

Abstract
Tubuloglomerular feedback (TGF) describes a causal and direct relationship between tubular NaCl concentration at the end of the ascending limb of the loop of Henle and afferent arteriolar tone. The use of genetically altered mice has led to an expansion of our understanding of the mechanisms underlying the functional coupling of epithelial, mesangial, and vascular cells in TGF. Studies in mice with deletions of the A or B isoform of NKCC2, and of ROMK indicate that NaCl uptake is required for response initiation. A role for transcellular salt transport is suggested by the inhibitory effect of ouabain in mutant mice with a ouabain-sensitive α1 Na,K-ATPase. No effect on TGF was observed in NHE2 and H/K-ATPase-deficient mice. TGF responses are abolished in A1 adenosine receptor-deficient mice, and studies in mice with null mutations in NTPDase1 or ecto-5′-nucleotidase indicate that adenosine involved in TGF is mainly derived from dephosphorylation of released ATP. Angiotensin II is a required co-factor for the elicitation of TGF responses as AT1 receptor or ACE deficiencies reduce TGF responses, mostly by reducing adenosine effectiveness. Overall, the evidence from these studies in genetically altered mice indicates that transcellular NaCl transport induces the generation of adenosine that in conjunction with angiotensin II elicits afferent arteriolar constriction.
Keywords: NaCl transport; macula densa; Na,K-ATPase; adenosine; ATP; angiotensin II
Introduction
Changes in NaCl concentration in the tubular lumen near the tubulo-vascular contact point at the distal end of the ascending loop of Henle elicit adjustments in glomerular arteriolar resistance, a phenomenon referred to as ‘tubuloglomerular feedback’ (TGF) 1. Since increases in NaCl concentration cause increases of afferent arteriolar resistance and a fall in glomerular filtration rate, the system is constructed as a negative feedback loop that serves to keep NaCl delivery into the distal parts of the nephron within narrow boundaries. The TGF response is complex, requiring coordinated functional changes in epithelial, mesangial, and smooth muscle cells, and delineation of the cellular mechanisms responsible for linking the NaCl input with the vascular endpoints has been relatively slow. Micropuncture has proven to be the most valuable tool in establishing the relationship between luminal NaCl concentration and glomerular filtration rate or glomerular capillary pressure, but this approach has major limitations in resolving the intermediate steps in the transmission pathway. The use of imaging and electrophysiological techniques in isolated perfused tubule/glomerulus preparations has provided an approach for the detailed study of the changes in epithelial function that result from changes in luminal composition, but the relationship between specific changes in epithelial function and the glomerular arteriolar endpoint has been difficult to study with these preparations 2, 3.
The use of gene-manipulated mice has generated a new venue to further explore the mechanisms responsible for TGF. Since micropuncture can be relatively easily adapted to this species, the effect of targeted deletions of gene products potentially involved in TGF can be studied without some of the uncertainties associated with pharmacologic interventions. In this review we are focusing on two areas where gene-manipulated mice have facilitated major progress in understanding. Substantial experimental evidence supports the notions that luminal NaCl concentration initiates TGF responses by changes in tubular NaCl transport, and that the signal arising from changes in NaCl transport is transmitted across the juxtaglomerular interstitium by the generation of paracrine messengers. The availability of animals with defined transport deficits and with targeted deficiencies in the generation or action of potential mediators has permitted new insights in both of these areas of TGF function.
NaCl transport
Apical NaCl uptake - NKCC2
There is general agreement that the primary mechanism mediating the transduction of luminal NaCl concentration into a propagated signal across the juxtaglomerular interstitium is activation of the Na,K,2Cl-cotransporter, NKCC2, in the apical membrane of macula densa cells. This basic tenet rests on the observation that a number of loop diuretics including furosemide, bumetanide, piretanide, ethacrynic acid, triflocin, or l-ozolinone 1, 4 produce complete TGF inhibition, and on the good quantitative agreement between the inhibitor concentrations causing half-maximal inhibition of transport and TGF 5. Furosemide also blocks TGF responses during retrograde perfusion, indicating that TGF inhibition does not depend upon a metabolic product of the thick ascending limb (TAL) transmitted to the MD cells 6. The direct evidence of a NaCl transport-dependency of renin secretion, the other endpoint of luminal NaCl concentration changes, is not as strong, but nevertheless highly suggestive 1. NKCC2 has been found to be expressed in MD cells, and its inhibition causes cellular hyperpolarization and reductions in cytosolic Na and Cl concentrations 2, 3.
The salt-losing phenotype of mice with deficiencies in the proximal fluid transporters NHE3 or AQP1 is relatively mild despite the fact that the reabsorption of a substantial amount of filtered NaCl and water depends upon these transport pathways 7, 8. TGF-mediated reductions of GFR and filtered NaCl have been observed in both NHE3-/- and AQP1-/- mice, and it has been surmised that reductions in filtered NaCl load by TGF are a major reason for the ability of mice with proximal transport defects to achieve Na balance 9. In contrast, mice with complete inactivation of the NKCC2 gene display the severe salt-losing phenotype of antenatal Bartter syndrome 10. Although TGF responses have not been directly assessed in these mice, inactivation of TGF is suggested by the apparent inability to respond to the elevated distal NaCl load with a reduction in GFR. In mice heterozygous for the NKCC2 null mutation NKCC2 protein expression was found to be normal, and mice were indistinguishable from wild type 11. Further functional exploration of the consequences of NKCC2 deficiency has become possible with the generation of mice with targeted disruption of single NKCC2 isoforms 12, 13. The existence of three different full length variants of NKCC2, first reported by Payne and Forbush in the rabbit kidney 14, has been confirmed in all species studied 15-17. These isoforms, called NKCC2B, NKCC2A, and NKCC2F, are derived from differential splicing of the variable exon 4 of the Slc12a1 gene, a short 96-bp exon that encodes for the second transmembrane domain and parts of the adjacent intracellular loop of the transporter 14. NKCC2 isoform expression shows a distinct distribution pattern with F found exclusively in the medullary thick ascending limb, A in both outer medulla and cortex, and B in the cortex 15, 17, 18. Macula densa cells of the mouse have been found to express both the B- and A-isoforms of the cotransporter 12. Marked differences in ion affinities have been identified in in vitro heterologous expression studies in Xenopus laevis oocytes: F was found to have much lower Na+ and Cl- affinities than the A or the B isoform 18. To create NKCC2B and NKCC2A-deficient mice, the alternate exons 4B or 4A were modified by the introduction of in-frame stop codons resulting in the premature termination of translation. Thus, these strains of isoform-specific knockout mice lack both the full length and the corresponding truncated NKCC2 isoforms.
In vivo microperfusion of loops of Henle showed that in NKCC2B-deficient mice Cl- reabsorption was significantly reduced at low flow rates 12, while the lack of NKCC2A resulted in reduced Cl- absorption at high perfusion rates 13. These in vivo data are in line with the notion that TAL reabsorption at low NaCl concentrations relies on the activity of the high Cl--affinity NKCC2B isoform while NKCC2A comes into play under when higher salt concentrations are achieved by high loop perfusion flow. Assessment of TGF responses has confirmed that macula densa signaling function depends on the successive engagement of NKCC2B and NKCC2A (Fig. 1). In the low flow range NKCC2B-deficient mice were less responsive than wild type animals whereas TGF responses of NKCC2A-/- mice were reduced at high flow rates. Thus, V1/2, the flow rate causing half maximum TGF responses, increased from 6.5 nl/min in mice expressing only NKCC2B to 15.5 nl/min in mice possessing only NKCC2A. These data suggest that the successive activation of the high Cl- affinity NKCC2B and the lower Cl- affinity NKCC2A is responsible for the surprisingly wide range of NaCl concentrations over which TGF operates.
Fig. 1.

Relationship between loop of Henle perfusion rate and the percentage reduction of stop flow pressure (± SEM), an expression of TGF responsiveness, in mice lacking NKCC2B (circles) or NKCC2A (dots). Dashed lines indicate position of V1/2, the flow rates causing half maximum reduction of PSF. Data are redrawn from references 45 and 46.
Apical NaCl uptake - ROMK
Whereas the inhibitory effect of luminal barium on TGF responses was diminished by a pronounced direct vascular constrictor action 19, retrograde application of the K+ channel blocker U37883A caused an almost complete inhibition of TGF responsiveness 20. This effect is mediated by ROMK type K+ channels since TGF responses were largely absent in mice with targeted ROMK deletion 21. The finding of a significantly reduced, but not abolished TGF response has subsequently been confirmed in mice in which selective breeding of surviving animals has generated ROMK-deficient mice with less compromised kidney function and reasonably well maintained blood pressure 22. As shown in Fig. 2, the mean TGF response of 11.3 ± 1.2 mm Hg in wild type mice was reduced to 2.2 ± 0.6 mm Hg in ROMK-deficient mice (p<0.001). The observation that inhibition of NKCC2 and ROMK has similar effects on TGF responses argues against a specific “sensor” function of the actual transport proteins suggesting instead a critical role of some consequence of MD NaCl transport. Since ambient distal K+ concentrations near the MD are close to the K+ affinity of the cotransporter variations in luminal K+ may regulate TGF response magnitude 20.
Fig. 2.

Maximum reductions of stop flow pressure (PSF) in ROMK+/+ (left) and ROMK-/- mice (right). Lines connect values from individual tubules taken at saturating flow rate (30 nl/min) bracketed by two measurements of PSF at zero loop flow. Mice were supplied by Tong Wang and Steve Hebert (Yale University, New Haven CT), and some of the characteristics of the new ROMK strain have been previously published 41.
Apical NaCl uptake - Na/H exchanger
Detailed studies in the isolated perfused JGA preparation of the rabbit have clearly documented the existence of Na/H exchange activity in both the apical and the basolateral membrane of macula densa cells. Increases in luminal NaCl concentration cause increased hydrogen efflux and subsequent alkalinization of the macula densa cell cytosol 23. It has been estimated that about 20% of total Na entry may be mediated by Na/H exchange. Immunocytochemical data indicate that the exchanger isoform in the apical membrane is NHE2 whereas NHE4 is the isoform of the basolateral membrane 24. Macula densa cells are thus distinct from TAL cells where NHE3 is the dominant variant of the exchanger 25. How Na/H exchange in macula densa cells could affect TGF has been unclear. One possibility is that the intracellular pH determines the magnitude of the response by disinhibiting macula densa nNOS and thereby augmenting the generation of nitric oxide. In fact, it has been reported that Na/H exchange inhibition with amiloride augments TGF responses by preventing cell alkalinization and thereby causing relative inhibition of nNOS 26, 27. In contrast to these observations in the isolated JGA preparation, there is no in vivo evidence that would support a major role of Na/H exchange activity in TGF responsiveness. Micropuncture studies in NHE2-deficient mice failed to show changes in TGF responses compared to wild type animals 28, and loop of Henle perfusion with amiloride or N-(isopropyl)-amiloride (EIPA) did not elicit measurable alterations of TGF response magnitude 4. Thus, a major TGF-modulating role of Na/H exchange-dependent variations of cytosolic pH in TAL or MD cells is not supported by in vivo observations currently available. TGF responses have also been found to be well maintained in NHE3-/- mice 7, and autoregulation of GFR and RBF was not altered in NHE3-/- mice with transgenic expression of NHE3 in the intestinal tract 29.
Basolateral NaCl extrusion - Na,K-ATPase
Whereas an effect of apical transport inhibition on TGF is generally accepted, there is still considerable uncertainty about the subsequent steps in the juxtaglomerular signaling cascade. One possibility is that one of the intracellular consequences of NKCC2-dependent NaCl uptake is directly coupled to the mediating step. Detailed and sophisticated studies in the isolated perfused rabbit JGA have identified depolarization, alkalinization, and various ionic compositional changes as results of an increased NaCl uptake, and it is therefore conceivable that one or more of these changes trigger the signaling events directly 3, 30.
A second possibility is that signal propagation is the consequence of transcellular NaCl transport, with apical uptake being the first step in the sequence. Transcellular NaCl transport across the renal tubular epithelium universally requires the support of Na,K-ATPase-dependent energy supply. Until recently, the available data about Na,K-ATPase in the macula densa made the transcellular transport hypothesis look less likely. First, cytochemical and immunological localization studies 31, 32 suggested that the expression of Na,K-ATPase in the basolateral membrane of MD cells was quite low. In addition, a microenzymatic determination of Na,K-ATPase activity in microdissected rabbit MD cells arrived at the conclusion that the activity levels were about 50fold lower than in neighboring TAL cells 33. However, the interpretation of these studies failed to take into account the important impact of basolateral membrane folding on enzyme density. For example, in the enzyme activity studies Na,K-ATPase was normalized to unit of cell volume rather than to membrane surface area, an approach that would underestimate a membrane-bound molecule by the membrane folding factor. Whereas the basolateral membrane of MD cells is typically non-folded, extensive infolding in the neighboring TAL cells may increase its basolateral membrane by a factor of 10 to 20 34. Thus, the difference in enzyme activity per unit cell surface area between MD and TAL cells may actually be at least 10fold less than assumed. Species-specific differences may also play a role 24, 35. Extensive studies in the rat using a well-defined antibody against α1 Na,K-ATPase and its co-localization with neuronal nitric oxide synthase, a macula densa cell marker, have clearly established robust presence of the enzyme in the basolateral membrane of MD cells 36, 37. In addition, MD cells also express the β1 subunit of the enzyme and a γ subunit that may be either γa or both γa and γb 36, 38-40. Again, staining of MD cells appeared somewhat less intense than in neighboring TAL cells, no doubt because of absence of basolateral infoldings in MD cells. Pharmacological studies examining the effect of Na,K-ATPase inhibition on TGF were also inconclusive. In rats Na,K-ATPase inhibition by ouabain, administered by luminal or peritubular microinfusion, did not elicit clear reductions of TGF responses (own unpublished data). However, the α1 subunit of Na,K-ATPase, by far the predominant isoform of the enzyme in the kidney, is largely resistant to ouabain in rats and mice. Mice homozygous for a complete α1 Na,K-ATPase null mutation are not viable while heterozygotes are essentially normal with respect to renal function 41.
Recently however, ingenious utilization of the possibilities of gene manipulation has brought important new information suggesting an important role of Na,K-ATPase in supporting TGF 28. TGF responses were determined in double mutant mice in which the normal ouabain-resistance pattern was reversed with α1 Na,K-ATPase mutated to be ouabain-sensitive and α2 Na,K-ATPase to be ouabain-resistant 42, 43. Single mutant mice with the natively ouabain-resistant α1 isoform and the mutated α2 isoform served as controls. In the double mutant mice, intravenous infusion of ouabain caused inhibition of TGF that increased over the course of 1-2 hours. No such effect was seen in the ouabain-resistant control mice indicating that the ouabain effect on TGF was not due to inhibition of a glycoside-sensitive ATPase other than Na,K-ATPase. Attenuation of TGF responses in sensitive, but not resistant mice was also seen with luminal ouabain administration. Maintenance of normal constrictor responses to direct A1AR activation indicates that the effect of ouabain was not a reflection of a general loss of vascular reactivity, but that it occurred at an earlier step in the signaling pathway. The implication that TGF signaling is an energy-consuming process is fully in accord with our earlier observations that ATP depletion by inhibitors of the respiratory chain or of oxidative phosphorylation interferes with TGF responsiveness (Briggs JP, Schnermann J; abstract; Pflugers Arch [Suppl] 1981; 389: R40). MD cells have numerous mitochondria scattered throughout all regions of the cell including the basal part. Presumably because of the absence of basolateral infoldings their membrane-associated alignment is less obvious than it is in cells of the proximal and distal convoluted tubule and TAL, and the distribution of mitochondria is more akin to that in principal and intercalated collecting duct cells. It is worth noting that involvement of Na,K-ATPase in TGF provides an immediate explanation for the inhibitory effect of furosemide. In contrast to the effect of ouabain alone, ouabain in combination with furosemide does not cause cell swelling or membrane depolarization of TAL cells indicating that as a result of the low intracellular Na concentration the Na pump becomes non-functional in the presence of the loop diuretic 44. Overall, studies exploring the relation between NaCl transport and TGF responses indicate that afferent arteriolar vasoconstriction is paralleled by increased epithelial ATP utilization, and they suggest a link between metabolic rate and the vascular endpoint.
Na extrusion - H(Na)/K-ATPase
An alternative model for the regulation of intracellular NaCl in macula densa cells has been suggested by the observations in the isolated rabbit JGA that the luminal administration of ouabain caused an elevation of intracellular Na and prevented the recovery of intracellular Na from the elevated levels resulting from high luminal NaCl 35. This observation was linked to the presence of a colonic H/K-ATPase (HKα2) in the apical membrane of macula densa cells based on immunocytochemistry and the finding that an increase in luminal K+ caused partial recovery of the cytosolic pH from the acidification resulting from luminal and basolateral NaCl removal 35, 45. The ability of H/K-ATPase to mediate active Na extrusion has previously been demonstrated in the distal colon 46. Nevertheless, in recent studies in H/K-ATPase-deficient mice TGF responses were found to be normal suggesting either that H/K-ATPase and its cytosolic effects do not play a major role in TGF or that H/K-ATPase in the mouse is not expressed in the cells activating the TGF pathway 28.
A word of caution may be appropriate at this point. Aside from anatomical plausibility there is no direct proof that the macula densa cells are in fact the cells responsible for the generation of the signal causing afferent arteriolar vasomotor responses. While retrograde in vivo microperfusion experiments and perfusion of dissected JGA specimen clearly point to an effect of a limited segment around the glomerulus, an influence of TAL cells within an about 50 μm segment upstream or downstream from the macula densa is entirely feasible. In fact, the clear effect of the undisputed TAL proteins NKCC2 and Na,K-ATPase and the lack of effect of the macula densa proteins H/K-ATPase and NHE2 on TGF responses may be interpreted as supporting this possibility. It is also worth recalling that amphibian species have been shown to possess mammalian-like TGF responses although they lack cells with the typical macula densa appearance 47, 48.
Signal Mediation
Adenosine
Control of glomerular arteriolar tone by tubular NaCl concentration is the result of changes in the concentration of several paracrine mediators within the confines of the juxtaglomerular interstitium. The nucleoside adenosine has been suggested to exert metabolic control of renal blood flow and GFR via TGF-mediated vasoconstriction 49, 50. In fact, inhibition of A1 adenosine receptors (A1AR) with receptor-specific antagonists largely prevented TGF-induced reductions of SNGFR and glomerular capillary pressure in the rat 51. Definitive support for a critical role of A1AR in TGF responsiveness came from two studies of TGF in A1AR-deficient mice generated independently by two laboratories 52, 53. In both experimental series TGF responses were completely abolished in the A1AR-deficient mice, indicating that adenosine, as the endogenous agonist of A1AR, is required for TGF responses to occur. In contrast to observations in wild type animals, SNGFR measured in proximal segments (with TGF interrupted) and in distal segments (with TGF intact) was not different in A1AR-/- mice indicating that the tonic suppression of GFR by ambient distal flows requires intact A1AR 54. Furthermore, TGF-induced oscillations of proximal tubular pressure were not observed in the A1AR-deficient mice 54. Since autoregulation of renal blood flow and GFR is mediated in part by TGF, one of the consequences of A1AR-deficiency may be a reduction in autoregulatory efficiency. In fact, steady-state autoregulation of RBF and GFR in response to a lowering of blood pressure was significantly impaired in A1AR-deficient mice 55. In addition, the dynamic autoregulatory response of A1AR-deficient mice to a step increase in blood pressure was reduced by 50% in the time frame mediated by TGF while total autoregulation was reduced by about 30% 56.
A direct vasoconstrictor action of adenosine has been demonstrated in perfused isolated afferent arterioles of the mouse, an effect that was not seen in vessels from A1AR-deficient mice thereby establishing the role of A1AR in the action of adenosine 57, 58. Adenosine induces vasoconstriction in mouse afferent arterioles by Gi-dependent activation of phospholipase C, release of Ca from intracellular stores, and subsequent entry of Ca++ through L-type Ca++ channels 57, 59. Consistent with the stable vasoconstriction elicited by TGF, the constrictor effect of adenosine in isolated vessels can be maintained for extended periods of time indicating absence of rapid receptor desensitization 57. TGF-mediated vasoconstriction by adenosine is noteworthy since in most vascular beds adenosine causes vasorelaxation through activation of Gs-coupled A2AR. In fact, the steady-state effect of administration of exogenous adenosine in the mouse kidney is vasodilatation due to an excess of the vasodilating A2AR receptor class in the entire renal vasculature. Thus, adenosine-induced vasoconstriction requires activation of A1AR without simultaneous stimulation of A2AR in other parts of the renal vasculature 60. This requirement appears to be met by generation of adenosine in the confines of the juxtaglomerular interstitium and its exclusive delivery to afferent arterioles in which A1AR expression dominates.
Angiotensin II has been shown to act as an important co-factor of the constrictor actions of adenosine 58, 61-63. Studies in mice with deletions of the angiotensin II receptor (AT1A) or of angiotensin-converting enzyme have shown that TGF responses are largely absent in these animals 64, 65. Furthermore, the fall in glomerular capillary pressure caused by the administration of the A1AR agonist cyclohexyl adenosine was markedly diminished in AT1A receptor-deficient mice suggesting that non-responsiveness to adenosine may importantly contribute to the attenuation of TGF in mice with a compromised renin-angiotensin system 66. In mice with deletions of tissue-bound ACE TGF responses were significantly reduced 67. This effect seems to be a result of absence of endothelial ACE since selective expression of ACE in the proximal tubule was unable to restore normal TGF responsiveness 68. The mechanism underlying the cooperation between A1AR and angiotensin II receptors has remained unclear. One possibility is that the activation of phospholipase C by Gβγ subunits released in the course of A1AR-dependent Gi-activation is synergistically enhanced by simultaneous stimulation of Gq proteins. Alternatively, adenosine has been shown to prevent angiotensin receptor desensitization by enhancing the Ca sensitivity of myosin light chain kinase 69. Enhanced TGF responses may also be the result of an angiotensin II-induced reduction in local levels of nitric oxide 70. This notion agrees with the demonstration that macula densa nNOS expression is elevated in AT1 receptor-deficient mice 71.
It has been proposed that adenosine is produced and released by macula densa cells as a by-product of increased NaCl transport and ATP utilization 49. Whereas this suggestion has not been completely excluded, the available experimental evidence suggests that a major part of the adenosine required for local A1AR activation is derived from the dephosphorylation of released ATP. In the perfused rabbit JGA an increase in luminal NaCl concentration has been shown to cause a rise of cytosolic Ca++ in biosensor cells positioned near the basolateral membrane of macula densa cells 72. Inhibition of this response by suramin suggests that it was elicited by activation of P2 receptors secondary to the release of ATP.
A direct role of ATP as a vasoconstrictor in the TGF signaling pathway may be possible, but the evidence for this is currently not compelling. Mice with a deletion of the P2X1 receptor that has previously been shown to be present in afferent arterioles 73 have impaired autoregulation of afferent arteriolar tone 74, but have largely normal TGF responses (Fig. 3). Mean maximum TGF responses of 7.6 ± 1 mm Hg in wild type mice (n=18) and of 5.4 ± 0.6 mm Hg in P2X1-deficient mice were not significantly different (p=0.073). Since the autoregulation studies were performed in juxtamedullary arterioles while the TGF evidence comes from superficial nephrons, regional differences in P2X1 receptor function may be responsible for these conflicting outcomes. Nevertheless, because of their rapid desensitization, P2X1 receptors would appear unsuited as mediators of a response that can last for extended time periods so that the role of other P2 receptors needs to be further investigated.
Fig. 3.

Maximum reductions of stop flow pressure (PSF) in wild type mice (left) and in P2X1-deficient mice (right). Lines connect values from individual tubules measured at saturating flow rate (30 nl/min) and bracketed by two measurements of PSF at zero loop flow. Mice originally generated by R. Evans (University of Leicester, UK) were supplied by E. Inscho (Medical College of Georgia, Atlanta, GA).
ATP appears more likely to act predominantly through generation of adenosine in the JG interstitium by extracellular breakdown by ecto-ATPases and nucleotidases. In fact, mice with deletion of NTDPase1, an extracellular ATPase with expression at the glomerular vascular pole 75, have significantly reduced TGF responses (see prepublished paper at http://www.ncbi.nlm.nih.gov/pubmed/1826308). Since one would expect NTPDase1-deficiency to be associated with elevated ATP levels, this finding may further argue against a direct role of ATP in TGF. Inhibition of ecto-5′-nucleotidase (CD 73) with alpha-beta-methyleneadenosine 5′-diphosphate (MADP) had been shown earlier to reduce the compensatory efficiency of TGF and the slope of the TGF function 76. Two groups of investigators using independently generated strains of ecto-5′-nucleotidase/CD73-deficient mice have reported significant attenuation of TGF responses 77, 78. Thus, successive dephosphorylation of ATP or ADP to AMP by NTPDase 1 and from AMP to adenosine by ecto-5′-nucleotidase appears to provide most of the adenosine required for TGF responsiveness.
Nitric oxide
The high levels of expression of neuronal NOS in macula densa cells has stimulated extensive investigations of the role of nitric oxide in juxtaglomerular signaling 79, 80. Luminal administration of inhibitors of NO synthases enhances TGF responses to elevations of loop of Henle flow rate 1. Nevertheless, the response of glomerular capillary pressure to changes in perfusion rate was found to be identical over the entire flow range in wild type and nNOS-deficient mice 81. In contrast, measurements of SNGFR showed a markedly increased proximal-distal SNGFR difference, due for the most part to a significantly lower SNGFR in distal tubules of nNOS-deficient mice 81. Thus, these data suggest that the chronic absence of a functional nNOS in macula densa cells is associated with an enhanced vasoconstrictor tone in the subnormal flow range, presumably a consequence of a proportional enhancement of pre-and postglomerular resistances. Since stimulation of NO generation by loop of Henle flow has been shown to usually occur in the supranormal flow range 1, it is conceivable that the reduced SNGFR in nNOS-/- mice is an indirect rather than direct effect of reduced NO availability. Nevertheless, it is noteworthy that direct measurements of NO concentrations in distal tubular fluid have shown an association of total NO release with reduced rather than enhanced NaCl transport 82. TGF responses have been found to be absent in mice with concurrent deficiencies in nNOS and A1AR indicating that nNOS deficiency does not overcome the lack of A1AR signaling (Fig. 4). This observation reaffirms the primacy of A1AR signaling and a modulating role of nitric oxide in the TGF pathway.
Fig. 4.

Decrease of stop flow pressure in response to a flow elevation to 30 nl/min in wild type mice (WT), A1AR-deficient mice (A1AR-/-), nNOS-deficient mice (nNOS-/-) and in mice without both A1AR and nNOS (A1AR/nNOS-/-). A1AR/nNOS double knockout mice were generated in the Schnermann/Briggs laboratory.
Conclusions
Taken together, evidence from studies in genetically modified mice has resulted in a fairly robust mechanistic framework of TGF operation. Metabolic activation of epithelial cells resulting from increased transcellular NaCl transport mediated by NKCC2, ROMK and Na,K-ATPase is coupled to ATP release and extracellular adenosine formation; this in turn leads to activation of A1AR and vasoconstriction (Fig. 5). Several other paracrine factors interact with this pathway to set vascular sensitivity. Since this scheme identifies a considerable number of testable (and still untested) hypotheses, we expect that the further utilization of gene-manipulated mice will remain a useful strategy to enhance the understanding of juxtaglomerular signaling mechanisms.
Fig. 5.
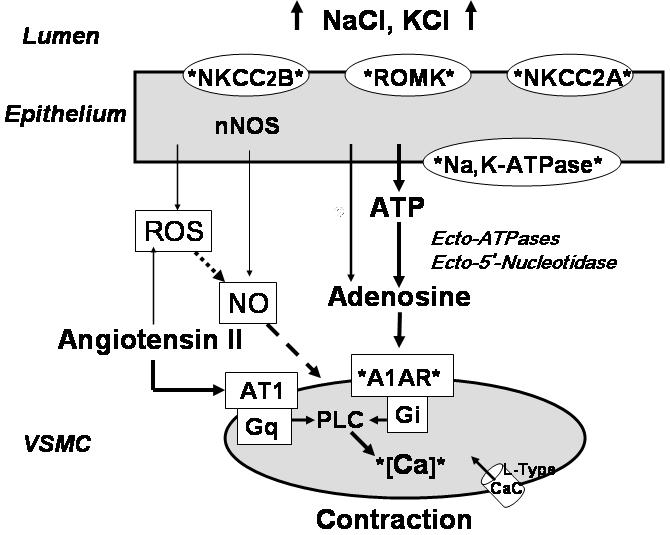
Scheme of TGF operation as supported by evidence mostly derived from studies in gene-manipulated mice. Solid arrows indicate positive/stimulatory, and broken arrows negative/inhibitory relationships. The depiction of cellular components is not meant to be complete, but to indicate those proteins for which a role in TGF is suggested by experimental evidence from gene-manipulated mice.
Acknowledgement
Research from the laboratory of the authors was supported by intramural funds of the National Institute of Diabetes, and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD.
References
- 1.Schnermann J, Briggs JP. Function of the juxtaglomerular apparatus: control of glomerular hemodynamics and renin secretion. In: Alpern RJ, Hebert SC, editors. The Kidney Physiology and Pathophysiology. Vol. 1. Elsevier Academic Press; Burlington-San Diego-London: 2008. pp. 589–626. [Google Scholar]
- 2.Lapointe JY, Bell PD, Cardinal J. Direct evidence for apical Na+:2Cl-:K+ cotransport in macula densa cells. Am J Physiol Renal Physiol. 1990;258:F1466–1469. doi: 10.1152/ajprenal.1990.258.5.F1466. [DOI] [PubMed] [Google Scholar]
- 3.Schlatter E, Salomonsson M, Persson AE, et al. Macula densa cells sense luminal NaCl concentration via furosemide sensitive Na+2Cl-K+ cotransport. Pflugers Arch. 1989;414:286–290. doi: 10.1007/BF00584628. [DOI] [PubMed] [Google Scholar]
- 4.Wright FS, Schnermann J. Interference with feedback control of glomerular filtration rate by furosemide, triflocin, and cyanide. J Clin Invest. 1974;53:1695–1708. doi: 10.1172/JCI107721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mason J, Takabatake T, Olbricht C, et al. The early phase of experimental acute renal failure. III. Tubologlomerular feedback. Pflugers Arch. 1978;373:69–76. doi: 10.1007/BF00581151. [DOI] [PubMed] [Google Scholar]
- 6.Schnermann J, Briggs JP. Concentration-dependent sodium chloride transport as the signal in feedback control of glomerular filtration rate. Kidney Int. 1982;22(suppl 12):S82–S89. [PubMed] [Google Scholar]
- 7.Lorenz JN, Schultheis PJ, Traynor T, et al. Micropuncture analysis of single-nephron function in NHE3-deficient mice. Am J Physiol Renal Physiol. 1999;277:F447–F453. doi: 10.1152/ajprenal.1999.277.3.F447. [DOI] [PubMed] [Google Scholar]
- 8.Schnermann J, Chou C-L, Ma T, et al. Defective proximal tubular fluid reabsorption in transgenic aquaporin-1 null mice. Proc. Natl. Acad. Sci. 1998;95:9660–9664. doi: 10.1073/pnas.95.16.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schnermann J. NaCl transport deficiencies--hemodynamics to the rescue. Pflugers Arch. 2000;439:682–690. doi: 10.1007/s004240000258. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi N, Chernavvsky DR, Gomez RA, et al. Uncompensated polyuria in a mouse model of Bartter’s syndrome. Proc Natl Acad Sci U S A. 2000;97:5434–5439. doi: 10.1073/pnas.090091297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi N, Brooks HL, Wade JB, et al. Posttranscriptional compensation for heterozygous disruption of the kidney-specific NaK2Cl cotransporter gene. J Am Soc Nephrol. 2002;13:604–610. doi: 10.1681/ASN.V133604. [DOI] [PubMed] [Google Scholar]
- 12.Oppermann M, Mizel D, Huang G, et al. Macula densa control of renin secretion and preglomerular resistance in mice with selective deletion of the B isoform of the Na,K,2Cl co-transporter. J Am Soc Nephrol. 2006;17:2143–2152. doi: 10.1681/ASN.2006040384. [DOI] [PubMed] [Google Scholar]
- 13.Oppermann M, Mizel D, Kim SM, et al. Renal function in mice with targeted disruption of the A isoform of the Na-K-2Cl co-transporter. J Am Soc Nephrol. 2007;18:440–448. doi: 10.1681/ASN.2006091070. [DOI] [PubMed] [Google Scholar]
- 14.Payne JA, Forbush B. Alternatively spliced isoforms of the putative renal Na-K-Cl cotransporter are differently distributed within the rabbit kidney. Proc.Nat.Acad.Sci. U.S.A. 1994;91:4544–4548. doi: 10.1073/pnas.91.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Igarashi P, Vanden Heuvel GB, Payne JA, et al. Cloning, embryonic expression, and alternative splicing of a murine kidney-specific Na-K-Cl cotransporter. Am J Physiol. 1995;269:F405–418. doi: 10.1152/ajprenal.1995.269.3.F405. [DOI] [PubMed] [Google Scholar]
- 16.Vargas-Poussou R, Feldmann D, Vollmer M, et al. Novel molecular variants of the Na-K-2Cl cotransporter gene are responsible for antenatal Bartter syndrome. Am. J. Hum. Genet. 1998;62:1332–1340. doi: 10.1086/301872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang T, Huang YG, Singh I, et al. Localization of bumetanide- and thiazide-sensitive Na-K-Cl cotransporters along the rat nephron. Am J Physiol. 1996;271:F931–939. doi: 10.1152/ajprenal.1996.271.4.F931. [DOI] [PubMed] [Google Scholar]
- 18.Gimenez I, Isenring P, Forbush B. Spatially distributed alternative splice variants of the renal Na-K-Cl cotransporter exhibit dramatically different affinities for the transported ions. J Biol Chem. 2002;277:8767–8770. doi: 10.1074/jbc.C200021200. [DOI] [PubMed] [Google Scholar]
- 19.Schnermann J. Effects of barium ions on tubuloglomerular feedback. Am J Physiol Renal Physiol. 1995;268:F960–966. doi: 10.1152/ajprenal.1995.268.5.F960. [DOI] [PubMed] [Google Scholar]
- 20.Vallon V, Osswald H, Blantz RC, et al. Potential role of luminal potassium in tubuloglomerular feedback. J Am Soc Nephrol. 1997;8:1831–1837. doi: 10.1681/ASN.V8121831. [DOI] [PubMed] [Google Scholar]
- 21.Lorenz JN, Baird NR, Judd LM, et al. Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter’s syndrome. J Biol Chem. 2002;277:37871–37880. doi: 10.1074/jbc.M205627200. [DOI] [PubMed] [Google Scholar]
- 22.Lu M, Wang T, Yan Q, et al. Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter’s) knockout mice. J Biol Chem. 2002;277:37881–37887. doi: 10.1074/jbc.M206644200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fowler BC, Chang YS, Laamarti A, et al. Evidence for apical sodium proton exchange in macula densa cells. Kidney Int. 1995;47:746–751. doi: 10.1038/ki.1995.114. [DOI] [PubMed] [Google Scholar]
- 24.Peti-Peterdi J, Chambrey R, Bebok Z, et al. Macula densa Na(+)/H(+) exchange activities mediated by apical NHE2 and basolateral NHE4 isoforms. Am J Physiol Renal Physiol. 2000;278:F452–463. doi: 10.1152/ajprenal.2000.278.3.F452. [DOI] [PubMed] [Google Scholar]
- 25.Amemiya M, Loffing J, Lotscher M, et al. Expression of NHE-3 in the apical membrane of rat renal proximal tubule and thick ascending limb. Kidney Int. 1995;48:1206–1215. doi: 10.1038/ki.1995.404. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Carretero OA, Garvin JL. Inhibition of apical Na+/H+ exchangers on the macula densa cells augments tubuloglomerular feedback. Hypertension. 2003;41:688–691. doi: 10.1161/01.HYP.0000048863.75711.B2. [DOI] [PubMed] [Google Scholar]
- 27.Liu R, Carretero OA, Ren Y, et al. Increased intracellular pH at the macula densa activates nNOS during tubuloglomerular feedback. Kidney Int. 2005;67:1837–1843. doi: 10.1111/j.1523-1755.2005.00282.x. [DOI] [PubMed] [Google Scholar]
- 28.Lorenz JN, Dostanic-Larson I, Shull GE, et al. Ouabain inhibits tubuloglomerular feedback in mutant mice with ouabain-sensitive alpha1 Na,K-ATPase. J Am Soc Nephrol. 2006;17:2457–2463. doi: 10.1681/ASN.2006040379. [DOI] [PubMed] [Google Scholar]
- 29.Noonan WT, Woo AL, Nieman ML, et al. Blood Pressure Maintenance in NHE3-Deficient Mice withTransgenic Expression of NHE3 in Small Intestine. Am J Physiol Regul Integr Comp Physiol. 2005;288:R685–691. doi: 10.1152/ajpregu.00209.2004. [DOI] [PubMed] [Google Scholar]
- 30.Bell PD, Lapointe JY, Peti-Peterdi J. Macula densa cell signaling. Annu Rev Physiol. 2003;65:481–500. doi: 10.1146/annurev.physiol.65.050102.085730. [DOI] [PubMed] [Google Scholar]
- 31.Beeuwkes R, Rosen S. Renal Na+-K+-ATPase: localization and quantitation by means of its K+-dependent phosphatase activity. In: EL B, editor. Current Topics in Membranes and Transport. Vol. 13. Academic Press; New York: 1980. pp. 343–354. [Google Scholar]
- 32.Kashgarian M, Biemesderfer D, Caplan M, et al. Monoclonal antibody to Na,K-ATPase: immunocytochemical localization along nephron segments. Kidney Int. 1985;28:899–913. doi: 10.1038/ki.1985.216. [DOI] [PubMed] [Google Scholar]
- 33.Schnermann J, Marver D. ATPase activity in macula densa cells of the rabbit kidney. Pflugers Arch. 1986;407:82–86. doi: 10.1007/BF00580725. [DOI] [PubMed] [Google Scholar]
- 34.Welling LW, Welling DJ. Surface areas of brush border and lateral cell walls in the rabbit proximal nephron. Kidney Int. 1975;8:343–348. doi: 10.1038/ki.1975.125. [DOI] [PubMed] [Google Scholar]
- 35.Peti-Peterdi J, Bebok Z, Lapointe JY, et al. Novel regulation of cell [Na(+)] in macula densa cells: apical Na(+) recycling by H-K-ATPase. Am J Physiol Renal Physiol. 2002;282:F324–329. doi: 10.1152/ajprenal.00251.2001. [DOI] [PubMed] [Google Scholar]
- 36.Wetzel RK, Sweadner KJ. Immunocytochemical localization of Na-K-ATPase alpha- and gamma-subunits in rat kidney. Am J Physiol Renal Physiol. 2001;281:F531–545. doi: 10.1152/ajprenal.2001.281.3.F531. [DOI] [PubMed] [Google Scholar]
- 37.Wetzel RK, Sweadner KJ. Phospholemman expression in extraglomerular mesangium and afferent arteriole of the juxtaglomerular apparatus. Am J Physiol Renal Physiol. 2003;285:F121–129. doi: 10.1152/ajprenal.00241.2002. [DOI] [PubMed] [Google Scholar]
- 38.Arystarkhova E, Wetzel RK, Sweadner KJ. Distribution and oligomeric association of splice forms of Na(+)-K(+)-ATPase regulatory gamma-subunit in rat kidney. Am J Physiol Renal Physiol. 2002;282:F393–407. doi: 10.1152/ajprenal.00146.2001. [DOI] [PubMed] [Google Scholar]
- 39.Farman N, Fay M, Cluzeaud F. Cell-specific expression of three members of the FXYD family along the renal tubule. Ann N Y Acad Sci. 2003;986:428–436. doi: 10.1111/j.1749-6632.2003.tb07225.x. [DOI] [PubMed] [Google Scholar]
- 40.Pu HX, Cluzeaud F, Goldshleger R, et al. Functional role and immunocytochemical localization of the gamma a and gamma b forms of the Na,K-ATPase gamma subunit. J Biol Chem. 2001;276:20370–20378. doi: 10.1074/jbc.M010836200. [DOI] [PubMed] [Google Scholar]
- 41.James PF, Grupp IL, Grupp G, et al. Identification of a specific role for the Na,K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol Cell. 1999;3:555–563. doi: 10.1016/s1097-2765(00)80349-4. [DOI] [PubMed] [Google Scholar]
- 42.Dostanic I, Lorenz JN, Schultz Jel J, et al. The alpha2 isoform of Na,K-ATPase mediates ouabain-induced cardiac inotropy in mice. J Biol Chem. 2003;278:53026–53034. doi: 10.1074/jbc.M308547200. [DOI] [PubMed] [Google Scholar]
- 43.Dostanic I, Schultz Jel J, Lorenz JN, et al. The alpha 1 isoform of Na,K-ATPase regulates cardiac contractility and functionally interacts and co-localizes with the Na/Ca exchanger in heart. J Biol Chem. 2004;279:54053–54061. doi: 10.1074/jbc.M410737200. [DOI] [PubMed] [Google Scholar]
- 44.Greger R. Ion transport mechanism in thick ascending limb of Henle’s loop of mammalian nephron. Physiol. Rev. 1985;65:760–797. doi: 10.1152/physrev.1985.65.3.760. [DOI] [PubMed] [Google Scholar]
- 45.Verlander JW, Moudy RM, Campbell WG, et al. Immunohistochemical localization of H-K-ATPase alpha(2c)-subunit in rabbit kidney. Am J Physiol Renal Physiol. 2001;281:F357–365. doi: 10.1152/ajprenal.2001.281.2.F357. [DOI] [PubMed] [Google Scholar]
- 46.Rajendran VM, Sangan P, Geibel J, et al. Ouabain-sensitive H,K-ATPase functions as Na,K-ATPase in apical membranes of rat distal colon. J Biol Chem. 2000;275:13035–13040. doi: 10.1074/jbc.275.17.13035. [DOI] [PubMed] [Google Scholar]
- 47.Persson BE, Marsh DJ. GFR regulation and flow-dependent electrophysiology of early distal tubule in Amphiuma. Am J Physiol Renal Physiol. 1987;253:F263–268. doi: 10.1152/ajprenal.1987.253.2.F263. [DOI] [PubMed] [Google Scholar]
- 48.Persson BE, Sakai T, Ekblom M, et al. Effect of bumetanide on tubuloglomerular feedback in Necturus maculosus. Acta Physiol Scand. 1989;137:93–99. doi: 10.1111/j.1748-1716.1989.tb08724.x. [DOI] [PubMed] [Google Scholar]
- 49.Osswald H, Nabakowski G, Hermes H. Adenosine as a possible mediator of metabolic control of glomerular filtration rate. Int J Biochem. 1980;12:263–267. doi: 10.1016/0020-711x(80)90082-8. [DOI] [PubMed] [Google Scholar]
- 50.Vallon V, Muhlbauer B, Osswald H. Adenosine and kidney function. Physiol Rev. 2006;86:901–940. doi: 10.1152/physrev.00031.2005. [DOI] [PubMed] [Google Scholar]
- 51.Schnermann J, Weihprecht H, Briggs JP. Inhibition of tubuloglomerular feedback during adenosine1 receptor blockade. Am J Physiol Renal Physiol. 1990;258:F553–561. doi: 10.1152/ajprenal.1990.258.3.F553. [DOI] [PubMed] [Google Scholar]
- 52.Brown R, Ollerstam A, Johansson B, et al. Abolished tubuloglomerular feedback and increased plasma renin in adenosine A1 receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1362–1367. doi: 10.1152/ajpregu.2001.281.5.R1362. [DOI] [PubMed] [Google Scholar]
- 53.Sun D, Samuelson LC, Yang T, et al. Mediation of tubuloglomerular feedback by adenosine: Evidence from mice lacking adenosine 1 receptors. Proc Natl Acad Sci U S A. 2001;98:9983–9988. doi: 10.1073/pnas.171317998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vallon V, Richter K, Huang DY, et al. Functional consequences at the single-nephron level of the lack of adenosine A1 receptors and tubuloglomerular feedback in mice. Pflugers Arch. 2004;448:214–221. doi: 10.1007/s00424-004-1239-8. [DOI] [PubMed] [Google Scholar]
- 55.Hashimoto S, Huang Y, Briggs J, et al. Reduced autoregulatory effectiveness in adenosine 1 receptor-deficient mice. Am J Physiol Renal Physiol. 2006;290:F888–891. doi: 10.1152/ajprenal.00381.2005. [DOI] [PubMed] [Google Scholar]
- 56.Just A, Arendshorst WJ. A novel mechanism of renal blood flow autoregulation and the autoregulatory role of A1 adenosine receptors in mice. Am J Physiol Renal Physiol. 2007;293:F1489–1500. doi: 10.1152/ajprenal.00256.2007. [DOI] [PubMed] [Google Scholar]
- 57.Hansen PB, Castrop H, Briggs J, et al. Adenosine Induces Vasoconstriction through Gi-Dependent Activation of Phospholipase C in Isolated Perfused Afferent Arterioles of Mice. J Am Soc Nephrol. 2003;14:2457–2465. doi: 10.1097/01.asn.0000086474.80845.25. [DOI] [PubMed] [Google Scholar]
- 58.Lai EY, Patzak A, Steege A, et al. Contribution of adenosine receptors in the control of arteriolar tone and adenosine-angiotensin II interaction. Kidney Int. 2006;70:690–698. doi: 10.1038/sj.ki.5001650. [DOI] [PubMed] [Google Scholar]
- 59.Hansen PB, Friis UG, Uhrenholt TR, et al. Intracellular signalling pathways in the vasoconstrictor response of mouse afferent arterioles to adenosine. Acta Physiol (Oxf) 2007;191:89–97. doi: 10.1111/j.1748-1716.2007.01724.x. [DOI] [PubMed] [Google Scholar]
- 60.Hansen PB, Hashimoto S, Oppermann M, et al. Vasoconstrictor and vasodilator effects of adenosine in the mouse kidney due to preferential activation of A1 or A2 adenosine receptors. J Pharmacol Exp Ther. 2005;315:1150–1157. doi: 10.1124/jpet.105.091017. [DOI] [PubMed] [Google Scholar]
- 61.Hall JE, Granger JP. Adenosine alters glomerular filtration control by angiotensin II. Am J Physiol Renal Physiol. 1986;250:F917–923. doi: 10.1152/ajprenal.1986.250.5.F917. [DOI] [PubMed] [Google Scholar]
- 62.Hall JE, Granger JP, Hester RL. Interactions between adenosine and angiotensin II in controlling glomerular filtration. Amer. J. Physiol. Renal Physiol. 1985;248:F340–F346. doi: 10.1152/ajprenal.1985.248.3.F340. [DOI] [PubMed] [Google Scholar]
- 63.Weihprecht H, Lorenz JN, Briggs JP, et al. Synergistic effects of angiotensin and adenosine in the renal microvasculature. Am J Physiol Renal Physiol. 1994;266:F227–239. doi: 10.1152/ajprenal.1994.266.2.F227. [DOI] [PubMed] [Google Scholar]
- 64.Schnermann JB, Traynor T, Yang T, et al. Absence of tubuloglomerular feedback responses in AT1A receptor-deficient mice. Am J Physiol Renal Physiol. 1997;273:F315–320. doi: 10.1152/ajprenal.1997.273.2.F315. [DOI] [PubMed] [Google Scholar]
- 65.Traynor T, Yang T, Huang YG, et al. Tubuloglomerular feedback in ACE-deficient mice. Am J Physiol Renal Physiol. 1999;276:F751–757. doi: 10.1152/ajprenal.1999.276.5.F751. [DOI] [PubMed] [Google Scholar]
- 66.Traynor T, Yang T, Huang YG, et al. Inhibition of adenosine-1 receptor-mediated preglomerular vasoconstriction in AT1A receptor-deficient mice. Am J Physiol Renal Physiol. 1998;275:F922–927. doi: 10.1152/ajprenal.1998.275.6.F922. [DOI] [PubMed] [Google Scholar]
- 67.Hashimoto S, Adams JW, Bernstein KE, et al. Micropuncture determination of nephron function in mice without tissue angiotensin converting enzyme. Am J Physiol Renal Physiol. 2005;288:F445–F452. doi: 10.1152/ajprenal.00297.2004. [DOI] [PubMed] [Google Scholar]
- 68.Kessler SP, Hashimoto S, Senanayake PS, et al. Nephron function in transgenic mice with selective vascular or tubular expression of Angiotensin-converting enzyme. J Am Soc Nephrol. 2005;16:3535–3542. doi: 10.1681/ASN.2005020151. [DOI] [PubMed] [Google Scholar]
- 69.Lai EY, Martinka P, Fahling M, et al. Adenosine restores angiotensin II-induced contractions by receptor-independent enhancement of calcium sensitivity in renal arterioles. Circ Res. 2006;99:1117–1124. doi: 10.1161/01.RES.0000249530.85542.d4. [DOI] [PubMed] [Google Scholar]
- 70.Ichihara A, Hayashi M, Koura Y, et al. Blunted tubuloglomerular feedback by absence of angiotensin type 1A receptor involves neuronal NOS. Hypertension. 2002;40:934–939. doi: 10.1161/01.hyp.0000041220.88322.6d. [DOI] [PubMed] [Google Scholar]
- 71.Kihara M, Umemura S, Sugaya T, et al. Expression of neuronal type nitric oxide synthase and renin in the juxtaglomerular apparatus of angiotensin type-1a receptor gene-knockout mice. Kidney Int. 1998;53:1585–1593. doi: 10.1046/j.1523-1755.1998.00904.x. [DOI] [PubMed] [Google Scholar]
- 72.Bell PD, Lapointe JY, Sabirov R, et al. Macula densa cell signaling involves ATP release through a maxi anion channel. Proc Natl Acad Sci U S A. 2003;100:4322–4327. doi: 10.1073/pnas.0736323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chan CM, Unwin RJ, Bardini M, et al. Localization of P2X1 purinoceptors by autoradiography and immunohistochemistry in rat kidneys. Am J Physiol. 1998;274:F799–804. doi: 10.1152/ajprenal.1998.274.4.F799. [DOI] [PubMed] [Google Scholar]
- 74.Inscho EW, Cook AK, Imig JD, et al. Physiological role for P2X1 receptors in renal microvascular autoregulatory behavior. J Clin Invest. 2003;112:1895–1905. doi: 10.1172/JCI18499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kishore BK, Isaac J, Fausther M, et al. Expression of NTPDase1 and NTPDase2 in murine kidney: relevance to regulation of P2 receptor signaling. Am J Physiol Renal Physiol. 2005;288:F1032–1043. doi: 10.1152/ajprenal.00108.2004. [DOI] [PubMed] [Google Scholar]
- 76.Thomson S, Bao D, Deng A, et al. Adenosine formed by 5′-nucleotidase mediates tubuloglomerular feedback. J Clin Invest. 2000;106:289–298. doi: 10.1172/JCI8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Castrop H, Huang Y, Hashimoto S, et al. Impairment of tubuloglomerular feedback regulation of GFR in ecto-5′-nucleotidase/CD73-deficient mice. J Clin Invest. 2004;114:634–642. doi: 10.1172/JCI21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang DY, Vallon V, Zimmermann H, et al. Ecto-5′-nucleotidase (cd73)-dependent and -independent generation of adenosine participates in the mediation of tubuloglomerular feedback in vivo. Am J Physiol Renal Physiol. 2006;291:F282–288. doi: 10.1152/ajprenal.00113.2005. [DOI] [PubMed] [Google Scholar]
- 79.Mundel P, Bachmann S, Bader M, et al. Expression of nitric oxide synthase in kidney macula densa cells. Kidney Int. 1992;42:1017–1019. doi: 10.1038/ki.1992.382. [DOI] [PubMed] [Google Scholar]
- 80.Wilcox CS, Welch WJ, Murad F, et al. Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc Natl Acad Sci U S A. 1992;89:11993–11997. doi: 10.1073/pnas.89.24.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vallon V, Traynor T, Barajas L, et al. Feedback Control of Glomerular Vascular Tone in Neuronal Nitric Oxide Synthase Knockout Mice. J Am Soc Nephrol. 2001;12:1599–1606. doi: 10.1681/ASN.V1281599. [DOI] [PubMed] [Google Scholar]
- 82.Levine DZ, Burns KD, Jaffey J, et al. Short-term modulation of distal tubule fluid nitric oxide in vivo by loop NaCl reabsorption. Kidney Int. 2004;65:184–189. doi: 10.1111/j.1523-1755.2004.00361.x. [DOI] [PubMed] [Google Scholar]