH, New York: 2007. [Google Scholar]
- Tirona RG, Leake BF, Wolkoff AW, Kim RB. Human Organic Anion Transporting Polypeptide-C (SLC21A6) Is a Major Determinant of Rifampin-Mediated Pregnane X Receptor Activation. J Pharmacol Exp Ther. 2003;304:223–228. doi: 10.1124/jpet.102.043026. [DOI] [PubMed] [Google Scholar]
- Vavricka SR, Van MJ, Ha HR, Meier PJ, Fattinger K. Interactions of Rifamycin SV and Rifampicin With Organic Anion Uptake Systems of Human Liver. Hepatology. 2002;36:164–172. doi: 10.1053/jhep.2002.34133. [DOI] [PubMed] [Google Scholar]
- Wagner M, Fickert P, Zollner G, Fuchsbichler A, Silbert D, Tsybrovskyy O, Zatloukal K, Guo GL, Schuetz JD, Gonzalez FJ, Marschall HU, Denk H, Trauner M. Role of Farnesoid X Receptor in Determining Hepatic ABC Transporter Expression and Liver Injury in Bile Duct-Ligated Mice. Gastroenterology. 2003;125:825–838. doi: 10.1016/s0016-5085(03)01068-0. [DOI] [PubMed] [Google Scholar]
- Wagner M, Halilbasic E, Marschall HU, Zollner G, Fickert P, Langner C, Zatloukal K, Denk H, Trauner M. CAR and PXR Agonists Stimulate Hepatic Bile Acid and Bilirubin Detoxification and Elimination Pathways in Mice. Hepatology. 2005;42:420–430. doi: 10.1002/hep.20784. [DOI] [PubMed] [Google Scholar]
- Zollner G, Wagner M, Moustafa T, Fickert P, Silbert D, Gumhold J, Fuchsbichler A, Halilbasic E, Denk H, Marschall HU, Trauner M. Coordinated Induction of Bile Acid Detoxification and Alternative Elimination in Mice: Role of FXR-Regulated Organic Solute Transporter-{Alpha}/Beta in the Adaptive Response to Bile Acids. Am J Physiol Gastrointest Liver Physiol. 2006;290:G923–G932. doi: 10.1152/ajpgi.00490.2005. [DOI] [PubMed] [Google Scholar]