

Abstract
An early event of cell migration is characterized as the rapid reorganization of the actin cytoskeleton. Recently we have demonstrated that rapamycin inhibits tumor cell motility. To understand the underlying mechanism, this study was set to determine whether rapamycin inhibition of cell motility is related to its prevention of F-actin reorganization. We found that rapamycin prevented type I insulin-like growth factor (IGF-I)-stimulated F-actin reorganization in human rhabdomyosarcoma (Rh30), Ewing sarcoma (Rh1), glioblastoma (U-373) and prostate carcinoma (PC-3) cells, and concurrently inhibited phosphorylation of focal adhesion proteins, including focal adhesion kinase (FAK), paxillin and p130Cas in the cells. The effect of rapamycin was blocked by expression of a rapamycin-resistant mutant of mTOR (mTORrr), but not a kinase-dead mTORrr. Downregulation of raptor mimicked the effect of rapamycin. Cells infected with a recombinant adenovirus expressing constitutively active and rapamycin-resistant mutant of p70 S6 kinase 1 (S6K1) conferred to resistance to rapamycin. Further, IGF-I failed to stimulate F-actin reorganization and phosphorylation of the focal adhesion proteins in the S6K1-downregulated cells. Expression of constitutively hypophosphoryated eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4E-BP1-5A) inhibited IGF-I-stimulated F-actin reorganization, but did not alter the cellular protein or phosphorylation levels of the focal adhesion proteins. The results suggest that rapamycin inhibits IGF-I-induced F-actin reorganization and phosphorylation of the focal adhesion proteins by disruption of mTOR-raptor complex. Both S6K1 and 4E-BP1 pathways, mediated by the mTOR-raptor complex, are involved in the regulation of IGF-I-stimulated F-actin reorganization, but only the former controls IGF-I-stimulated phosphorylation of the focal adhesion proteins.
Keywords: Rapamycin, mammalian target of rapamycin, F-actin, lamellipodia, focal adhesion kinase
Introduction
The mammalian target of rapamycin (mTOR), a serine/threonine (S/T) kinase, lies downstream of phosphatidylinositol 3′-kinase (PI3K), and is a central controller of cell proliferation and growth (Wullschleger et al., 2006). Two mTOR complexes (TORC1 and TORC2) have recently been identified (Guertin and Sabatini, 2007). TORC1 consists of mTOR, mLST8 (also termed G-protein β-subunit-like protein, GβL, a yeast homolog of LST8), PRAS40 (proline-rich Akt substrate 40 kDa) and raptor (regulatory associated protein of mTOR), and is sensitive to rapamycin (Fonseca et al., 2007; Hara et al., 2002; Kim et al., 2002; 2003; Loewith et al., 2002; Sancak et al., 2007; Vander Haar et al., 2007). TORC2 is composed of mTOR, mLST8, mSin1 (mammalian stress-activated protein kinase (SAPK)-interacting protein 1), rictor (rapamycin-insensitive companion of mTOR) and protor (protein observed with rictor), and is insensitive to rapamycin (Frias et al., 2006; Jacinto et al., 2004; 2006; Pearce et al., 2007; Sarbassov et al., 2004; Yang et al., 2006). TORC1 regulates phosphorylation of p70 S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4E-BP1) (Hara et al., 2002; Kim et al., 2002; Loewith et al., 2002), and TORC2 phosphorylates Akt at S473 (Sarbassov et al., 2005). Though the cellular functions of the mTOR complexes remain to be determined, current data indicate that the PI3K-mTOR signaling pathway regulates many cellular events, such as cell proliferation and growth, differentiation, survival and motility (Guertin and Sabatini, 2007; Wullschleger et al., 2006).
A variety of transformed or tumor cells with dysregulated mTOR signaling have shown higher susceptibility to inhibitors of mTOR than normal cells, forming a rationale for mTOR inhibitors as potential tumor-selective therapeutic agents. Clinical trials have demonstrated that rapamycin analogs CCI-779, RAD001 and AP23573 (designated rapamycins) are promising anticancer drugs. They specifically block the function of mTOR, inhibiting growth of numerous solid tumors (renal, breast, prostate, colon and brain cancers) with only mild side effects. Recently, the U. S. Food and Drug Administration approved CCI-779 (temsirolimus, Torisel™, Wyeth) for the treatment of advanced renal cell carcinoma, further revealing the potentiality of rapamycins in cancer therapy. However, to date, the anticancer mechanism of rapamycins remains to be defined. While intensive studies have focused on the anti-proliferative activity of rapamycins, their inhibitory effect on cell motility is only beginning to be recognized. Recently, inhibition of motility and invasion by rapamycin has been described in numerous cell lines in culture (ref. Liu et al., 2006). In vivo, rapamycin also potently inhibits metastases of transplanted tumors derived from murine adenocarcinoma cells (CT-26) (Guba et al., 2002), human renal cancer cells (RCC 786-O) (Luan et al., 2003), murine non-small cell lung cancer cells (KLN-205) (Boffa et al., 2004) and osteosarcoma (K7M2) (Wan et al., 2005) in mice. The data indicate that rapamycin is a potent inhibitor not only for cell proliferation and growth, but also for cell motility.
The type I insulin-like growth factor (IGF-I) receptor (IGF-IR) and its ligands, IGF-I and IGF-II, play critical roles in the regulation of cellular proliferation, survival and transformation (Guertin and Sabatini, 2007; Wullschleger et al., 2006). Increasing evidence indicates that IGF-I signaling is also pivotal to cell motility and invasion (LeRoith and Roberts, 2003). IGF-I-induced cell motility has been characterized in various cell types (Kadowaki et al., 1986; Leventhal et al., 1997; Guvakova et al., 1999). The earliest detectable morphological change induced by IGF-I is the reorganization of the actin cytoskeleton associated with the formation of lamellipodia (Kadowaki et al., 1986; Leventhal et al., 1997; Guvakova et al., 1999). Activation of the PI3K pathway by IGF-I is essential for this process, which is regulated through focal adhesion proteins, such as focal adhesion kinase (FAK), paxillin and p130Cas (Kadowaki et al., 1986; Leventhal et al., 1997; Guvakova and Surmacz, 1999). IGF-I induces rapid tyrosine phosphorylation of FAK, paxillin and p130Cas in human neuroblastoma cells (SH-SY5Y) and Swiss 3T3 cells (Leventhal et al., 1997; Guvakova et al., 1999), though disputation exists (Casamassima et al., 1998; Konstantopoulous and Clark, 1996). However, whether and how mTOR is involved in the regulation of F-actin reorganization is poorly understood, which links to the mechanism by which rapamycin inhibits cell motility. To address this issue, we studied whether rapamycin inhibition of cell motility is related to its prevention of F-actin reorganization. Here we show that rapamycin inhibits IGF-I-induced F-actin reorganization in a panel of tumor cell lines, such as human rhabdomyosarcoma (Rh30) and Ewing sarcoma (Rh1), glioblastoma (U-373) and prostate carcinoma (PC-3) cells. This may be related to rapamycin inhibition of IGF-I induced phosphorylation of focal adhesion proteins (FAK, paxillin and p130Cas), which is dependent on mTOR kinase activity. Disruption of mTOR-raptor complex, mimicking the effect of rapamycin, prevented IGF-I-stimulated F-actin reorganization and phosphorylation of the focal adhesion proteins. Both S6K1 and 4E-BP1 pathways are involved in the regulation of F-actin reorganization, but only the former controls phosphorylation of the focal adhesion proteins.
Results
Rapamycin inhibits IGF-I-stimulated F-actin reorganization in an mTOR kinase activity dependent manner
Cell migration is a highly concerted, cyclical, and multi-step process, including cell polarization/protrusion, adhesion and de-adhesion (Ridley et al., 2003). Inhibition of any of these steps may impair cell motility. Recently we have demonstrated that rapamycin inhibits IGF-I-stimulated cell motility (Liu et al., 2006). To further understand the underlying mechanism, this study first examined whether rapamycin inhibition of cell motility is related to its prevention of cell polarization/protrusion (F-actin reorganization). Consistent with a previous report (Liu et al., 2006), we found that rapamycin inhibited basal and IGF-I-stimulated motility of rhabdomyosarcoma (Rh30), glioblastoma (U-373), prostate cancer (PC-3), and Ewing sarcoma (Rh1) cells (Fig. 1A). Of interest, in response to stimulation with IGF-I (10 ng/ml), the actin cytoskeleton in Rh30 cells (Upper panel, Fig. 1B) underwent rapid reorganization. In the absence of IGF-I, F-actin was randomly distributed across Rh30 cells, as revealed by staining with FITC-conjugated phalloidin. Some F-actin was condensed in dot-like structures at the margins of the cells. However, within 1 h of stimulation with IGF-I, more F-actin was condensed at the leading edge within structures resembling fans or protrusions (termed lamellipodia, see arrows), compared to non-treated control cells. Rapamycin did not obviously alter the pattern of basal F-actin distribution. In contrast, treatment with rapamycin (100 ng/ml) for 2 h dramatically reduced IGF-I-stimulated F-actin reorganization at the leading edge. To examine whether this is dependent on cell-type, we also examined the effect of rapamycin on IGF-I-stimulated lamellipodia formation in other three highly motile cell lines, including U-373, PC-3 and Rh1 cells. Similar results were observed in U-373, PC-3 (Fig. 1B) and Rh1 cells (data not shown). The data suggest that rapamycin inhibition of cell motility is correlated to its prevention of F-actin reorganization.
Figure 1.

Rapamycin inhibits cell motility and prevents IGF-I-induced F-actin reorganization, which is independent of cell types. (A) Motility of the indicated cell lines was determined by the single cell motility assay, as described in Materials and methods. Results are means ± SD of 3–4 independent experiments. *P < 0.05, difference with control group. #P < 0.05, difference with IGF-I group. (B) Serum-starved (for 24 h) Rh30, U-373, and PC-3 cells grown on glass coverslips in 6-well-plates were pretreated with or without rapamycin (100 ng/ml) for 2 h, followed by stimulation with or without IGF-I (10 ng/ml) for 1 h. F-actin was stained as described in Materials and Methods. Actin filaments (F-actin) were visualized and photographed with a Nikon TE300 digital inverted microscope. Left panel shows representative photos. Formation of lamellipodia is shown (arrows). Bar = 20 μm. Right panel shows percentage of cells with lamellipodia formation. Results are means ± SD of 3 independent experiments. *P < 0.05, difference with control group. #P < 0.05, difference with IGF-I group.
To determine the role of mTOR kinase activity in IGF-I-stimulated lamellipodia formation, Rh1 and Rh30 clones expressing empty vector (pcDNA), rapamycin resistant mutant mTOR (mTOR-rr), and kinase-dead (mTOR-SIDA) were pretreated with or without rapamycin for 2 h, followed by stimulation with or without IGF-I for 1 h. We found that rapamycin failed to inhibit IGF-I-induced F-actin reorganization in Rh30 cells stably expressing rapamycin-resistant mutant mTOR (Middle panel, Fig. 2A). In contrast, cells stably expressing an empty vector (pcDNA3) (Upper panel, Fig. 2A) or a kinase-dead mTORrr (mTOR-SIDA) remained sensitive to rapamycin (Bottom panel, Fig. 2A). Similar data were observed in Rh1 cells (data not shown). The results suggest that IGF-I-induced F-actin reorganization is regulated in an mTOR kinase activity dependent manner.
Figure 2.

Rapamycin prevents IGF-I-induced F-actin reorganization in an mTOR kinase activity-dependent manner. (A) Serum-starved (for 24 h) Rh30/pcDNA, Rh30/mTORrr and Rh30/mTOR-SIDA cells grown on glass coverslips in 6-well-plates were pretreated with or without rapamycin (100 ng/ml) for 2 h, followed by stimulation with or without IGF-I (10 ng/ml) for 1 h. F-actin was stained, as described in Figure 1. Formation of lamellipodia is shown (arrows). Bar = 20 μm. (B) Lentiviral shRNA to mTOR, but not the control shRNA to GFP, downregulated mTOR and inhibited the basal or IGF-I-stimulated phosphorylation of S6K1 in Rh30 cells, as detected by Western blot analysis. (C) Lentiviral shRNA to mTOR blocked IGF-I-stimulated lamellipodia formation in Rh30 cells. Bar = 20 μm.
We also confirmed the above finding using RNA interference (RNAi). As shown in Fig. 2B, lentiviral shRNA to mTOR silenced expression of mTOR protein by ~90% in Rh30 cells, comparing with the control shRNA (to GFP). Downregulation of mTOR, mimicking the effect of rapamycin, dramatically decreased the mTOR kinase activity, since the basal or IGF-I-stimulated phosphorylation of S6K1 at T389, routinely used as an indicator of mTOR kinase activity (Guertin and Sabatini, 2007; Wullschleger et al., 2006), was not detectable by Western blot (Fig. 2B). As expected, downregulation of mTOR obviously prevented IGF-I-simulated F-actin reorganization (Fig. 2C).
mTOR-raptor complex controls F-actin reorganization
Two mTOR complexes (TORC1, mTOR-raptor, and TORC2, mTOR-rictor) have been identified recently (Guertin and Sabatini, 2007). Disruption of TORC2 prevents cell spreading and F-actin polymerization of NIH 3T3 cells (Jacinto et al., 2004), and inhibits cell motility (Liu et al., 2006). However, TORC2 is rapamycin-insensitive (Jacinto et al., 2004; Sarbassov et al., 2004). As described above, we have found that rapamycin inhibits F-actin reorganization in an mTOR-dependent manner. Therefore, we reasoned involvement of TORC1 in the regulation of such cellular event as well. To this end, lentiviral shRNA to raptor was used to downregulate raptor in Rh30 cells. Lentiviral shRNA to rictor was set as a positive control. shRNAs to raptor and rictor, but not the control shRNA to GFP, downregulated the protein levels of raptor and rictor by 90% and 85%, respectively (vs. control) in the cells (Fig. 3A). Consistent with previous findings (Hara et al., 2002; Kim et al., 2002; Jacinto et al., 2004; Sarbassov et al., 2004; 2005), downregulation of raptor, but not rictor, blocked mTOR-mediated phosphorylation of S6K1 and 4E-BP1, mimicking the effect of rapamycin, whereas downregulation of rictor inhibited phosphorylation of Akt (Fig. 3A). Of interest, downregulation of rictor dismantled F-actin stress fibers, induced F-actin condensation (white dots) in the cells, and abolished IGF-I-stimulated actin reorganization (Fig. 3B). However, downregulation of raptor only suppressed IGF-I-stimulated formation of lamellipodia, without influence on the actin stress fibers in Rh30 cells (Fig. 3B). The above data suggest that both TORC1 and TORC2 are involved in the regulation of F-actin reorganization, but only disruption of TORC1 mimicked the effect of rapamycin treatment. Also, the two mTOR complexes may regulate F-actin reorganization through different mechanisms.
Figure 3.
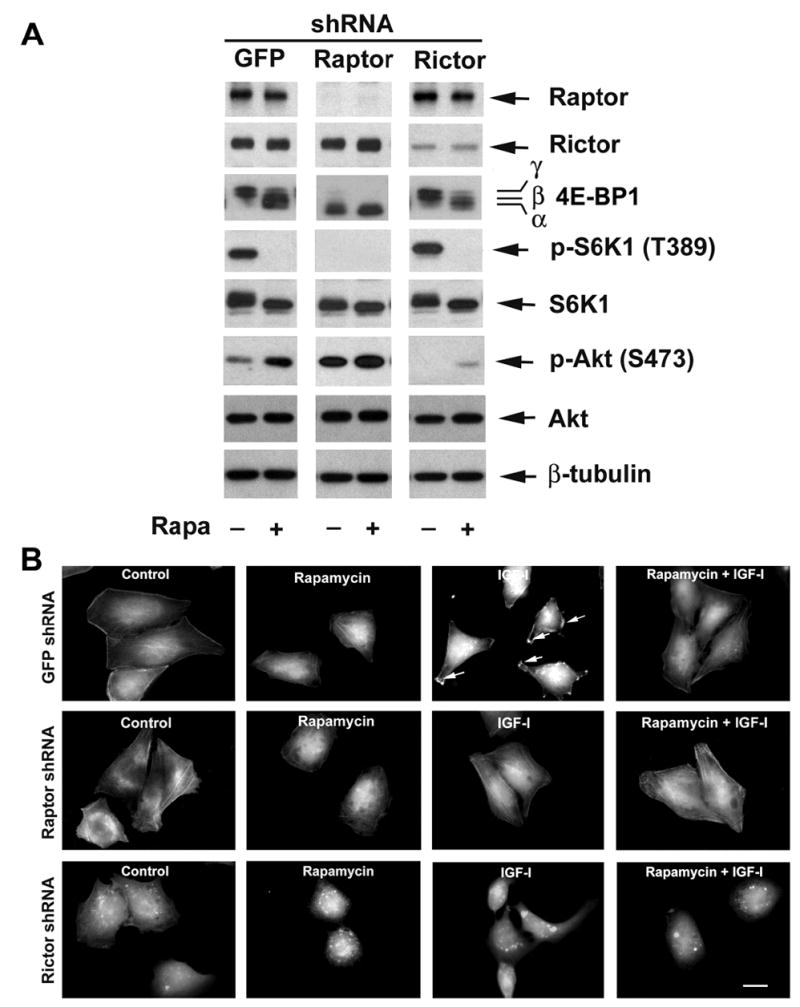
Downregulation of raptor or rictor blocks IGF-I-stimulated F-actin reorganization. (A) Lentiviral shRNA to raptor or rictor, but not GFP, downregulated raptor or rictor in Rh30 cells, respectively. Downregulation of raptor prevented IGF-I-stimulated phosphorylation of S6K1 (T389) and 4E-BP1 in Rh30 cells in serum-free medium, mimicking the effect of rapamycin, whereas downregulation of rictor only attenuated IGF-I-stimulated phosphorylation of Akt (S473), as determined by Western blotting. (B) F-actin was stained in raptor, rictor, and GFP downregulated Rh30 cells, respectively, as described in Figure 1. Formation of lamellipodia is shown (arrows). Bar = 20 μm.
Both S6K1 and 4E-BP1 pathways are essential for the regulation of F-actin reorganization
Phosphorylation of S6K1 has been reported to co-localize with F-actin arc at the leading edge in NIH 3T3 cells (Berveen et al., 2004). However, whether rapamycin inhibits F-actin reorganization through TORC1-mediated S6K1 and/or 4E-BP1 pathways is not clear. To address this question, expression of constitutively active and rapamycin-resistant S6K1 (S6K1-ca) and constitutively hypophosphorylated 4E-BP1 (4E-BP1-5A) was performed by infection of Rh30 cells with Ad-S6K1-ca and Ad-4EBP1-5A, respectively (Fig. 4A). Expression of S6K1-ca, but not GFP alone, induced F-actin reorganization even without stimulation with IGF-I, and conferred to resistance to rapamycin inhibition of formation of lamellipodia (Fig. 4B). In contrast, expression of 4E-BP1-5A, which binds eIF4E and quenches the function of eIF4E (Liu et al., 2006), inhibited lamellipodia formation in the presence of IGF-I (Fig. 4B).
Figure 4.
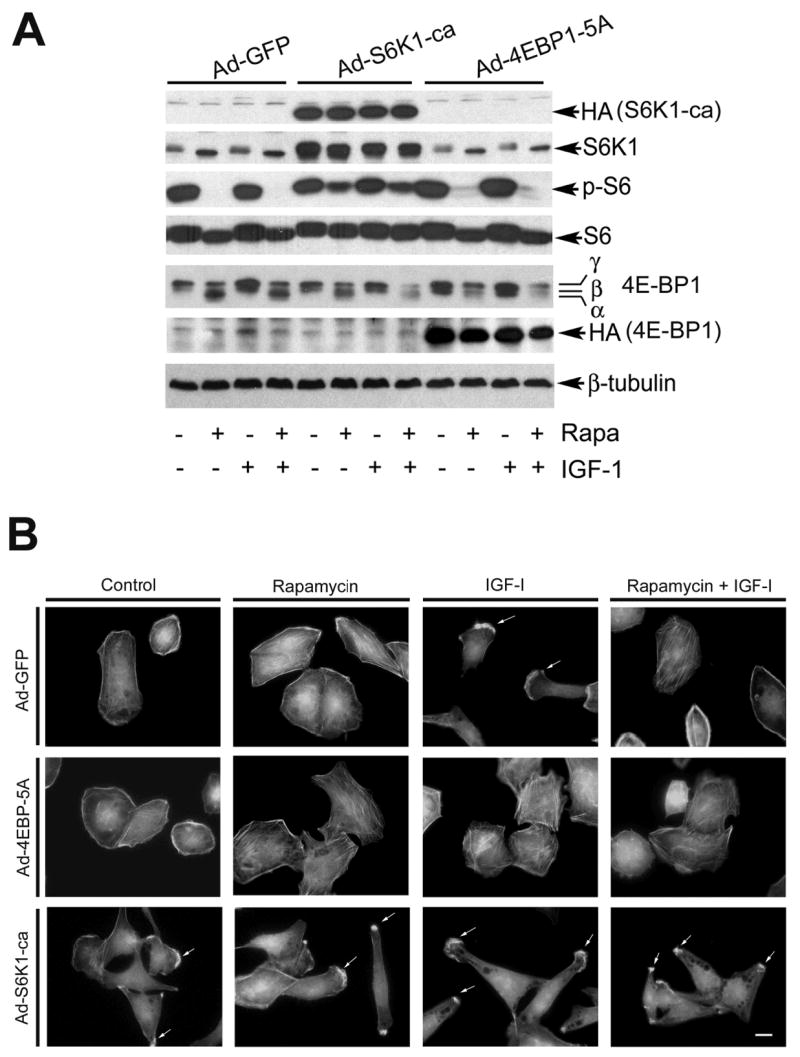
Expression of rapamycin-resistant and constitutively active S6K1 (S6K1-ca) induces lamellipodia formation and confers to resistance to rapamycin inhibition of F-actin reorganization, whereas expression of constitutively hypophosphorylated 4E-BP1 (4E-BP1-5A) directly prevented IGF-I-stimulated F-actin reorganization. (A) Serum-starved Rh30 cells, infected with Ad-S6K1-ca, Ad-4EBP1-5A or Ad-GFP (control), were pretreated with or without rapamycin (100 ng/ml) for 2 h, followed by stimulation with or without IGF-I (10 ng/ml) for 1 h. Expression of HA-tagged S6K1-ca and 4E-BP1-5A were detected Western blotting with antibodies to HA. The function of rapamycin-resistant and constitutively active S6K1 (S6K1-ca) was confirmed by detecting the phosphorylation of ribosomal protein S6, the substrate of S6K1. β-tubulin was used for loading control. (B) F-actin was stained in Ad-S6K1-ca, Ad-4EBP1-5A or Ad-GFP infected Rh30 cells, respectively, as described in Figure 1. Formation of lamellipodia is shown (arrows). Bar = 20 μm.
We also confirmed the above findings using RNAi technology. As shown in Fig. 5A, lentiviral shRNAs to 4E-BP1 and S6K1 downregulated protein expression of 4E-BP1 and S6K1 by ~90%, respectively. When S6K1 was downregulated by lentiviral shRNA to S6K1, Rh30 cells failed to form lamellipodia in the presence of IGF-I (Fig. 5B). On the other hand, when 4E-BP1 was downregulated by lentiviral shRNA to 4E-BP1, formation of lamellipodia occurred at the leading edge of the cells even without stimulation with IGF-I. Also, Rh30 cells were highly resistant to rapamycin treatment in the formation of lamellipodia (Fig. 5B). Consistent with the results obtained with raptor shRNA (Fig. 3), expression of 4E-BP1-5A (Fig. 4) or knockdown of S6K1 (Fig. 5) did not show obvious impact on the cytoskeletal structure of actin stress fibers. The findings reveal that both S6K1 and 4E-BP1 pathways are necessary for IGF-I-stimulated F-actin reorganization.
Figure 5.
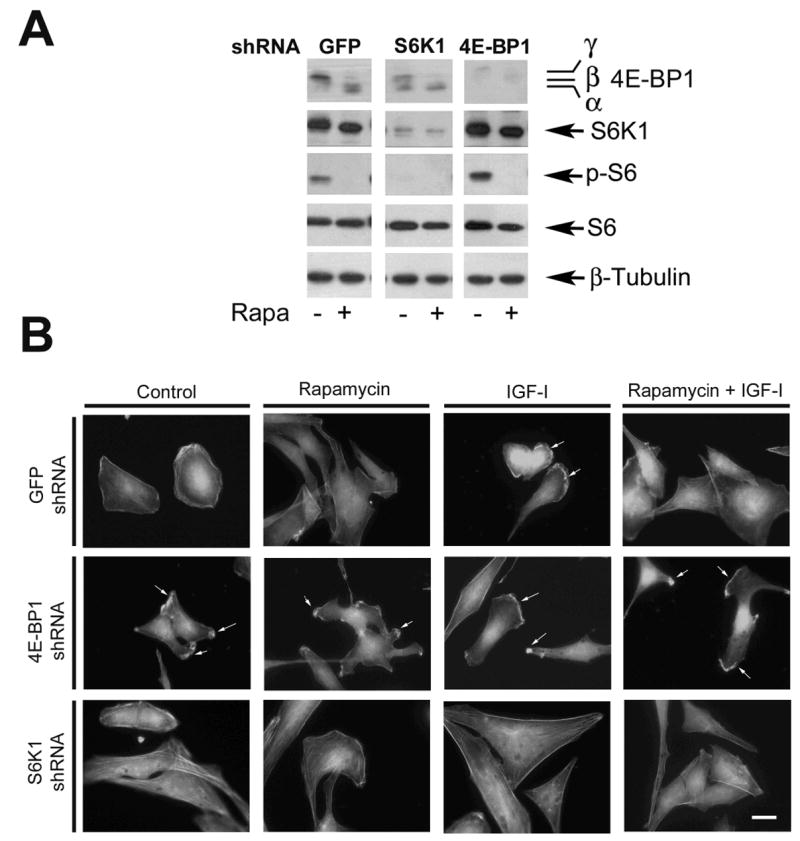
Downregulation S6K1 inhibits IGF-I-induced lamellipodia formation, whereas downregulation of 4E-BP1 induces F-actin reorganization. (A) S6K1 and 4E-BP1 were downregulated in Rh30 cells by lentiviral shRNAs to S6K1 and 4E-BP1, respectively, as detected by Western blotting. Note: Downregulation of S6K1 also inhibited phoshorylation of its substrate, ribosomal protein S6. (B) F-actin was stained in Rh30 cells, treated with lentiviral shRNAs to S6K1, 4E-BP1 and GFP, respectively, as described in Figure 1. Formation of lamellipodia is shown (arrows). Bar = 20 μm.
Inhibition of mTOR suppresses phosphorylation of the focal adhesion proteins
A close relationship between F-actin reorganization and tyrosine phosphorylation of focal adhesion proteins has been described (Leventhal et al., 1997; Guvakova et al., 1999; Leopoldt et al., 2001). Since IGF-I stimulation very quickly induced F-actin reorganization, next we examined whether IGF-I impacts tyrosine phosphorylation of the focal adhesion proteins in Rh1 and Rh30 cells. As shown in Fig. 6, stimulation with IGF-I (10 ng/ml) within 1 h did not alter the protein levels of FAK, paxillin and p130Cas (Fig. 6A), but rapidly increased tyrosine phosphorylation of FAK, paxillin and p130Cas in Rh1 cells (Fig. 6B). Robust tyrosine phosphorylation of FAK and paxillin was detectable within 5–10 min, and that of p130Cas within 30–60 min. Similar data were observed in Rh30 cells (data not shown). Because rapamycin prevents IGF-I-induced change of F-actin reorganization in Rh1 (not shown) and Rh30 cells (Fig. 1), we have further examined the effect of rapamycin on IGF-I-stimulated tyrosine phosphorylation of the focal adhesion proteins. As shown in Fig. 6C, pretreatment of Rh1 cells with rapamycin for 2 h markedly inhibited IGF-I-stimulated tyrosine phosphorylation of FAK, p130Cas and paxillin. Furthermore, we found that rapamycin failed to inhibit IGF-I-induced phosphorylation of the focal adhesion proteins in the cells stably expressing rapamycin-resistant mutant mTOR (S2035I) (Fig. 6D). In contrast, cells stably expressing an empty vector (pcDNA3) (data not shown) or a kinase-dead mTORrr (mTOR-SIDA) remained sensitive to rapamycin (Fig. 6D). The results suggest that rapamycin inhibits IGF-I-induced F-actin reorganization probably in part through inhibition of phosphorylation of the focal adhesion proteins, which is dependent on mTOR kinase activity.
Figure 6.
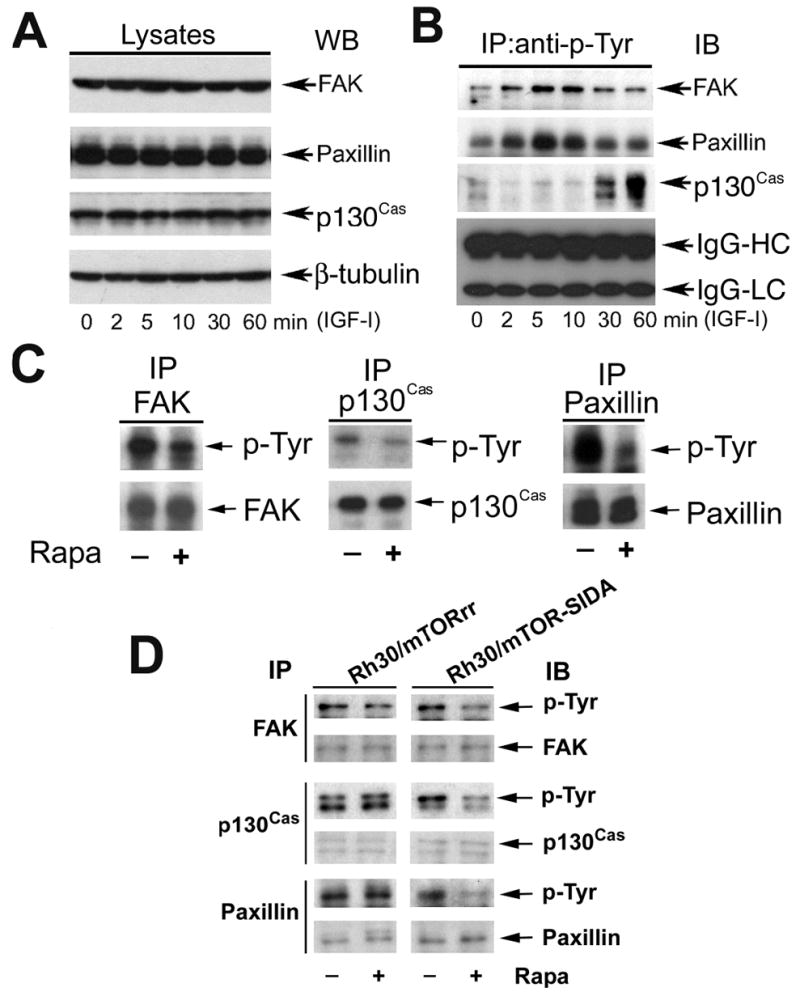
Rapamycin inhibits IGF-I-stimulated tyrosine phosphorylation of FAK, paxillin and p130Cas. (A) and (B) Serum-starved Rh1 cells were stimulated with IGF-I (10 ng/ml) for the indicated time. Equivalent amounts of protein (lysates) was used for Western blot with antibodies to paxillin, FAK, p130Cas, and β-tubulin (loading control) (A). Immunoprecipitation was performed by incubation of cell lysates (500 mg) with mouse monoclonal antibody to phosphotyrosine (p-Tyr) (5 μg), plus protein A/G-agarose beads overnight at 4°C, followed by immunoblotting with antibodies to FAK, paxillin, or p130Cas. IgG-heavy chain (IgG-HC) and light chain (IgG-LC) from each immunoprecipitated sample are shown as loading control (B). (C) Serum-starved Rh1 cells, or (D) Rh30/mTORrr and Rh30/mTOR-SIDA cells were pretreated with or without rapamycin (100 ng/ml) for 2 h, and then stimulated with IGF-I (10 ng/ml) for 10 min (for FAK and paxillin) or 1 h (for p130Cas). Immunoprecipitation was performed by incubation of cell lysates (500 mg) with antibodies to FAK, paxillin or p130Cas, followed by immunoblotting with antibodies to phosphotyrosine (p-Tyr), FAK, p130Cas and paxillin, respectively.
Downregulation of raptor mimicks the effect of rapamycin, inhibiting phosphorylation of the focal adhesion proteins
Since TORC1 regulates F-actin reorganization, next we examined whether the TORC1 regulates phosphorylation of the focal adhesion proteins. Lentiviral shRNA to raptor were used to disrupt TORC1. Because TORC2 has been reported to regulate phosphorylation of paxillin (Jacinto et al., 2004), shRNA to rictor was used as a control. As shown in Fig. 7A, downregulation of raptor, but not GFP (control), inhibited IGF-I-stimulated tyrosine phosphorylation of FAK and paxillin, mimicking the effect of rapamycin. As expected, downregulation of rictor inhibited phosphorylation of paxillin and FAK as well (Fig. 7A). Consistent with previous findings (Hara et al., 2002; Kim et al., 2002), rapamycin treatment only disrupted TORC1 (Fig. 7B). Therefore, we conclude that both TORC1 and TORC2 control tyrosine phosphorylation of FAK and paxillin, and rapamycin inhibits IGF-I-stimulated phosphorylation of the focal adhesion proteins through targeting TORC1.
Figure 7.
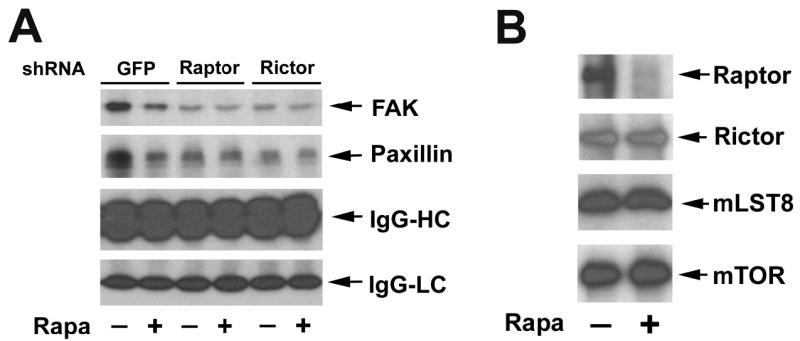
Rapamycin-sensitive mTOR-raptor complex regulates phosphorylation of FAK and paxillin. (A) Downregulation of raptor or rictor inhibited phosphorylation of FAK and paxillin. Serum-starved Rh30 cells that were infected with lentiviral shRNAs to GFP, raptor and rictor, respectively, were pretreated with or without Rapa (100 ng/ml) for 2 h, and then stimulated with IGF-I (10 ng/ml) for 10 min, followed by immunoprecipitation with mouse monoclonal antibody to phosphotyrosine (p-Tyr) antibody, and immunoblotting with antibodies to FAK and paxillin, respectively. IgG heave chain (IgG-HC) and light chain (IgG-LC) were served as loading control. (B) Rapamycin disrupts mTOR-raptor complex, but not mTOR-rictor complex. Serum-starved Rh30 cells were pretreated with Rapa (100 ng/ml) for 2 h, and then stimulated with IGF-I (10 ng/ml) for 1 h, followed by immunoprecipitation with anti-mTOR antibody, and immunoblotting with antibodies to raptor, rictor, mLST8 and mTOR, respectively.
TORC1-mediated S6K1, but not 4E-BP1 pathway, regulates phosphophorylation of the focal adhesion proteins
Since S6K1 and 4E-BP1 are the best characterized effectors of TORC1 (Guertin and Sabatini, 2007), we further determined which of them is responsible for mTOR-controlled phosphorylation of FAK and paxillin. First, the S6K1 pathway was studied. Rh30 cells were infected with recombinant adenoviruses expressing HA-tagged constitutively active and rapamycin-resistant S6K1 mutant (F5A-E389-R3A) (Ad-S6K1-ca) and Ad-GFP (as controls). As shown in Fig. 8A, overexpression of S6K1-ca, but not GFP, abrogated the inhibitory effect of rapamycin on tyrosine phosphorylation of FAK and paxillin. In contrast, downregulation of S6K1 by lentiviral shRNA mimicked rapamycin treatment, decreasing the IGF-I-stimulated tyrosine phosphorylation of FAK and paxillin (Fig. 8B). The results indicate that S6K1 is involved in the regulation of phosphorylation of the focal adhesion proteins.
Figure 8.
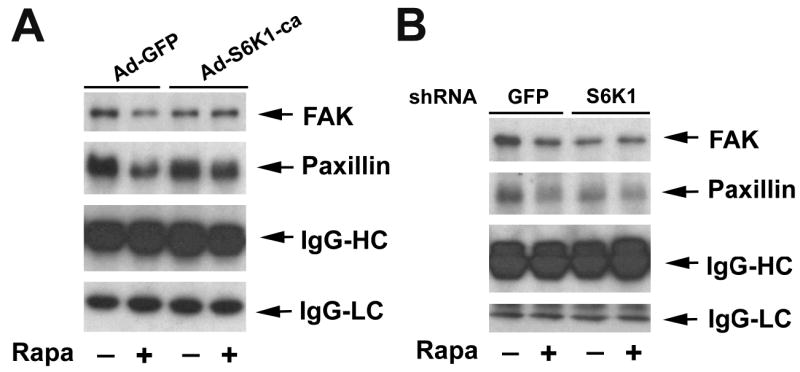
S6K1 pathway controls phosphorylation of FAK and paxillin. (A) Expression of constitutively active and rapamycin-resistant S6K1 (S6K1-ca) renders cells resistant to rapamycin inhibition of IGF-I-stimulated phosphorylation of FAK and paxillin, whereas (B) downregulation of S6K1 mimicks the inhibitory effect of rapamycin on IGF-I-stimulated phosphorylation of FAK and paxillin. Serum-starved Rh30 cells, infected with Ad-GFP and Ad-S6K1-ca, respectively (A), or with lentiviral shRNAs to GFP and S6K1, respectively (B), were pretreated with or without Rapa (100 ng/ml) for 2 h, and then stimulated with IGF-I (10 ng/ml) for 10 min, followed by immunoprecipitation with mouse monoclonal antibody to phosphotyrosine (p-Tyr) antibody, and immunoblotting with antibodies to FAK and paxillin, respectively.
Next, 4E-BP1 pathway was investigated. Since 4E-BP1 pathway affects F-actin reorganization, we expected that downregulation of 4E-BP1 would result in resistance to rapamycin inhibition of phosphorylation of FAK and paxillin. However, to our surprise, downregulation of 4E-BP1 by RNAi failed to affect IGF-I-stimulated phosphorylation of FAK and paxillin in Rh30 cells (Fig. 9A), regardless of pretreatment with or without rapamycin. Similar data were observed in Rh1 cells (data not shown). Furthermore, ectopic expression of 4E-BP1-5A by infection of the cells with Ad-4EBP1-5A did not obviously alter the cellular protein levels of FAK and paxillin (Fig. 9B), and IGF-I-stimulated phosphorylation of the focal adhesion proteins (Fig. 9C). The data imply that TORC1-mediated 4E-BP1 pathway does not participate in the regulation of tyrosine phosphorylation of the focal adhesion proteins.
Figure 9.
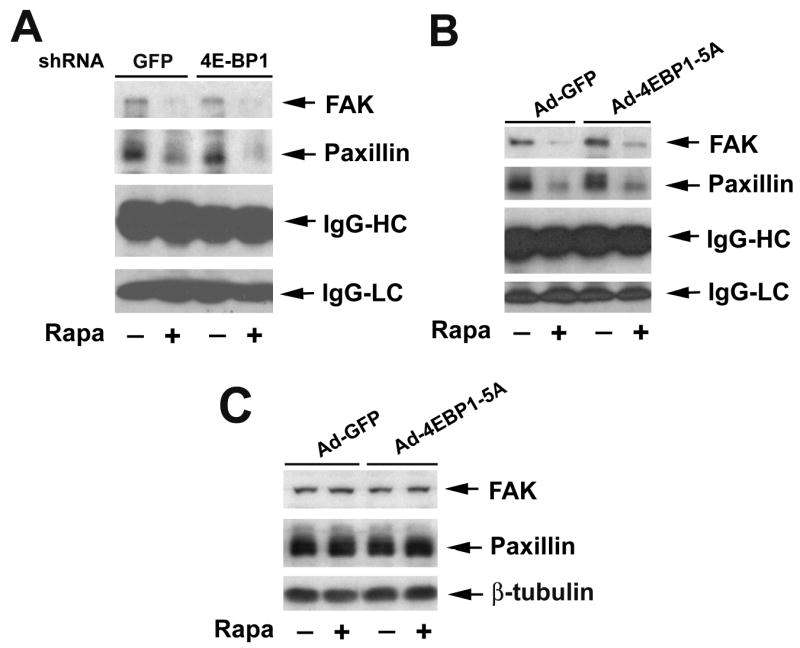
4E-BP1 pathway does not control phosphorylation of FAK and paxillin. (A) Downregulation of 4E-BP1 or (B) expression of constitutively hypophosphorylated 4E-BP1 (4E-BP1-5A) does not affect rapamycin inhibition of IGF-I-stimulated phosphorylation of FAK and paxillin. Serum-starved Rh30 cells, infected with lentiviral shRNAs to GFP and 4E-BP1, respectively (A), or with Ad-GFP and Ad-4EBP1-5A, respectively (B, C), were pretreated with or without Rapa (100 ng/ml) for 2 h, and then stimulated with IGF-I (10 ng/ml) for 10 min, followed by immunoprecipitation with mouse monoclonal antibody to phosphotyrosine (p-Tyr) antibody, and immunoblotting with antibodies to FAK and paxillin, respectively (A, C), or Western blot analysis of cell lysates using the indicated antibodies (B).
Discussion
Recently we have shown that rapamycin inhibits cell motility (Liu et al., 2006). Since cell migration is a multi-step cellular event, including cell polarization/protrusion, adhesion and de-adhesion (Ridley et al., 2003), disturbance of any of these steps may affect cell motility. To further understand the underlying mechanism, this study was set to examine whether rapamycin inhibition of cell motility is related to its prevention of F-actin reorganization. Here we show that rapamycin inhibits IGF-I-stimulated F-actin reorganization in a panel of tumor cell lines, such as human rhabdomyosarcoma (Rh30), Ewing sarcoma (Rh1), glioblastoma (U-373) and prostate carcinoma (PC-3) cells, revealing that rapamycin may inhibit cell motility, at least by impairing cell polarization/protrusion. Furthermore, our data indicate that rapamycin may inhibit F-actin reorganization by suppression of mTOR-mediated phosphorylation of focal adhesion proteins, including FAK, paxillin and p130Cas.
It is well known that rapamycin inhibits proliferation and growth (Guertin and Sabatini, 2007; Wullschleger et al., 2006), or induces apoptosis of tumor cells under certain conditions (Hosoi et al., 1999; Huang et al., 2003) by inhibiting the kinase activity of mTOR. However, studies have also shown that rapamycin inhibits differentiation of C2C12 cells (Erbay and Chen, 2001), which is independent of the kinase activity of mTOR, though this remains controversial (Erbay and Chen, 2001; Shu et al., 2002). This prompted us to study whether rapamycin inhibits F-actin reorganization and phosphorylation of the focal adhesion proteins in an mTOR-kinase activity dependent manner. We found that rapamycin failed to inhibit IGF-I-induced F-actin reorganization and phosphorylation of the focal adhesion proteins in the mTORrr-transfected cells, but not in control cells (empty-vector or mTOR-SIDA-transfected cells), suggesting the kinase activity of mTOR is essential for IGF-I-stimulated F-actin reorganization and phosphorylation of the focal adhesion proteins.
Previous studies have demonstrated that disruption of TORC2 prevents cell spreading, F-actin polymerization and phosphorylation of paxillin in NIH 3T3 cells (Jacinto et al., 2004). TORC2 is rapamycin-insensitive (Guertin and Sabatini, 2007; Wullschleger et al., 2006). Since rapamycin inhibited IGF-I-induced lamellipodia formation and phosphorylation of the focal adhesion proteins, we therefore hypothesized that the rapamycin-sensitive complex, TORC1, regulates F-actin reorganization and phosphorylation of the focal adhesion proteins as well. Our RNAi data support this notion. Of interesting, looking through the data (Fig. 2), we found that TORC1 and TORC2 regulate F-actin reorganization in different ways, though both of them controls phosphorylation of the focal adhesion proteins. Downregulation of raptor, mimicking the effect of rapamycin, inhibited IGF-I-stimulated phosphorylation of S6K1 and 4E-BP1, and lamellipodia formation, but did not impact F-actin stress fibers. However, downregulation of rictor not only inhibited IGF-I-stimulated lamellipodia formation, but also dismantled F-actin stress fibers.
We also noticed that inhibition of mTOR by rapamycin or downregulation of raptor increased phosphorylation of Akt (Fig. 2A). This is consistent with recent findings (Tzatsos et al., 2006), and is probably the consequence of S6K1 inhibition. It has been reported that S6K1 phosphorylates insulin receptor substrate 1 (IRS-1), promoting its degradation (Tremblay et al., 2001). Inhibition of S6K1 activity prevents phosphorylation of IRS-1 (S636/639), resulting in accumulation of IRS-1 and activation of its downstream kinases, such as PI3K and Akt, by a feedback regulating mechanism (O’Reilly et al., 2006; Tremblay et al., 2001; Tzatsos et al., 2006; Wan et al., 2007). Increased phosphorylation of Akt by raptor shRNA in the absence of rapamycin did not induce formation of lamellipodia in Rh30 cells (Fig. 2B), implying that Akt may not be involved in the regulation of lamellipodia formation, or at least Akt activity itself is not enough for lamellipodia formation, if TORC1-mediated S6K1 and 4E-BP1 pathways are blocked.
Our studies indicate that rapamycin may inhibit IGF-I-induced F-actin reorganization by targeting TORC1-mediated S6K1 and 4E-BP1 pathways. However, only S6K1 pathway was found to be involved in the regulation of phosphorylation of the focal adhesion proteins (FAK and paxillin). This is supported by the following findings: (1) Cells infected with an adenovirus expressing constitutively active and rapamycin-resistant mutant of S6K1 conferred to resistance to rapamycin inhibition of IGF-I-stimulated F-actin reorganization and phosphorylation of FAK and paxillin. (2) IGF-I failed to stimulate F-actin reorganization and phosphorylation of FAK and paxillin in the S6K1-downregulated cells. (3) Expression of constitutively hypophosphoryated 4E-BP1 (4E-BP1-5A) inhibited IGF-I-stimulated F-actin reorganization, but did not alter phosphorylation of FAK and paxillin. Our data are in contrast to previous findings that FAK regulates phosphorylation of S6K1 (Gan et al., 2006; Malik et al., 1996). This is probably related to different cell types or approaches used. Gan et al. (2006) used 293T cells overexpressing FAK and S6K1. In fact, they found that overexpression of wild-type FAK did not significantly impact phosphorylation of S6K1 in 293T cells (Gan et al., 2006). Only overexpression of a kinase-dead FAK (Gan et al., 2006) in 293T cells or a dominant negative FAK (FRNK) (Malik et al., 1996) in chick embryo cells reduced phosphorylation of S6K1. Whether FAK really regulates S6K1 is challenged by other findings (Govindarajan et al., 2000) as well, because overexpression of FRNK did not affect phosphorylation of S6K1 in rat aortic vascular smooth muscle cells. The findings from this study and others (Jacinto et al., 2004) indicate that mTOR may regulate phosphorylation of the focal adhesion proteins, including FAK, paxillin and p130Cas. Clearly, more studies are needed to elucidate the relationship between FAK and mTOR/S6K1. For example, early findings suggested that Akt fuctions as an upstream kinase, phosphorylating mTOR (Ser2448) (Nave et al., 1999; Sekulic et al., 2000). However, latest data have demonstrated that mTOR (TORC2) phosphorylates Akt (Ser473) (Sarbassov et al., 2005).
It remains unclear how 4E-BP1 pathway regulates F-actin reorganization. 4E-BP1 primarily functions as a suppressor of eIF4E (Guertin and Sabatini, 2007; Wullschleger et al., 2006). Hypophosphorylated 4E-BP1 tightly binds to eIF4E, and prevents association of eIF4E with eIF4G and formation of the eIF4F initiation complex, thereby inhibiting cap-dependent translation of mRNA (Guertin and Sabatini, 2007; Mothe-Satney et al., 2000; Wullschleger et al., 2006;). In our studies, expression of 4E-BP1-5A, which sequesters the eIF4E (Liu et al., 2006), did not alter the cellular protein levels or phosphorylation levels of FAK and paxillin. We have further studied whether 4E-BP1 pathway may affect synthesis of other proteins, such as small GTPases (RhoA, Cdc42 and RacI), atypical PKC, and integrins, which are associated with F-actin reorganization. Our new data suggest that 4E-BP1 pathway at least plays an important role in controlling protein synthesis of small GTPases. This is evidenced by the findings that expression of constitutively hypophosphorylated 4E-BP1 (4E-BP1-5A), mimicking the effect of rapamycin, severely inhibits protein synthesis of RhoA, Rac1 and Cdc42 (our unpublished observations). More studies are on the way to understand how 4E-BP1 pathway controls F-actin reorganization and cell motility.
In conclusion, we have shown that rapamycin inhibits IGF-I-induced F-actin reorganization and phosphorylation of the focal adhesion proteins by blocking the kinase activity of mTOR. This is attributed to rapamycin disruption of mTOR-raptor complex. Furthermore, both S6K1 and 4E-BP1 pathways, mediated by the mTOR-raptor complex, are involved in the regulation of IGF-I-stimulated F-actin reorganization. However, only S6K1 pathway controls IGF-I-stimulated phosphorylation of the focal adhesion proteins.
Materials and Methods
Cell lines and cultures
Cell lines from human rhabdomyosarcoma (Rh30) (Huang et al., 2003) and Ewing sarcoma (Rh1) (Smith et al., 2006) (gifts from Dr. Peter J. Houghton, St. Jude Children’s Research Hospital, Memphis, TN) were grown in antibiotic-free RPMI 1640 medium (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT) at 37°C and 5% CO2. Human prostate carcinoma (PC-3) and glioblastoma (U-373) cells (American Type Culture Collection, Manassas, VA) were grown in antibiotic-free Dulbecco’s Modified Eagle Medium (DMEM) (Mediatech, Herndon, VA) supplemented with 10% FBS at 37°C and 5% CO2. Human embryonic kidney (HEK) 293 (American Type Culture Collection, Manassas, VA) and 293TD cells (Invitrogen, Carlsbad, CA) were grown in antibiotic-free DMEM supplemented with 10% heat-inactivated FBS at 37°C and 5% CO2. For experiments where cells were deprived of serum, cell monolayers were washed with phosphate-buffered saline (PBS), and incubated in the serum-free Dulbecco’s modified Eagle medium (DMEM) (Mediatech, Herndon, VA). Rh1 and Rh30 clones expressing pcDNA3 (empty vector, Invitrogen, Carlsbad, CA), AU1-tagged mTORrr and mTOR-SIDA (Sekulic et al., 2000) were generated as described (Hosoi et al., 1999). mTORrr represents the entire mTOR gene with the site mutation (S2035I) that has intact mTOR kinase activity, but prevents FKBP12-rapamycin complex binding and is rapamycin-resistant (Hosoi et al., 1999). mTOR-SIDA stands for a catalytically inactive (kinase dead) mTORrr mutant (D2038A) (Sekulic et al., 2000). Clones were expanded in growth medium containing G418 (500 μg/ml). Individual clones were screened for expression of the mutant proteins by Western blot analysis with anti-AU1 antibodies (Bethyl Laboratories, Montgomery, TX). The functions of mTORrr and mTORSIDA in the cells were conformed as described previously (Liu et al., 2006).
Recombinant adenoviral constructs and infection of cells
Constitutively hypophosphorylated mutant 4E-BP1 (Thr36A, Thr45A, Ser64A, Thr69A, and Ser82A) (4E-BP1-5A) (Mothe-Satney et al., 2000) and constitutively active and rapamycin-resistant S6K1 (pRK7/HA-S6K1-F5A-E389-R3A) (Schalm et al., 2005) were gifts from Dr. John Lawrence (University of Virginia, Charlottesville, VA) and Dr. John Blenis, Harvard Medical School, Boston, MA), respectively. The recombinant adenoviruses encoding HA-tagged 4E-BP1-5A (Ad-4EBP1-5A), HA-tagged constitutively active and rapamycin-resistant S6K1 (Ad-S6K1-ca), and the control virus encoding the green fluorescence protein (GFP) alone (Ad-GFP) were generated, titrated and used, as described (Liu et al., 2006).
Lentiviral shRNA cloning, production, and infection
To generate lentiviral shRNAs to 4E-BP1 and S6K1, oligonucleotides containing the target sequences were synthesized, annealed and inserted into FSIPPW lentiviral vector (Kanellopoulou et al., 2005) via the EcoR1/BamH1 restriction enzyme site. Oligonucleiotides used were: 4E-BP1 sense: 5′-AATTCCCATTCCTGATGGAGTGTCGGTGCAAGAGACCGACACTCCATCAGGAATTTTTTG-3′, anti-sense: 5′-GATCCAAAAAATTCCTGATGGAGTGTCGGTCTCTTGCACCGACACTCCATCAGGAATGGG-3′; S6K1 sense: 5′-AATTCCCGGGAGTTGGACCATATGAACTTGCAAGAGAAGTTCATATGGTCCAACTCCCTTTTTG-3′, anti-sense: 5′-GATCCAAAAAGGGAGTTGGACCA TATGAACTTCTCTTGCAAGTTCATATGGTCCAACTCCCGGG-3′. Lentiviral shRNA constructs targeting raptor and rictor (Liu et al., 2006) were gifts from Dr. David M Sabatini (Massachusetts Institute of Technology, Cambridge, MA). Lentiviral shRNA constructs were made and used, as described (Liu et al., 2006).
Western blot analysis
Western blot was performed, as described previously (Liu et al., 2006). The primary antibodies used included antibodies to AU1, raptor, rictor (Bethyl Laboratories, Montgomery, TX), HA, phospho-S6K1 (Thr389), S6K1, eIF4E, p-Akt (S473), Akt, mTOR, β-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA), 4E-BP1 (Zymed Laboratories, South San Francisco, CA), phospho-S6 ribosomal protein (Ser235/236), S6 ribosomal protein (Cell Signaling, Beverly, MA), phosphotyrosine, FAK, paxillin, and p130Cas (BD Biosciences, Lexington, KY). To semi-quantitate the intensities of the bands, NIH ImageJ was used.
Single Cell Motility Assay
The single cell motility assay was performed as described (Liu et al., 2006).
F-actin staining
Serum-starved cells grown on glass coverslips in 6-well-plates were pretreated with or without rapamycin (100 ng/ml) for 2 h, followed by stimulation with or without IGF-I (10 ng/ml) for ~1 h. Cells were then fixed with 4% ice cold paraformaldehyde in PBS for 20 min at 4°C and permeabilized with 0.2% Triton X-100 in PBS for 5 min. Cells were stained with FITC-conjugated phalloidin (1 μg/ml, Sigma) for 30 min. Actin filaments (F-actin) were visualized and photographed with a Nikon TE300 digital inverted microscope. Percentage of cells with lamellipodia formation was determined by randomly counting at least 200 cells under the fluorescence microscope using × 600 magnification.
Detection of phosphorylation of the focal adhesion proteins
Serum-starved cells were pretreated with or without rapamycin (100 ng/ml) for 2 h, and then stimulated with IGF-I (10 ng/ml) for 10 min. Cells were lysed in RIPA buffer, as described (Liu et al., 2006). Immunoprecipitation was performed by incubation of cell lysates (500 μg) with mouse monoclonal antibody to phosphotyrosine (p-Tyr) (5 μg) (BD Pharmingen), plus protein A/G-agarose beads (Santa Cruz Biotechnology) overnight at 4°C, followed by immunoblotting with antibodies to FAK, paxillin and p130Cas, respectively. IgG-heavy chain (IgG-HC) and light chain (IgG-LC) from each immunoprecipitated sample are shown as loading control. In some cases, immunoprecipitation was performed by incubation of cell lysates (500 mg) with antibodies to FAK, paxillin and p130Cas, respectively, followed by immunoblotting with antibodies to phosphotyrosine (p-Tyr), FAK, paxillin, and p130Cas, respectively.
Statistical Analysis
Data were expressed as means ± SD and statistically subjected to Student’s unpaired t-test. A level of P < 0.05 was considered to be significant.
Acknowledgments
We thank Drs. Robert T. Abraham, Peter J. Houghton, John C. Lawrence, John Blenis and David M. Sabatini for providing cell lines or constructs. This work was supported in part NIH grant R01 CA115414 (S. H.), Louisiana Board of Regents [LEQSF(2006-09)-RD-A-18] (S. H.), and the Feist-Weiller Cancer Center Award (S. H.), Louisiana State University Health Sciences Center, Shreveport, LA.
References
- Berven LA, Willard FS, Crouch MF. Role of the p70S6K pathway in regulating the actin cytoskeleton and cell migration. Exp Cell Res. 2004;296:183–195. doi: 10.1016/j.yexcr.2003.12.032. [DOI] [PubMed] [Google Scholar]
- Boffa DJ, Luan F, Thomas D, Yang H, Sharma VK, Lagman M, et al. Rapamycin inhibits the growth and metastatic progression of non-small cell lung cancer. Clin Cancer Res. 2004;10:293–300. doi: 10.1158/1078-0432.ccr-0629-3. [DOI] [PubMed] [Google Scholar]
- Casamassima A, Rozengurt E. Insulin-like growth factor I stimulates tyrosine phosphorylation of p130Cas, focal adhesion kinase, and paxillin. Role of phosphatidylinositol 3′-kinase and formation of a p130Cas Crk complex. J Biol Chem. 1998;273:26149–26156. doi: 10.1074/jbc.273.40.26149. [DOI] [PubMed] [Google Scholar]
- Erbay E, Chen J. The mammalian target of rapamycin regulates C2C12 myogenesis via a kinase-independent mechanism. J Biol Chem. 2001;276:36079–36082. doi: 10.1074/jbc.C100406200. [DOI] [PubMed] [Google Scholar]
- Fonseca BD, Smith EM, Lee VH, Mackintosh C, Proud CG. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem. 2007;282:24514–24524. doi: 10.1074/jbc.M704406200. [DOI] [PubMed] [Google Scholar]
- Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, Sabatini DM. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16:1865–1870. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Gan B, Yoo Y, Guan JL. Association of focal adhesion kinase with tuberous sclerosis complex 2 in the regulation of s6 kinase activation and cell growth. J Biol Chem. 2006;281:37321–37329. doi: 10.1074/jbc.M605241200. [DOI] [PubMed] [Google Scholar]
- Govindarajan G, Eble DM, Lucchesi PA, Samarel AM. Focal adhesion kinase is involved in angiotensin II-mediated protein synthesis in cultured vascular smooth muscle cells. Circ Res. 2000;87:710–716. doi: 10.1161/01.res.87.8.710. [DOI] [PubMed] [Google Scholar]
- Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8:128–135. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Guvakova MA, Surmacz E. The activated insulin-like growth factor I receptor induces depolarization in breast epithelial cells characterized by actin filament disassembly and tyrosine dephosphorylation of FAK, Cas, and paxillin. Exp Cell Res. 1999;251:244–255. doi: 10.1006/excr.1999.4566. [DOI] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- Hosoi H, Dilling MB, Shikata T, Liu LN, Shu L, Ashmun RA, et al. Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res. 1999;59:886–894. [PubMed] [Google Scholar]
- Huang S, Shu L, Dilling MB, Easton J, Harwood FC, Ichijo H, et al. Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21Cip1. Mol Cell. 2003;11:1491–1501. doi: 10.1016/s1097-2765(03)00180-1. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Koyasu S, Nishida E, Sakai H, Takaku F, Yahara I, et al. Insulin-like growth factors, insulin, and epidermal growth factor cause rapid cytoskeletal reorganization in KB cells. Clarification of the roles of type I insulin-like growth factor receptors and insulin receptors. J Biol Chem. 1986;261:16141–16147. [PubMed] [Google Scholar]
- Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, et al. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Konstantopoulos N, Clark S. Insulin and insulin-like growth factor-1 stimulate dephosphorylation of paxillin in parallel with focal adhesion kinase. Biochem J. 1996;314:387–390. doi: 10.1042/bj3140387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopoldt D, Yee HF, Jr, Rozengurt E. Calyculin-A induces focal adhesion assembly and tyrosine phosphorylation of p125Fak, p130Cas, and paxillin in Swiss 3T3 cells. J Cell Physiol. 2001;188:106–119. doi: 10.1002/jcp.1102. [DOI] [PubMed] [Google Scholar]
- LeRoith D, Roberts CT., Jr The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127–137. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- Leventhal PS, Shelden EA, Kim B, Feldman EL. Tyrosine phosphorylation of paxillin and focal adhesion kinase during insulin-like growth factor-I-stimulated lamellipodial advance. J Biol Chem. 1997;272:5214–5218. doi: 10.1074/jbc.272.8.5214. [DOI] [PubMed] [Google Scholar]
- Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25:7029–7040. doi: 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Luan FL, Ding R, Sharma VK, Chon WJ, Lagman M, Suthanthiran M. Rapamycin is an effective inhibitor of human renal cancer metastasis. Kidney Int. 2003;63:917–926. doi: 10.1046/j.1523-1755.2003.00805.x. [DOI] [PubMed] [Google Scholar]
- Malik RK, Parsons JT. Integrin-dependent activation of the p70 ribosomal S6 kinase signaling pathway. J Biol Chem. 1996;271:29785–29791. doi: 10.1074/jbc.271.47.29785. [DOI] [PubMed] [Google Scholar]
- Mothe-Satney I, Yang D, Fadden P, Haystead TA, Lawrence JC., Jr Multiple mechanisms control phosphorylation of PHAS-I in five (S/T)P sites that govern translational repression. Mol Cell Biol. 2000;20:3558–3567. doi: 10.1128/mcb.20.10.3558-3567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344:427–431. [PMC free article] [PubMed] [Google Scholar]
- O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LR, Huang X, Boudeau J, Pawlowski R, Wullschleger S, Deak M, et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. 2007;405:513–522. doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Schalm SS, Tee AR, Blenis J. Characterization of a conserved C-terminal motif (RSPRR) in ribosomal protein S6 kinase 1 required for its mammalian target of rapamycin-dependent regulation. J Biol Chem. 2005;280:11101–11106. doi: 10.1074/jbc.M413995200. [DOI] [PubMed] [Google Scholar]
- Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, et al. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60:3504–3513. [PubMed] [Google Scholar]
- Shu L, Zhang X, Houghton PJ. Myogenic differentiation is dependent on both the kinase function and the N-terminal sequence of mammalian target of rapamycin. J Biol Chem. 2002;277:16726–16732. doi: 10.1074/jbc.M112285200. [DOI] [PubMed] [Google Scholar]
- Smith MA, Morton CL, Phelps D, Girtman K, Neale G, Houghton PJ. SK-NEP-1 and Rh1 are Ewing family tumor lines. Pediatr Blood Cancer. 2006 2006, Dec 7; doi: 10.1002/pbc.21099. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem. 2001;276:38052–38060. doi: 10.1074/jbc.M106703200. [DOI] [PubMed] [Google Scholar]
- Tzatsos A, Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol. 2006;26:63–76. doi: 10.1128/MCB.26.1.63-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- Wan X, Mendoza A, Khanna C, Helman LJ. Rapamycin inhibits ezrin-mediated metastatic behavior in a murine model of osteosarcoma. Cancer Res. 2005;65:2406–2411. doi: 10.1158/0008-5472.CAN-04-3135. [DOI] [PubMed] [Google Scholar]
- Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]