

Abstract
Understanding angiogenesis and growth control is central for elucidating prostate tumorigenesis. However, the mechanisms of activation of the angiogenic gene, vascular endothelial growth factor (VEGF) are complex and its regulation in prostate cancer is not well understood. In previous studies, VEGF expression levels were correlated with altered levels of the zinc finger transcription factor, WTl. Since the VEGF promoter has several potential WTl binding sites and WT1 regulates many growth control genes, here we assessed whether WTl might also regulate VEGF transcription. Using transfection and DNA binding assays, functional WTl binding sites were localized within the proximal VEGF promoter. Transfection of the DDS-WTl (R394W) zinc finger mutant had no significant effect on VEGF-luciferase reporter activity, suggesting that an intact zinc finger DNA binding domain was required. Interestingly, WTl-mediated regulation of VEGF reporter constructs varied in different cell types. In androgen-responsive, LNCaP prostate cancer cells, hormone treatment enhanced WTl-mediated activation of the VEGF promoter constructs. Overall, these results suggest that WTl transcriptionally regulates VEGF through interaction of its zinc finger DNA binding domain with the proximal GC-rich VEGF promoter. These findings may shed light on the role of WTl in angiogenesis and prostate cancer progression.
Keywords: Transcription, Wilms tumor, WTl, Vascular Endothelial Growth Factor, VEGF, prostate cancer, androgen, transfection, Electrophoretic Mobility Shift Assay, EMSA
2. INTRODUCTION
In American men, prostate cancer is the most common cancer and second leading cause of cancer death (1). A central problem with current treatment regimens is the development of androgen-independent metastases. In order to better understand prostate tumor growth and metastasis, research has centered on the regulation of angiogenesis, and in particular, vascular endothelial growth factor (VEGF), a mitogen essential for tumor angiogenesis (2, 3). VEGF is secreted by tumor cells and is necessary for tumor growth greater than 1-3 mm3 (4). Therefore, a better understanding of prostate cancer progression requires examination of the mechanisms of regulation of VEGF.
VEGF regulation is complex and occurs at both transcriptional and post-transcriptional levels (5, 6). While the VEGF promoter lacks a TATA-binding site, it contains a GC-rich core promoter region, and additional distal enhancer sites (such as, the hypoxia response elements that bind HIFl-alpha). Although hormone-responsive (7-10), the VEGF promoter lacks any classical consensus androgen receptor (AR) or estrogen receptor (ER) binding sites. However, a distal ER/GC-box composite site has been identified, where ER-alpha physically interacts with the zinc finger transcription factors SP1 and SP3 to regulate VEGF levels in hormone-responsive endometrial and breast cancer cells (10, 11). An AR/GC site has not yet been identified, but the VEGF GC-rich core promoter contains multiple overlapping sites for: SpI/SP3 (12, 13), Egr-1 (early growth response-1), AP2 (14), and potentially the zinc finger transcription factor, WT1 (Figure 1A). Since WT1 transcriptionally regulates GC-rich promoters and forms a complex with ER-□ □ □ □ □ to regulate insulin-like growth factor I receptor (IGF-1R) (15), we hypothesized that WT1 might regulate the hormone responsive VEGF promoter.
Figure 1.
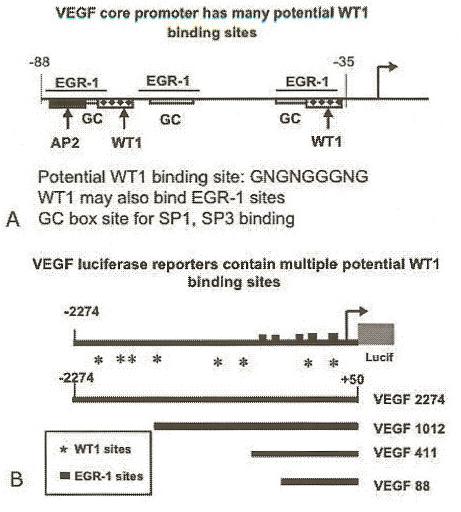
The VEGF promoter contains multiple potential WT1 binding sites. Panel A: The TATA-less, GC-rich VEGF core promoter has multiple potential binding sites for zinc-finger transcription factors of SP1 and EGR-1 families (map not drawn to scale). Potential WT1 binding sites were identified by in silico analyses using Genomatix MatInspector Software, as described in text. Panel B: The full-length VEGF promoter contains multiple potential WT1 binding sites and EGR-1 sites to which WT1 can also bind (map not drawn to scale). Four overlapping luciferase reporter constructs spanning the VEGF promoter region (12) were tested for activity in transient transfection assays as described in the text.
The functions of the WT1 gene product vary depending upon the isoform of WT1 and reflect its structural domains. The four major isoforms of WT1 are formed by alternative splicing at two sites resulting in the inclusion or exclusion of 1) exon V and/or 2) a tripeptide (KTS) in exon 9 that alters the zinc finger DNA binding structure (and the WT1(A) isoform lacks both). The carboxyl-terminus has four Cys2-His2 zinc fingers (16, 17) that bind a common consensus sequence, GNGNGGGNG, as well as the related Egr-1 sites (18). The importance of the zinc finger domain for DNA binding can be observed in the congenital syndromes associated with naturally occurring WT1 mutations, such as the Denys-Drash Syndrome (DDS), characterized by renal mesangial sclerosis, genital anomalies and elevated risk of Wilms tumor nephroblastoma (19). The most common mutation for DDS-WT1 occurs in codon 394 in which an arginine is replaced with a tryptophan (R394W) (20) resulting in altered DNA binding ability.
The transcriptionally active isoform of the Wilms’ tumor suppressor gene, WT1(A), regulates a large family of genes involved in growth control, sex determination, and genitourinary development, (for reviews see 20-23). We and others have demonstrated that WT1 regulates genes important in prostate cancer growth control pathways; both growth promoting pathways, IGF axis (24, 25) and androgen signaling (26, 27), and growth suppressing/apoptotic pathways (28-32). Previously we have characterized the effect of over-expression of WT1 in LNCaP prostate cancer cells engineered to express either the wild-type WT1 or the DDS-WT1 mutant (R394W) (33, 34). Microarray studies indicated that VEGF, among other genes, was differentially expressed in stably transfected WT1-LNCaP cells (34) and led to the hypothesis tested here, that WT1 would bind the VEGF promoter and regulate its transcription. Transfection assays using VEGF deletion constructs (Figure 1B) were performed in several cell lines to identify the WT1 responsive region. Once identified, electrophoretic mobility shift assays (EMSAs) were performed to identify functional WT1 binding sequences. These results verified that WT1 regulated VEGF promoter activity and in androgen-responsive LNCaP prostate cancer cells, hormone treatment strongly enhanced WT1-mediated regulation.
3. MATERIALS AND METHODS
3.1. Cell Culture and Transfections
LNCaP prostate cancer cells (ATCC CRL 1740 from the American Type Culture Collection, Rockville, MD) and PC3 (ATCC CRL 1435), an androgen insensitive cell line were grown in RPMI-1640 media (Invitrogen; Carlsbad, CA) with 10% fetal calf serum (FCS) and 100 IU/ml penicillin and streptomycin. HEK-293 cells (ATCC CRL 1573), a kidney cell line, were maintained in DME media supplemented with 10% FCS. The cultures were maintained in a 5% CO2 humidified incubator at 37°C. In preparation for the transfections, the cells were cultured in 12-well plates. When the cells reached 80% confluency they were transfected as described (35) using lipofectamine 2000 (Invitrogen; Carlsbad CA) in serum- and antibiotic-free media. For hormone induction, cells were cultured in charcoal-dextran stripped (ChS) serum. The synthetic androgen, R1881 (methyltrienolone) was used for these studies because of its strong affinity for the androgen receptor (36).
The cytomegalovirus (CMV) promoter-driven pCB6+WT1(A) expression construct (lacking both KTS insertion and exon 5) and the murine wild-type pCMV-WT1(A) and mutant DDS-WT1(R384W) expression plasmids were previously described (18, 19, 26, 27). The pGL3-VEGF promoter-luciferase reporter constructs (VEGF 88, 411, 1012, 2274) (Figure 1B) were obtained from Dr. K. Xie (12). All DNA was purified by the Qiagen plasmid Maxi Kit (Qiagen, Carlsbad CA) and transfections were performed as described (35). Briefly, 250 ng of the stated VEGF promoter-luciferase reporter construct was cotransfected along with 5ng of pRL-null, the promoterless-Renilla luciferase normalizer (Promega; Madison, Wisconsin), and increasing concentrations of the WTl(A) or DDS-WT1(R384W) expression constructs (0, 250, 500 ng). DNA levels were held constant by the addition of the appropriate empty CMV expression vector, pCB6+ or pCMV 4 (Promega; Madison, Wisconsin). For LNCaP cells, the medium was removed after 5-6 hours and replaced with fresh RPMI with either 10% ChS FCS or full “unstripped” FCS. For those plates that received ChS, half of the wells were treated with 0nM and half with 5nM R1881. For PC3 and HEK-293 cells, media was removed after 5 hours or 18 hrs, respectively, and replaced with media containing 10% FCS. Cells were harvested at 48 hrs (293) or 72 hrs (LNCaP and PC3).
3.2. Reporter assays
Initially both firefly and Renilla luciferase activities of the cellular extracts were measured as per manufacturer’s recommendations using the Dual-Luciferase Reporter Assay System (Promega; Madison, Wisconsin) and a 20/20n luminometer (Turner; Sunnyvale, California). Since WT1 activated the pRL-null expression vector, as previously described (35), luciferase activity was not normalized using Renilla luciferase activity, but rather by cellular protein concentration. Cellular protein concentration reflects cell viability and controls for variability in numbers of transfected cells (37). The protein concentration of cell extracts was determined using the Micro BCA Protein Assay Reagent kit (Pierce; Rockford, Illinois), and absorbance was read at 570nm on a Dynex Technologies MRX Revelation plate reader (Chantilly, VA). Cellular protein concentrations were relatively constant varying <25% between samples. Average protein concentration was determined using a BSA standard and normalized luciferase activity was reported relative to the protein concentration of the cell extracts.
3.3. Statistical analysis
Each transfection was performed in triplicate (293 and PC3) or quadruplicate (LNCaP) and repeated at least three times. Standard errors of the mean were determined using the GraphPad InStat statistical software program (San Diego, California). Significance was determined by one-way analysis of variance (ANOVA) followed by the Tukey-Kramer Multiple Comparison Test. Two-way analysis of variance (ANOVA) (GraphPad Prism; San Diego, CA) was used to determine significance of the interaction between WT1 and R1881.
3.4. Electrophoretic Mobility Shift Assay (EMSA)
Forward and reverse oligonucleotides of at least 30 bp and containing potential WT1 binding sites were annealed in TEK, (0.01M Tris, 0.001M EDTA, 0.1M KCl) (Table 1). Five pmol of double stranded DNA was labeled with gamma-32P-ATP using T4 kinase (Invitrogen; Carlsbad, CA). After a 30-minute incubation at 37°C, the labeled probes were separated from free gamma-32P-ATP by centrifugation using a MicroSpin G-50 column (Amersham Biosciences; Piscataway, NJ) and radioactivity was determined in a Beckman LS 6000TA liquid scintillation counter.
Table 1.
Oligonucleotides tested for WT1 binding activity
| (Promoter Region)1 | Oligonucleotides Sequence2 |
|---|---|
| 2310-2342 (V88) | GAGCCATGCGCCCCCCCTTTTTTTTTTAAAAG |
| 2294-2330 (V88) | GGGTCCCGGCGGGGCGGAGCCATGCGCCCCCCCCTTT |
| 2281-2310 (V88) | CGGGCCGGGGGCGGGGTCCCGGCGGGGCGG |
| 2109-2138 (V411) | GGACAGAGTTTCCGGGGGCGGATGGGTAAT |
| 575-605 (V2274) | GGAGGGTTGGGGTGGGTGGGAGCCAGCCCTT |
| EGR-13 | GGCCCGGCGCGGGGGCGAGGGCG |
| SP14 | ATTCGATCGGGGCGGGGCGAGC |
Brackets indicate VEGF promoter construct in which the oligonucleotide is located
Potential WT1 binding site(s) are in bold. Potential SP1 binding sites (italics) overlap WT1 sites.
Consensus EGR-1 oligonucleotide as described (26)
Consensus SP1 oligonucleotide (Promega, Madison, WI).
EMSAs were performed using an in vitro translated WT1 protein, containing the zinc finger region as described (26) or 0.2 μg of recombinant SP1 protein (Promega, Madison WI). Binding assays were performed in buffer containing Tris (pH 7.5), 6.5% glycerol, 90mMKCl, 0.2mMDTT, 1mg/ml BSA, 100uMZnCl2, 1 microgram of poly (dI-dC), and where stated, 1 microgramWT1 protein and 20-100-fold molar excess cold competitor oligonucleotide. Supershifts were performed using 400 ng of WT1 C-19 antibody (Santa Cruz Biotechnology; Santa Cruz, CA) or 400 ng of normal rabbit IgG control sera. Approximately 125 fmol of 32P-labeled probe was added to the mixture and incubated for 20 minutes at room temperature prior to electrophoresis. The DNA-protein complexes were electrophoresed on a 5% polyacrylamide:bis (37.5:1) gel for 1 hour at 250 volts, dried on a Bio Rad gel dryer and visualized by autoradiography with Hyperfilm (Amersham) developed in a Konica SRX-101 film processor.
4. RESULTS
4.1. Identification and characterization of WT1 Responsive Regions in the VEGF Promoter
Transfection assays were performed to determine if WT1 directly modulated VEGF expression. LNCaP cells were co-transfected with pCB6+WT1(A) expression construct, and/or empty vector pCB6+ (to maintain constant DNA levels), along with one of several VEGF promoter constructs (VEGF 88, VEGF 411, VEGF 1012, VEGF 2274). Cultures were incubated in RPMI-10% FCS and transfected as described (35). Luciferase activity was normalized by cellular protein concentration, controlling for variability in cell numbers as described in methods. Transfection of 500 ng of WT1 enhanced the normalized luciferase activity of the VEGF 88 minimal core promoter 4.7-fold (Figure 2). Significance was determined by ANOVA (p<0.0001) and verified by the Tukey-Kramer Multiple Comparison post-test. Similar results were obtained in LNCaP cells co-transfected with the slightly larger VEGF promoter construct (VEGF 411), and 500 ng WT1 increased normalized luciferase activity 3.3-fold (Figure 2). Significance was determined by ANOVA (p=0.0005) and was confirmed using the Tukey-Kramer Multiple Comparison Test. In LNCaP cells co-transfected with 500 ng WT1 and the larger VEGF constructs containing the distal promoter regions, luciferase activity was increased only slightly (1.6-fold) (Figure 2). Although this activation was determined to be significant (p=0.0038 for VEGF 1012 and p=0.0052 for VEGF 2274 ANOVA), it was less than two-fold, and therefore, unlikely to be biologically relevant. Thus, the WT1 responsive region appears to be located within the proximal 400 base pairs of the VEGF promoter.
Figure 2.
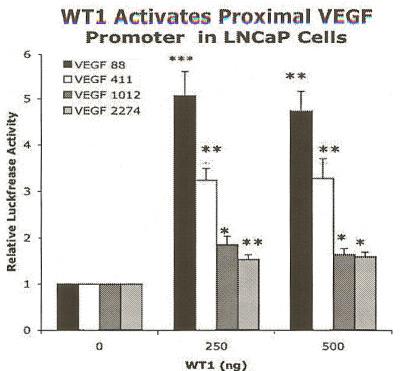
WT1 increased VEGF promoter activity in LNCaP cells. Cells were co-transfected with WT1(A) expression constructs and pGL3-VEGF promoter constructs: VEGF 88 (black), VEGF 411 (white), VEGF 1012 (dark gray), or VEGF 2274 (light gray). Total DNA levels were held constant by addition of empty vector pCB6+. The luciferase values were normalized by protein concentration as described in the text. Each experiment was performed in quadruplicate and repeated three times. Results are given as relative activation by WT1 ± SEM and normalized luciferase activity is shown relative to CB6+ vector control (0 ng WT1). WT1 greatly increased activity of the proximal promoters VEGF 88 and VEGF 411, as determined by ANOVA (p<0.0001 and p=.0005, respectively). Asterisks indicate significant differences between the mean luciferase activities of transfected vector control and WT1 as determined by the Tukey-Kramer Multiple Comparison post-test (* p< 0.05, **p<0.01, ***p<0.001).
Transfections of the pCMV4-DDS-WT1(R384W) mutant were performed to determine whether the zinc finger DNA binding domain played a critical role in regulating transcription of the VEGF promoter (Figure 3). The DDS-WT1 (R384W) mutant has an altered DNA binding domain and was not expected to bind DNA. Indeed, the DDS-WT1 (R384W) mutant had no significant effect on the transcriptional regulation of VEGF in LNCaP cells (Figure 3). While these results suggested that the DNA binding domain of WT1 plays an essential role in the regulation of VEGF, the WT1 mutant pCMV4-DDS-WT1(R384W) expression construct was derived from a murine WT1 gene, so it was necessary to confirm that the wild-type murine WT1 expression construct pCMV4-WT1(A) functioned similarly in human LNCaP prostate cancer cells. A similar up-regulation of the proximal VEGF promoter constructs was observed when the murine WT1 was transfected into LNCaP cells. WT1 (500 ng) increased activity 4.3-fold and 5.5-fold in LNCaP cells transfected with VEGF 88 and VEGF 411, respectively (data not shown). WT1 also increased activity, though to a lesser extent, of the larger VEGF promoter constructs that included the distal regions. However, activation of all four VEGF promoter constructs was considered significant using ANOVA. Since the murine WT1 gene behaved similarly to the human gene, this strengthened the conclusion that the zinc finger mutation in the DDS-WT1(R384W) expression construct prevents activation of the VEGF promoter constructs in LNCaP cells. The inactivity of the DDS-WT1(R384W) construct was confirmed in two other cell lines (HEK-293 and PC3), as well (data not shown). Overall, this suggested that the DNA binding domain of WT1 plays an essential role in the regulation of VEGF.
Figure 3.
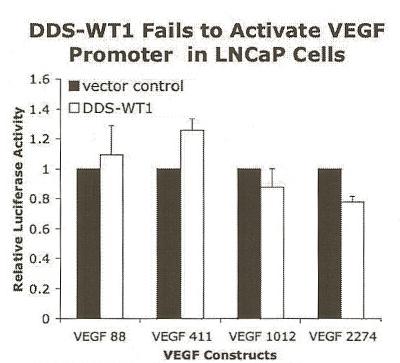
DDS-WT1 did not enhance VEGF promoter activity in LNCaP cells. Cells were co-transfected with 500 ng of DDS-WT1(R384W) (white) or pCMV4 vector control (black) with a VEGF promoter construct, as listed in Figure 2. The luciferase values were normalized by protein concentration as described and results are given as relative activation by DDS-WT1 ± SEM and compared to pCMV4+ vector control (black). Each experiment was performed in quadruplicate and repeated three times. DDS-WT1 did not significantly affect the VEGF promoter constructs.
To determine whether WT1 regulated the VEGF promoter similarly in different cellular contexts, we also transfected an immortalized human embryonic kidney cell line (HEK-293). Surprisingly, the WT1 expression construct repressed transcription of the proximal VEGF promoters (VEGF 88 and VEGF 411) in HEK-293 cells (Figure 4). Transfection of 500 ng of pCB6+WT1(A) decreased the normalized luciferase activity of the VEGF 88 and VEGF 411 promoter constructs 2.7-fold and 4.9-fold, respectively. Significance was determined by ANOVA (p=0.0067 for VEGF 88, p=0.0002 for VEGF 411) and verified by the Tukey-Kramer Multiple Comparison post-test. These results suggested that WT1 regulated transcription of the VEGF promoter differently in different cell lines. In HEK-293 cells co-transfected with 500 ng WT1 and the larger VEGF constructs containing the distal promoter regions, the normalized luciferase activity remained relatively constant (decreased only 1.2-fold, or 1.4-fold for V1012 and VEGF 2274, respectively) and was not significantly affected by WT1 (p=0.4786 for VEGF 1012 and p=0.0313 for VEGF 2274) (Figure 4). Overall the HEK-293 data confirmed the location of the WT1 responsive region, showing that WT1 acted strongly on the proximal VEGF promoter. Interestingly, the regulatory effect in 293 cells was the inverse of that in LNCaP (where WT1 strongly activated the proximal VEGF promoter).
Figure 4.
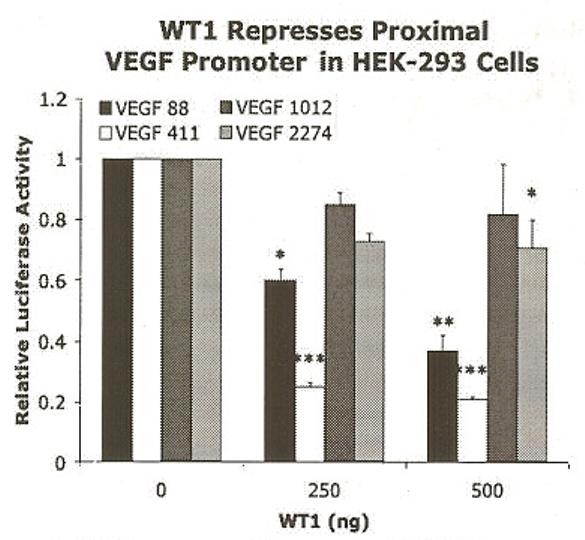
WT1 repressed the proximal VEGF promoters in HEK-293 cells. Cells were co-transfected with WT1(A) expression constructs and pGL3-VEGF promoter constructs: VEGF 88 (black), VEGF 411 (white), VEGF 1012 (dark gray), or VEGF 2274 (light gray). DNA levels were held constant and luciferase values were normalized as described in Figure 2. Results are given as relative activation by WT1 ± SEM and shown relative to CB6+ vector control (0 ng WT1). Each experiment was performed in triplicate and repeated 5 times. Significant differences were determined by ANOVA. WT1 significantly decreased normalized luciferase activity of VEGF 88 (p=0.0067) and VEGF 411 (p=0.0002), with little or no effect on VEGF 1012 (p=0.4786) and VEGF 2274 (p=0.0313). Asterisks indicate significant differences determined by the Tukey-Kramer Multiple Comparison Test as described in Figure 2.
4.2. WT1 up-regulates the proximal VEGF promoter in the presence or absence of androgen
The effects of hormone treatment on WT1-mediated regulation of the VEGF promoter were tested by transfecting LNCaP cells as described above, but culturing in RPMI ChS FCS with or without 5nM R1881 (Figure 5A and B). In the presence of hormone, WT1 strongly activated both proximal promoters. Cotransfection of 500 ng pCB6+WT1(A) increased the normalized luciferase activity of VEGF 88 and VEGF 411 by 4.1 1-fold or 4.96-fold, respectively (Figure 5A). Significance was determined by ANOVA (p=0.0025 for VEGF 88, p<0.0001 for VEGF 411) and verified by the Tukey-Kramer Multiple Comparison post-test. Conversely, in the absence of hormone, WT1 activated the proximal promoter region modestly (Figure 5B). In LNCaP cells cotransfected with 500 ng pCB6+WT1 (A) and VEGF 88 or VEGF 411 normalized luciferase activity was increased only 2.5-fold or 2.2-fold, respectively. Significance was determined by ANOVA (p=0.0009 for VEGF 88, p=0.0015 for VEGF 411) and verified by the Tukey-Kramer Multiple Comparison post-test. While WT1 up-regulated the proximal VEGF promoters, it had no significant effect on the larger VEGF 2274 promoter construct whether in the presence or absence of hormone. This was surprising since the VEGF 2274 promoter construct contained the ER-□ □ □ □ □ composite-site. Thus, we have confirmed the WT1 responsive region is in the proximal VEGF promoter and that WT1-mediated activation of this promoter region is enhanced by hormone. Due to the location of the WT1 responsive region, the ER-□ □ □ □ □ composite-site is unlikely to contribute to androgen enhancement of WT1 activation.
Figure 5.
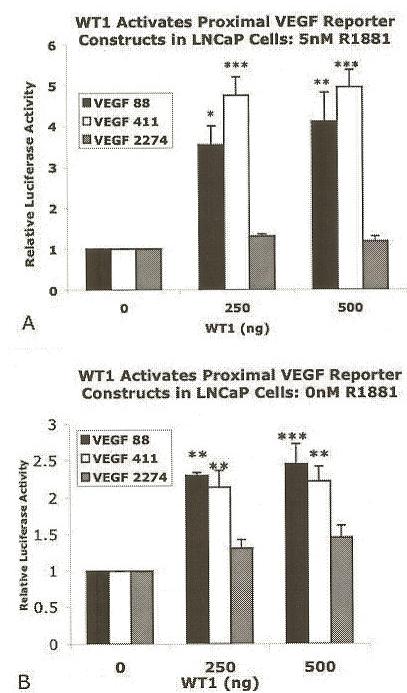
Hormone enhanced WT1-mediated activation of the proximal VEGF promoter. LNCaP cells were cultured in RPMI-10% ChS with 5nM R1881 (Panel A) or without (0nM) R1881 (Panel B) following co-transfection of WT1(A) with pGL3-VEGF promoter constructs: VEGF 88 (black), VEGF 411 (white), or VEGF 2274 (gray). DNA levels were held constant and luciferase values were normalized as described in Figure 2. Results are given as relative activation by WT1 ± SEM and shown relative to CB6+ vector control (0 ng WT1). Each experiment was performed in quadruplicate and repeated at least 4 times. Significant differences were determined by ANOVA (p=0.0025 for VEGF 88, p<0.0001 for VEGF 411) for co-transfected cells treated with 5nMR1881 in RPMI-10% ChS (Panel A) or (p=0.0009 for VEGF 88, p=0.0015 for VEGF 411) for untreated cells (0nM R1881) (Panel B). Asterisks indicate significant differences determined by the Tukey-Kramer Multiple Comparison Test as described in Figure 2.
To validate the apparent hormone effect on WT1-mediated activation, LNCaP cells were treated with 5nM R1881 to determine the androgen response of the VEGF reporter constructs (first, in the absence of the WT1 expression construct and then with WT1 co-transfection). As shown in Table 2, hormone treatment (5 nM R1881) induced the full-length VEGF promoter (VEGF 2274) 6.3-fold, whereas the proximal VEGF constructs VEGF 88 and VEGF 411 were up-regulated only 3.1- and 1.8-fold, respectively. Although the distal promoter lacks classical androgen responsive elements, these results are consistent with the notion that hormone may activate the VEGF promoter through the distal ER-alpha composite-site. Conversely, to determine the effect of WT1 expression independent of any hormone induction, we cultured WT1 transfected LNCaP cells in the absence of hormones, using charcoal-dextran-stripped FCS (ChS-FCS) to supplement the media. Transfection of WT1 in the absence of hormone, modestly induced VEGF luciferase activity 2.5-fold and 2.2-fold in LNCaP cells co-transfected with VEGF 88 and VEGF 411, respectively (Table 2), but failed to significantly activate the full-length VEGF 2274 promoter construct.
Table 2.
Fold activation of VEGF luciferase by WT1 in the presence or absence of R18811
| WT1(ng) | VEGF 88 | VEGF 411 | VEGF 2274 | |||
|---|---|---|---|---|---|---|
| 0 nM2 | 5 nM3 | 0 nM2 | 5 nM3 | 0 nM2 | 5 nM3 | |
| 0 | 1 | 3.1 | 1 | 1.8 | 1 | 6.3 |
| 500 | 2.5 | 12.6 | 2.2 | 8.7 | 1.5 | 7.5 |
Fold activation is expressed relative to average normalized luciferase activity in the absence of both hormone and WT1
LNCaP cells cultured in RMPI without R1881
LNCaP cells cultured in RMPI with 5nM R1181
In contrast to the modest activation of the proximal promoters by WT1 in the absence of hormone, luciferase activities of VEGF 88 and VEGF 411 were strongly up-regulated (12.6- and 8.7-fold respectively) in the presence of both WT1 and 5nM R1881 (Table 2). These results suggested that WT1 and hormone are working together to enhance VEGF luciferase. Two-way ANOVA was used to determine the likelihood that an interaction between WT1 and hormone contributed to VEGF promoter activation by WT1 in hormone treated cells. Analysis of the interaction between WT1 and hormone by two-way ANOVA confirmed that it was significant (p=0.0067 for VEGF 88 and p<0.0001 for VEGF 411). In contrast, two-way ANOVA indicated that hormone alone had a significant effect on activation of the full-length VEGF promoter construct (p<0.0001), but the potential interaction of WT1 and hormone was not significant (p=0.5050) nor was the contribution by WT1 likely significant (p=0.2086). Overall, these results suggest that the combination of WT1 and hormone activates transcription of the proximal VEGF promoter region to an extent greater than either factor alone, and that this interaction of WT1 and androgen contributes significantly to the up-regulation of the VEGF proximal promoter region in LNCaP cells. The mechanism of this interaction is unknown.
Since WT1 activation of VEGF promoter was hormone responsive in LNCaP cells, we asked whether WT1 would also activate VEGF in androgen insensitive PC3 cell line. Transfection of 500 ng pCB6+WT1 (A) failed to activate the proximal VEGF promoters in PC3 cells (Table 3). The full-length VEGF 2274 promoter construct was slightly repressed by WT1 (decreased 1.4-fold), similar to that seen in HEK-293 cells. By comparing relative luciferase activities of the VEGF reporters in different cell lines co-transfected with WT1, we have observed differential effects of transcriptional regulation by WT1. This comparison clearly indicates that regulation of VEGF by WT1 is dependent upon cellular context.
Table 3.
WT1-mediated regulation of VEGF promoter depends upon cellular context
| Construct | Fold-activation of VEGF reporters by WT11 | ||
|---|---|---|---|
| LNCaP | PC3 | 293 | |
| VEGF-88 | 4.5 | 1.0 | 0.3 |
| VEGF-411 | 3.5 | 0.9 | 0.4 |
| VEGF-2274 | 1.5 | 0.7 | 0.7 |
Fold-activation of VEGF promoter constructs by WT1(A), relative to pCB6+ vector control DNA. LNCaP, PC3, and 293 cells were transfected and luciferase activity normalizedas described in text.
4.3. Identification of WT1 binding sites in the VEGF promoter
DNA binding assays were performed to determine whether WT1 regulation of the VEGF promoter was mediated by DNA binding. Genomatix MatInspector software (38) was used to analyze the VEGF 2kb promoter region (Sequence Accession Number AF095785) to locate potential WT1 and other transcription factor binding sites. Based on the in silico analysis, we chose potential binding sites that mapped to the functional domains identified by the reporter assays. The G-rich oligonucleotides that were selected for electrophoretic mobility shift assays (EMSA) (Table 1) contained potential WT1 or EGR-1 binding sites (Figure 1). EGR-1 sites were included as control WT1 binding sites, since WT1 has been documented to bind to these sequences (16). Initially we focused on the VEGF core promoter (VEGF 88) and systematically tested the region using three overlapping double-stranded oligonucleotides (Table 1). Additionally, we examined a binding site unique to the VEGF 411 region and one site within the distal promoter region (575-605) (numbered as per AF095785).
EMSA were performed using in vitro translated WT1 protein containing the zinc finger region (-KTS form) previously described (18, 26). The WT1 protein bound to only one of the probes tested from the VEGF 88 core promoter, the 2310-2343 sequence (Figure 6), and not the 2281-2310 or 2294-2330 oligonucleotides (data not shown). The identity of the protein in the observed complexes (black arrows) was confirmed by supershift assay using the polyclonal WT1 antibody (Ab) C-19 (Santa Cruz, CA) (Panel A lane 4). The WT1 Ab shifted the complexes upward (black arrow), whereas the normal rabbit IgG control did not diminish or alter the migration of the complexes (Panel A lane 5). Specificity of WT1 binding was confirmed by competition assays using a canonical EGR-1 site known to bind WT1 (26) (Figure 6 Panel B). Competition with 20-fold (L, panel Blane 3) and 80-fold (H, panel Blane 4) molar excess of unlabeled EGR1 oligonucleotide decreased binding of WT1 to the labeled probe. Competition with an 80-fold molar excess of a non-specific oligonucleotide (the consensus GATA binding site) failed to decrease the signal intensity of the WT1 complex (data not shown). Note that the WT1 protein/DNA complex migrated as two bands (black arrows) above the free probe (dashed gray arrow). Since both bands were diminished by antibody supershift and cold oligonucleotide competition assays, this suggested that the WT1 protein may have formed a multimeric complex. We also asked whether the zinc finger transcription factor, SP1, known to regulate the VEGF core promoter region, could bind to the same sequences as WT1. No SP1 site was identified by MatInspector, but surprisingly, 200 ng of recombinant SP1 protein (Promega, Madison, WI) bound to the 2310-2343 sequence (Figure 6 Panel C lanes 1-3). Competition with 20-fold (L, Panel C lane 2) and 80-fold (H, Panel C lane 3) molar excess of unlabeled SP1 oligonucleotide (Promega, Madison, WI) decreased binding of SP1 to the labeled probe. The SP1 complex appeared larger than the WT1 complex as the in vitro translated WT1 protein was not full-length.
Figure 6.
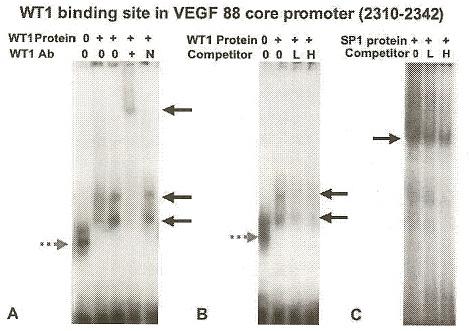
A functional WT1 binding site was identified in the VEGF core promoter (VEGF 88). Electrophoretic mobility shift assays (EMSA) showed that WT1 protein bound VEGF oligonucleotide 2310-2342 (Table 1) and specificity was demonstrated by Ab supershift (Panel A) and unlabeled control probe competition (Panel B). This oligonucleotide also bound SP1 (Panel C). Radio-labeled oligonucleotide 2310-2342 from the VEGF 88 core promoter (Table 1) was incubated with 1 microgram recombinant WT1(-KTS) protein (+, Panel A lanes 2-5 and Panel B lanes 2-4), or 200 ng of recombinant SP1 protein (Promega, Madison WI) (+, Panel C lanes 1-3). DNA-protein complexes were electrophoresed, and visualized by autoradiography (black arrows) as described in the text. Panel A, Addition of WT1 antibody (+, lane 4) shifted the complex upward, whereas addition of normal rabbit serum (N, lane 5) did not alter complex migration. Panel B, Competition with 20-fold (L, lane 3) and 80-fold (H, lane 4) molar excess of unlabeled control EGR-1 oligonucleotide (Table 1), known to bind WT1 (26), decreased WT1 binding to labeled VEGF oligonucleotide. Panel C. Specific binding by SP1 was demonstrated by competition with 20-fold (L, lane 2) and 80-fold (H, lane 3) molar excess of unlabeled control SP1 oligonucleotide (Table 1). Labeled oligonucleotide without protein formed a nonspecific band (gray dashed arrow) (Lane 1, Panels A and B).
The reporter assays also indicated a functional WT1 site existed in the VEGF 411 region (5’ of the core promoter), so the 2109-2138 oligonucleotide, was also tested for binding activity (Figure 7). As observed for the core promoter site, the identity of the WT1 protein was confirmed by supershift assay (Figure 7 Panel B) using WT1 Ab (Panel B lanes 1 and 2) compared to control sera (Panel Blane 3) and specificity was confirmed by competition (Figure 7 panel A) with 25-fold (L, panel A, lane 1) and 100-fold (panel A, lane 2) molar excess of unlabeled EGR1 oligonucleotide. Again the appearance of two complexes (black arrows) suggested a multimeric complex. Since an overlapping SP1 binding site was identified in this sequence (Table 1), we confirmed that 200 ng recombinant SP1 protein would bind and verified specificity by competition with 20- and 80-fold molar excess of unlabeled SP1 oligonucleotide (Promega, Madison, WI) as described for Figure 6. As expected a single band was observed (data not shown).
Figure 7.
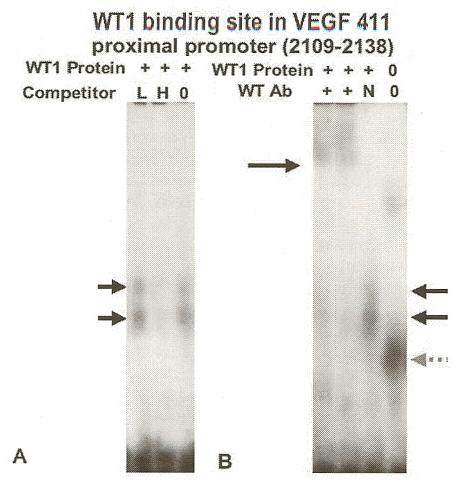
A functional WT1 binding site was identified in the VEGF proximal promoter (VEGF 411). EMSA showed that WT1 protein bound VEGF oligonucleotide 2109-2138 (Table 1) and specificity was demonstrated by unlabeled probe competition (Panel A) and Ab supershift (Panel B). Radio-labeled oligonucleotide 2109-2138 from the proximal promoter region (Table 1) was incubated with 1 microgram recombinant WT1(-KTS) protein (+, lanes 1-3 Panels A and B) and formed DNA-protein complexes (black arrows) as described in Figure 6. Panel A, Competition with 25-fold (L, lane 1) and 100-fold (H, lane 2) molar excess of unlabeled EGR-l oligonucleotide (Table 1) decreased binding. Panel B, Addition of WT1 antibody (+, lanes 1 and 2) shifted the complex upward, whereas addition of normal rabbit serum (N, lane 3) did not alter migration. Labeled oligonucleotide without protein formed a nonspecific band (gray dashed arrow) (Lane 4).
Overall, these results indicated that transcriptional regulation of the VEGF promoter was likely mediated by WT1 binding at the 2109-2138 and/or the 2310-2343 sites within the VEGF proximal promoter. Conversely, potential WT1 sites that were unlikely to contribute to this regulation included the 575-605 site in the distal promoter region, and the 2281-2310 and 2294-2330 sites in VEGF 88 region. Potential competition between WT1 and SP1 binding was suggested by the presence of a canonical SP1 site at 2109-2138 overlapping the WT1 site (Table 1) and observed SP1 binding at both sites.
5. DISCUSSION
The results presented here are a continuation of earlier studies using microarray analyses to identify physiologically relevant WT1 target genes in prostate cancer cells (34). A primary result of the previous study was that VEGF was identified as a potential WT1 target gene in LNCaP cells, a finding supported by the results of the transfection and DNA binding assays described here. The identification of two functional WT1 binding sites within the GC-rich proximal VEGF promoter region supports a novel mechanism of transcriptional regulation of VEGF that contributes to our understanding of angiogenesis.
Additionally, these results suggested a possible explanation for recent studies of angiogenesis in murine kidney cultures that demonstrate WT1 stimulates growth and nephrogenesis by inducing vegfa in metanephric mesenchyme (39). WT1 is an essential regulator of nephrogenesis (40-42) and has been suggested to play an important regulatory role in angiogenesis (34, 39, 43). WT1 and VEGF are co-expressed in normal podocytes, but also in some Wilms’ tumors (42-44). In addition to the kidney, WT1 is expressed in many other organs (20), including hematopoietic tissues such as the spleen, fetal liver, and bone marrow (17, 45, 46) where tightly regulated VEGF expression is required. For all these reasons, it seemed likely that VEGF was a physiologically relevant target of WT1 regulation in several different tissues, including prostate. VEGF expression is elevated in prostate cancer cells (47) and WT1 mRNA has been observed in cultured prostate cancer cells (27, 48) and in some prostate cancer sections (Brown K, and Fraizer G, unpublished). Interestingly, a truncated WT1 variant has been identified in a metastatic prostate cancer cell line (49). The finding that WT1 regulated VEGF in prostate cancer cells is consistent with its presence in some prostate cells and its ability to regulate growth control pathways important in prostate cancer (24-32).
Although WT1-mediated transcriptional regulation of VEGF expression was demonstrated in these studies, VEGF is regulated at many levels, both transcriptionally and post-transcriptionally. Indeed, many of the growth factors that affect VEGF levels are themselves regulated by WT1; such as platelet-derived growth factor (PDGF) (50, 51), transforming growth factor-beta 1 (TGF-b) (52), insulin-like growth factor II (IGFII) (24), insulin-like growth factor I receptor (IGF-IR) (25), and epidermal growth factor receptor (EGFR) (53). Thus, in addition to the direct transcriptional regulation demonstrated here, WT1 could also indirectly regulate VEGF expression. Furthermore, WT1 has been demonstrated to act as both a transcriptional repressor and an activator, depending upon the gene target, the cells transfected and expression vector utilized (54). The varied responses of the VEGF proximal promoter to WT1 expression in LNCaP prostate cancer cells versus the EIA-immortalized human embryonic kidney HEK-293 cells could be explained by differences in cellular co-factors that determine whether WT1 acts as an activator or a repressor. Amongst other functions, the WT1 zinc finger domain serves as a protein interaction domain for other transcription factors or as a DNA binding domain for coactivator/co-repressor proteins (28, 55-60). This is important for understanding VEGF regulation, as WT1 may be a part of a larger complex that binds and regulates the VEGF promoter in LNCaP cells. Indeed, the cAMP responsive element binding protein (CREB)-binding protein (CBP), important in both androgen signaling (61) and angiogenesis (62, 63), forms a complex with WT1 that activates transcription of amphiregulin (59). Thus, the regulation of VEGF by WT1 will be influenced by the presence of co-factors found within prostate cancer cells.
Based on previous microarray expression results of a stably transfected WT-LNCaP cell line, we had originally predicted that WT1 would repress transcription of VEGF promoter constructs (34). The expected results were obtained in transient transfection assays of HEK-293 cells, but not LNCaP cells, where WT1 activated the VEGF proximal promoter. This was unexpected as previous results showed VEGF transcripts reduced in cells engineered to express WT1 (relative to cells stably transfected with vector control). Similarly, while LNCaP reporter assays demonstrated hormone responsive activation, treatment with R1881 did not significantly affect VEGF mRNA (measured by quantitative real-time-PCR) in the WT-LNCaP line (34). One explanation for the differences between these results is that the former expression studies were performed on a stably transfected, G418-selected LNCaP line that may not respond to WT1 and R1881 treatments in the same manner as does transiently transfected LNCaP cells. However, endogenous VEGF expression in LNCaP cells is hormone responsive (7, 64), so in some respects the transient assays are a better model system to study mechanisms of VEGF regulation.
Overall, transfection of hormone-responsive LNCaP cells showed that WT1 strongly enhanced VEGF promoters in the presence of hormone and only modestly activated them in its absence. Similarly, WT1 only weakly modulated VEGF reporters in the androgen-independent PC3 prostate cancer cell line. This finding is consistent with the notion that WT1 may interact with components of the androgen signaling pathway in responsive cells (26, 27) and with the observation that both SP1 and WT1 can modulate the estrogen signaling pathway in breast cancer cells by forming complexes with ER-alpha (10, 11, 15). One intriguing possibility is that hormone responsive coactivators present in LNCaP cells are absent or inactive in the unresponsive PC3 and 293 cells. This possibility is consistent with our recent findings that both VEGF promoter and pRL-null control constructs (35) are up-regulated (5- and 6-fold, respectively) by R1881 treatment of WT1 transfected LNCaP cells (compared to untreated cells). The interaction between WT1 and androgen is a current focus of our studies aimed at delineating how WT1 might enhance transcription of hormone responsive genes.
6. ACKNOWLEDGMENTS
We gratefully acknowledge receipt of the VEGF luciferase constructs from Dr. K. Xie, the DDS-WT1 constructs from Dr. J. Pelletier, the pCB6+WT1 constructs and recombinant WT1 protein from Dr. F. Rauscher. We thank Yasmeen Tandon and Jennifer Cash for their excellent technical assistance and Dr. D. Stroup for helpful comments. These studies were supported by the NIH (RI5CA113360) and the Kent State University Research Council, and are dedicated to the memory of Dr. Grady F. Saunders.
Abbreviations
- VEGF
vascular endothelial growth factor
- WT1
Wilms tumor gene
- DDS
Denys-Drash syndrome
- Egr-1
early growth response-1
- AR
androgen receptor
- ER
estrogen receptor
- IGF-1R
insulin-like growth factor I receptor
- EMSA
electrophoretic mobility shift assay
- ChS
charcoal-dextran stripped
- FCS
fetal calf serum
7. REFERENCES
- 1.Jemal A, Murray T, Ward E, Samuels A, Tiwari R, Ghafoor A, Feuer EJ, Thun MJ. Cancer Statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J, Shing Y. Angiogenesis. The Journal of Biological Chemistry. 1992;267:10931–10934. [PubMed] [Google Scholar]
- 3.Breier G. Functions of the VEGF/VEGF receptor system in the vascular system. Seminars in thrombosis and hemostasis. 2000;26:553–559. doi: 10.1055/s-2000-13212. [DOI] [PubMed] [Google Scholar]
- 4.Fox WD, Higgins B, Maiese KM, Drobnjak M, Cordon-Cardo C, Scher HI, Agus DB. Antibody to vascular endothelial growth factor slows growth of an androgen-independent xenograft model of prostate cancer. Clinical Cancer Research. 2002;8:3226–3231. [PubMed] [Google Scholar]
- 5.Loureiro RMB, D′Amore PA. Transcriptional regulation of vascular endothelial growth factor in cancer. Cytokine and Growth Factor Reviews. 2005;16:77–89. doi: 10.1016/j.cytogfr.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocrine Reviews. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- 7.Sordello S, Bertrand N, Plouet J. Vascular Endothelial Growth Factor is Up-Regulated in Vitro and in Vivo by Androgens. Biochem Biophys Res Commun. 1998;251 doi: 10.1006/bbrc.1998.9328. [DOI] [PubMed] [Google Scholar]
- 8.Joseph IBJK, Nelson JB, Denmeade SR, Isaacs JT. androgens Regulate Vascular Endothelial Growth Factor Content in Normal and Malignant Prostatic Tissue. Clin Cancer Res. 1997;3:2507–2511. [PubMed] [Google Scholar]
- 9.Aslan G, Cimen S, Yorukoglu K, Tuna B, Sonmez D, Mungan U, Celebi I. Vascular endothelial growth factor expression in untreated and androgen-deprived patients with prostate cancer. Pathology-Research and Practice. 2005;201:593–598. doi: 10.1016/j.prp.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Mueller MD, Vigne J-L, Minchenko A, Lebovic DI, Leitman DC, Taylor RN. Regulation of Vascular Endothelial Growth Factor (VEGF) Gene Transcription by Estrogen Receptors a and b. PNAS. 2000;97:10972–10977. doi: 10.1073/pnas.200377097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stoner M, Wormke M, Saville B, Samudio I, Qin C, Abdelrahim M, Safe S. Estrogen regulation of vascular endothelial growth factor gene expression in ZR-75 breast cancer cells through interaction of estrogen receptor a and SP proteins. Oncogene. 2004;23:1052–1063. doi: 10.1038/sj.onc.1207201. [DOI] [PubMed] [Google Scholar]
- 12.Shi Q,LX, Abbruzzese JL, Peng Z, Qian C-N, Tang H, Xiong Q, Wang B, Li X-C, Xie K. Constitutive Sp1 Activity is Essential for Differential Constitutive Expression of Vascular Endothelial Growth Factor in Human Pancreatic Adenocarcinoma. Cancer Research. 2001;61:4143–4154. [PubMed] [Google Scholar]
- 13.Schafer G, Cramer T, Suske G, Kemmner W, Wiedenmann B, Hocker M. Oxidative Stress Regulates Vascular Endothelial Growth Factor-A Gene Transcription through Sp1- and Sp3-dependent Activation of Two Proximal GC-rich Promoter Elements. The Journal of Biological Chemistry. 2003;278:8190–8198. doi: 10.1074/jbc.M211999200. [DOI] [PubMed] [Google Scholar]
- 14.Ruiz M, Pettaway C, Song R, Stoeltzing O, Ellis L, Bar-Eli M. Activator Protein 2a Inhibits Tumorigenicity and Represses Vascular Endothelial Growth Factor Transcription in Prostate Cancer Cells. Cancer Research. 2004;64:631–638. doi: 10.1158/0008-5472.can-03-2751. [DOI] [PubMed] [Google Scholar]
- 15.Reizner N, Maor S, Sarfstein R, Abramovitch S, Welshons WV, Curran EM, Lee AV, Werner H. The WT1 Wilms’ tumor suppressor gene product interacts with estrogen receptor-a and regulates IGF-I receptor gene transcription in breast cancer cells. Journal of Molecular Endocrinology. 2005;35:135–144. doi: 10.1677/jme.1.01761. [DOI] [PubMed] [Google Scholar]
- 16.Madden SL, Cook DM, Morris JF, Gashler A, Sukhatme VP, Rausher FJ., III Transcription Repression Mediated by the WT1 Wilms Tumor Gene Product. Science. 1991;253:1550–1553. doi: 10.1126/science.1654597. [DOI] [PubMed] [Google Scholar]
- 17.Renshaw J, King-Underwood L, Pritchard-Jones K. Differential Splicing of Exon 5 of the Wilms Tumour (WT1) Gene. Genes Chromosomes Cancer. 1997;19:256–266. doi: 10.1002/(sici)1098-2264(199708)19:4<256::aid-gcc8>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 18.Rauscher FJ, Morris JF, Touruay OE, Cook DM, Curran T. Binding of the Wilms’ tumor locus zinc finger protein to the EGR-1 consensus sequence. Science (Washington DC) 1990;250:1259–1262. doi: 10.1126/science.2244209. [DOI] [PubMed] [Google Scholar]
- 19.Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC, Striegel JE, Houghton DC, Junien C, Habib R, Fouser L. Germline Mutations in the Wilms′ Tumor Suppressor Gene are Associated with Abnormal Urogenital Development in Denys-Drash Syndrome. Cell. 1991;67:437–447. doi: 10.1016/0092-8674(91)90194-4. [DOI] [PubMed] [Google Scholar]
- 20.Mrowka C, Schedl A. Wilms′ Tumor Suppressor Gene WT1: From Structure to Renal Pathophysiologic Features. J Am Soc Nephrol. 2000;11:S106–S115. [PubMed] [Google Scholar]
- 21.Rivera MN, Haber DA. Wilms′ Tumor: Connecting tumorigenesis and organ development in the kidney. Nature Rev Cancer. 2005;5:699–712. doi: 10.1038/nrc1696. [DOI] [PubMed] [Google Scholar]
- 22.Roberts S. Transcriptional regulation by WT1 in development. Current Opinion in Genetics & Development. 2005;15:542–547. doi: 10.1016/j.gde.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Loeb DM, Sukumar S. The role of WT1 in oncogenesis: tumor suppressor or oncogene. International Journal of Hematology. 2002;76:117–126. doi: 10.1007/BF02982573. [DOI] [PubMed] [Google Scholar]
- 24.Drummond IA, Madden SL, Rohwer-Nutter P, Bell GI, Sukhatme VP, Rauscher FJ., III Repression of the insulin-like growth factor II gene by the Wilms′ tumor suppressor WT1. Science (Washington DC) 1992;257:674–678. doi: 10.1126/science.1323141. [DOI] [PubMed] [Google Scholar]
- 25.Werner H, Rauscher FJ, Sukhatme VP, Drummond IA, Roberts CT, Leroith D. Transcriptional repression of the insulin-like growth factor I receptor (IGF-IR) gene by the tumor suppressor WT1 involves binding to sequences both upstream and downstream of the IGF-I-R gene transcription start site. J. Biol. Chem. 1994;269:12577–12582. [PubMed] [Google Scholar]
- 26.Shimamura R, Fraizer GC, Trapman J, Lau YC, Saunders GF. The Wilms′ Tumor Gene WT1 can Regulate Genes Involvd in Sex Determination and Differentiation: SRY, Mullerian-inhibiting Substance, and the Androgen Receptor. Clin Cancer Res. 1997;3:2571–2580. [PubMed] [Google Scholar]
- 27.Zaia A, Fraizer GC, Piantanelli L, Saunders GF. Transcriptional Regulation of the Androgen Signaling Pathway by the Wilms′ Tumor Suppressor Gene WT1. Anticancer Res. 2001;21:1–10. [PubMed] [Google Scholar]
- 28.Cheema SK, Mishra SK, Rangnekar VM, Tari AM, Kumar R, Lopez-Berestein G. Par-4 Transcriptionally Regulates Bcl-2 Through WT1-binding Site on the Bcl-2 Promoter. J Biol Chem. 2003;278:19995–20005. doi: 10.1074/jbc.M205865200. [DOI] [PubMed] [Google Scholar]
- 29.Hewitt S, Hamada S, McDonnell T, Rauscher F, Saunders GF. Regulation of the Proto-oncogenes bcl-2 and c-myc by the Wilms′ Tumor Suppressor Gene WT1. Cancer Res. 1995;55:5386–5389. [PubMed] [Google Scholar]
- 30.Heckman C, Mochon E, Arcinas M, Boxer L. The WT1 Protein is a Negative Regulator of the Normal bcl-2 Allele in t (14;18) Lymphomas. J Bioi Chem. 1997;272:19609–14. doi: 10.1074/jbc.272.31.19609. [DOI] [PubMed] [Google Scholar]
- 31.Morrison DJ, English MA, Licht JD. WT1 induces apoptosis through transcriptional regulation of the proapoptotic Bcl-2 family member Bak. Cancer Res. 2005;15:8174–8182. doi: 10.1158/0008-5472.CAN-04-3657. [DOI] [PubMed] [Google Scholar]
- 32.Johnstone RW, See RH, Sells SF, Wang J, Muthukkumar S, Englert C, Haber DA, Licht JD, Sugrue SP, Roberts T, Rangnekar VM, Shi Y. A Novel Repressor, par-4, Modulates Transcription and Growth Suppression Functions of the Wilms′ Tumor Suppressor WT1. Mol Cell Biol. 1996;16:6945–56. doi: 10.1128/mcb.16.12.6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraizer GC, Leahy R, Priyadarshini S, Graham K, Delacerada J, Diaz M. Suppression of Prostate Tumor Cell Growth in vivo by WT1, the Wilms′ Tumor Suppressor Gene. Int J Oncol. 2004;24:461–471. [PubMed] [Google Scholar]
- 34.Graham K, Li W, Williams BRG, Fraizer G. Vascular endothelial growth factor (VEGF) is suppressed in WT1-transfected LNCaP cells. Gene Expression. 2006;13:1–14. doi: 10.3727/000000006783991953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanson J, Reese J, Gorman J, Cash J, Fraizer G. Hormone treatment enhances WT1 activation of Renilla luciferase constructs in LNCaP cells. Frontiers in Bioscience. 2007;12:1387–1394. doi: 10.2741/2155. [DOI] [PubMed] [Google Scholar]
- 36.Brown TR, Rothwell SW, Migeon CJ. Comparison of methyltrienolone and dihydrotestosterone binding and metabolism in human genital skin fibroblasts. Journal of Steroid Biochemistry. 1981;14:1013–1022. doi: 10.1016/0022-4731(81)90209-0. [DOI] [PubMed] [Google Scholar]
- 37.Sims RJ, III, Liss AJ, Gottlieb PD. Normalization of luciferase reporter assays under conditions that alter internal controls. Biotechniques. 2003;34:938–940. doi: 10.2144/03345bm07. [DOI] [PubMed] [Google Scholar]
- 38.Quandt K, French K, Karas H W, Wingender E, Werner T. MatInd and MatInspector-New fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Research. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao X, Chen X, Taglienti M, Rumballe B, Little M, Kreidberg J. Angioblast-mesenchyme induction of early kidney development is mediated by WT1 and Vegfa. Development. 2005;132:5437–5449. doi: 10.1242/dev.02095. [DOI] [PubMed] [Google Scholar]
- 40.Hammes A, Guo JK, Lutsch G, Leheste JR, Landrock D, Ziegler U, Gubler MC, Schedl A. Two Splice Variants of the Wilms′ Tumor 1 Gene have Distinct Functions During Sex Determination and Nephron Formation. Cell. 2001;106:319–329. doi: 10.1016/s0092-8674(01)00453-6. [DOI] [PubMed] [Google Scholar]
- 41.Kreidberg J, Sariola H, Loring J, Maeda M, Pelletier J, Housman D, Jaenisch R. WT1 is Required for Early Kidney Development. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- 42.Armstrong JF, Pritchard-Jones K, Bickmore WA, Hastie ND, Bard JBL. The expression of the Wilms’ tumor gene, WT1, in the developing mammalian embryo. Mech. Develop. 1992;40:85–97. doi: 10.1016/0925-4773(93)90090-k. [DOI] [PubMed] [Google Scholar]
- 43.Baudry D, Faussillon M, Cabanis M, Rigolet M, Zucker J, Patte C, Sarnacki S, Boccon-Gibod L, Junien C, Jeanpierra C. Changes in WT1 Splicing are Associated with a Specific Gene Expression Profile in Wilms′ Tumour. Oncogene. 2002;21:5566–5573. doi: 10.1038/sj.onc.1205752. [DOI] [PubMed] [Google Scholar]
- 44.Karth J, Ferrer F, Perlman E, Hanrahan C, Simons JW, Gearhart JP, Rodriguez R. Coexpression of Hypoxia-Inducible Factor 1-alpha and Vascular Endothelial Growth Factor in Wilms′ Tumor. Journal of Pediatric Surgery. 2000;35:1749–1753. doi: 10.1053/jpsu.2000.19241. [DOI] [PubMed] [Google Scholar]
- 45.Fraizer GC, Patmasiriwat P, Zhang XH, Saunders GF. Expression of the tumor suppressor gene WT1 in both human and mouse bone marrow. Blood. 1995;86:4704–4707. [PubMed] [Google Scholar]
- 46.Algar E. A review of the Wilms′ tumor 1 gene (WT1) and its role in hematopoiesis and leukemia. J Hematother Stem Cell Res. 2002;11:589–599. doi: 10.1089/15258160260194749. [DOI] [PubMed] [Google Scholar]
- 47.Mazzucchelli R, Montironi R, Santinelli A, Lucarinia G, Pugnaloni A, Biagini G. Vascular Endothelial Growth Factor Expression and Capillary Architecture in High-Grade PIN and Prostate Cancer in Untreated and Androgen-Ablated Patients. Prostate. 2000;45:72–79. doi: 10.1002/1097-0045(20000915)45:1<72::aid-pros9>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 48.Dong G, Rajah T, Vu T, Hoffman A, Rosenfeld R, Roberts C, Peehl D, Cohen P. Decreased Expression of Wilms′ Tumor Gene WT-1 and Elevated Expression of Insulin Growth Factor-II and Type 1 IGF Receptor Genes in Prostatic Stromal Cells from Patients with Benign Prostatic Hyperplasia. J Clin Endocrin and Metabolism. 1997;82:2198–2203. doi: 10.1210/jcem.82.7.4067. [DOI] [PubMed] [Google Scholar]
- 49.Dechsukhum C, Ware JL, Ferreira-Gonzalez A, Wilkinson DS, Garrett CT. Detection of a novel truncated WT1 transcript in human neoplasia. Mol Diagn. 2000;5:117–128. doi: 10.1007/BF03262030. [DOI] [PubMed] [Google Scholar]
- 50.Gashler AL, Bonthron DT, Madden SL, Rauscher FJ, Collins T, Sukhatme VP. Human platelet-derived growth factor A chain is transcriptionally repressed by the Wilms tumor suppressor WT1. Proc Natl Acad Sci USA. 1992;89:10984–10988. doi: 10.1073/pnas.89.22.10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang ZY, Madden SL, Deuel TF, Rauscher FJ., III The Wilms’ tumor gene product, WT1, represses transcription of the platelet-derived growth factor A-chain gene. J Biol Chern. 1992;267:21999–22002. [PubMed] [Google Scholar]
- 52.Dey BR, Sukhatme VP, Roberts AB, Sporn MB, Rauscher FJ, Kim S-J. Repression of the transforming growth factor-beta-1 gene by the Wilms’ tumor suppressor WT1 gene product. Mol Endocrinol. 1994;8:595–602. doi: 10.1210/mend.8.5.8058069. [DOI] [PubMed] [Google Scholar]
- 53.Englert C, Hou X, Maheswaran S, Bennett P, Ngwu C, Re GG, Garvin AJ, Rosner MR, Haber DA. WT1 suppresses synthesis of the epidermal growth factor receptor and induces apoptosis. Embo J. 1995;14:4662–4675. doi: 10.1002/j.1460-2075.1995.tb00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reddy JL, Licht J. The WT1 Wilms’ tumor suppressor gene: how much do we really know. BBA. 1996;1287:1–28. doi: 10.1016/0304-419x(95)00014-7. [DOI] [PubMed] [Google Scholar]
- 55.Roberts S. The modulation of WT1 transcription by co factors. Biochem. Soc. Symp. 2006;73:191–201. doi: 10.1042/bss0730191. [DOI] [PubMed] [Google Scholar]
- 56.Nachtigal MW, Hirokawa Y, Enyeart-VanHouten DL, Flanagan IN, Hammer GD, Ingraham HA. Wilms′ Tumor 1 and Dax-l Modulate the Orphan Nuclear Receptor SF-1 in Sex-specific Gene Expression. Cell. 1998;93:445–454. doi: 10.1016/s0092-8674(00)81172-1. [DOI] [PubMed] [Google Scholar]
- 57.Hossain A, Saunders GF. Role of WT1 in the Transcriptional Regulation of the Mullerian Inhibiting Substance Promoter. Biology of Reproduction. 2003;69:1808–1814. doi: 10.1095/biolreprod.103.015826. [DOI] [PubMed] [Google Scholar]
- 58.Idelman G, Glaser T, Roberts CT, Jr., Werner H. WT1-p53 Interactions in Insulin-like Growth Factor-I Receptor Gene Regulation. J BioI Chern. 2003;278:3474–3482. doi: 10.1074/jbc.M211606200. [DOI] [PubMed] [Google Scholar]
- 59.Wang W, Lee SB, Palmer R, Ellisen LW, Haber DA. A Functional Interaction with CBP Contributes to Transcriptional Activation by the Wilms Tumor Suppressor WT1. J Biol Chern. 2001;276:16810–6. doi: 10.1074/jbc.M009687200. [DOI] [PubMed] [Google Scholar]
- 60.Richard D, Schumacher V, Royer-Pokora B, Roberts S. Par4 is a coactivator for Splice Isoform-specific Transcriptional Activation Domain in WT1. Genes Dev. 2001;15:328–339. doi: 10.1101/gad.185901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Culig Z, Comuzzi B, Steiner H, Bartsch G, Hobisch A. Expression and function of androgen receptor coactivators in prostate cancer. J Steroid Biochem Mol Biol. 2004;92:265–271. doi: 10.1016/j.jsbmb.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 62.Gray MJ, Zhang J, Ellis LM S, Semenza GL, Evans DB, Watowich SS, Gallick GE. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24:3110–3120. doi: 10.1038/sj.onc.1208513. [DOI] [PubMed] [Google Scholar]
- 63.Oike Y, Takakura N, Hata A, Kaname T, Akizuki M, Yamaguchi Y, Yasue H, Araki K, Yamamura K, Suda T. Mice homozygous for a truncated form of CREB-binding protein exhibit defects in hematopoiesis and vasculo-angiogenesis. Blood. 1999;93:2771–2779. [PubMed] [Google Scholar]
- 64.Stewart RJ, Panigrahy D, Flynn E, Folkman J. Vascular Endothelial Growth Factor Expression and Tumor Angiogenesis are Regulated by Androgens in Hormone Responsive Human Prostate Carcinoma: Evidence for Androgen Dependent Destabilization of Vascular Endothelial Growth Factor Transcripts. J Ural. 2001;165:688–693. doi: 10.1097/00005392-200102000-00095. [DOI] [PubMed] [Google Scholar]