

Abstract
Previously we have shown that intestinal cells efficiently take up oxidized fatty acids (OxFAs) and that atherosclerosis is increased when animals are fed a high cholesterol diet in the presence of oxidized linoleic acid. Interestingly, we found that in the absence of dietary cholesterol, the oxidized fatty acid fed low-density lipoprotein (LDL) receptor negative mice appeared to have lower plasma triglyceride (TG) levels as compared to animals fed oleic acid. In the present study, we fed C57BL6 mice a normal mice diet supplemented with oleic acid or oxidized linoleic acid (at 18 mg/animal/day) for 2 weeks. After the mice were sacrificed, we measured the plasma lipids and collected livers for the isolation of RNA. The results showed that while there were no significant changes in the levels of total cholesterol and high-density lipoprotein cholesterol (HDLc), there was a significant decrease (41.14%) in the levels of plasma TG in the mice that were fed oxidized fatty acids.
The decreases in plasma TG levels were accompanied by significant increases (P < 0.001) in the expressions of APOA5 and acetyl-CoA oxidase genes as well as a significant (P < 0.04) decrease in APOClll gene expression. Oxidized lipids have been suggested to be ligands for peroxisome proliferator-activated receptor (PPARα). However, there were no increases in the mRNA or protein levels of PPARα in the oxidized linoleic acid fed animals. These results suggest that oxidized fatty acids may act through an APOA5/APOClll mechanism that contributes to lowering of TG levels other than PPARα induction.
Keywords: Fatty acids, PPARα, Atherosclerosis, Lipoproteins, Inflammation
1. Introduction
Dietary oxidized lipids have been suggested to contribute to atherosclerosis [1]. Increased peroxide levels in chylomicronsVLDL/LDL and increased atherosclerosis have been observed in animals fed an atherogenic diet that included oxidized lipids. We have previously shown that the intestine efficiently absorbs oxidized linoleic acid. We have also shown that atherosclerosis is increased in animals fed a high cholesterol diet in the presence of oxidized fatty acids (OxFAs). Animals fed non-atherogenic diet did not develop atherosclerosis even in the presence of oxidized fatty acids [2]. Oxidized lipids have been shown to be ligands for PPARs [3]. Feeding rats oxidized fatty acids have been shown to activate hepatic PPARα and PPARα-regulated genes [4]. Fenofibrate decreases plasma TG through the modulation of hepatic expression of PPARα responsive genes, increases PPARα mRNA expression and lowers apolipoproteins B and C-III [5]. It is now recognized that high TG levels and low HDL-cholesterol levels are independent risk factors for coronary artery disease (CAD) and that the metabolic syndrome increases risk at any LDL-cholesterol level [6]. There is also evidence that low HDL-cholesterol and high TG levels modulate the capacity of statins to decrease cardiovascular risk in primary and secondary prevention of CHD. The evidence also shows that by increasing HDL-cholesterol levels in patients with both low HDL-cholesterol and LDL-cholesterol, it decreases the risk for CHD in secondary prevention [7–9]. APOA5 is identified as a member of the apolipoprotein gene family, which belongs to the APOA1/APOClll/A4 gene cluster on chromosome 11. It is expressed predominantly in the liver and excreted into plasma. APOA5 is associated with low TG and increased high-density lipoprotein (HDL) levels. Contrary to APOClll, transgenic mice expressing the hAPOA5 gene have significantly decreased triglyceride concentrations. Whereas the APOA5 knockout mouse has increased plasma TG concentrations compared with wild-type mice [10,11]. In the current study, we have examined the effect of dietary oxidized fatty acids on plasma lipoproteins and its relation to genes involved in TG metabolism.
2. Materials and methods
Linoleic acid and soybean lipoxidase (Type V) were purchased from Sigma–Aldrich, St. Louis, MO. All primers, Trizol reagent and SuperScriptTM III First-Strand Synthesis SuperMix for qRT-PCR kit (cDNA preparation and mRNA quantification kit) were obtained from Invitrogen, Carlsbad, CA, USA. The polyclonal antibody to mouse PPARα was purchased from Cayman Chemical Company, Ann Arbor, MI, USA. Other antibodies were obtained from General Electric Company, Fairfield, CT, USA. C57 Black mice were obtained from The Jackson Laboratory, Bar Harbor, ME. Standard mouse chow was provided by Harlan Teklad (Cat: 8640, 22/5 rodent diet), Harlan Teklad Laboratory, Madison, WI, USA.
2.1. Animals, diet administration, and collection of the samples
Ohio State University Animal Care Committee approved the protocol and animals were treated in compliance with the University Animal Committee regulations. The research was conducted in conformity with the PHS policy.
Eighteen male C57BL6 mice weighing 18−21 g were used in the study. The animal diet was prepared by mixing standard mouse (Table 1) with oleic acid or oxidized linoleic acid (13-HPODE). Animals were divided into two groups. Control group was fed 18 mg/day of oleic acid (n = 9) and the experimental group was fed 13-HPODE (n = 9). Both groups were fed the customized mouse chow for 2 weeks. Animals were housed under controlled light and temperature conditions (12-h light–dark cycle for 15 days). At the end of the feeding period, animal chow was removed and mice were fasted over night; blood was collected into tubes containing heparin, plasma was removed after blood centrifugation, and stored at −80 °C until processed for lipid analysis. The liver was immediately harvested, placed in Trizol reagent and kept at −80 °C processed for RNA extraction.
Table 1.
Standard rodent chow nutrients composition
| Protein | 22.58% |
| Fat | 5.23% |
| Fiber | 3.94% |
| Ash | 7.06% |
| Nitrogen-free extract | 51.19% |
| Gross energy | 3.82 kcal/g |
| Digestible energy | 3.38 kcal/g |
| Metabolizable energy | 3.11 kcal/g |
| Amino acids | |
| Arginine | 1.55% |
| Methionine | 0.37% |
| Cystine | 0.37% |
| Histidine | 0.52% |
| Isoleucine + valine | 1.34% |
| Leucine + tryptophane | 1.17% |
| Lysine + threonine | 0.72% |
| Phenylalanine + tyrosine | 1.04% |
| Minerals | |
| Calcium + phosphorus | 1.01% |
| Sodium + potassium | 1.40% |
| Chlorine | 0.67% |
| Magnesium | 0.24% |
| Iron | 348.75 mg/kg |
| Manganese | 104.19 mg/kg |
| Zinc + cobalt | 90.89 mg/kg |
| Copper | 24.07 mg/kg |
| Iodine + selenium | 2.95 mg/kg |
| Vitamins | |
| Vitamin A + D3 | 18.93 IU/g |
| Vitamin E | 109.54 IU/kg |
| Choline | 2.39 mg/g |
| Nicotinic acid | 65.61 mg/kg |
| Pantothenic acid + B1 | 55.13 mg/kg |
| Vitamin B6 + vitamin B2 | 23.01 mg/kg |
| Menadione (vitamin K3) | 5.22 mg/kg |
| Folic acid + biotin | 3.61 mg/kg |
| Vitamin B12 | 54.60 mcg/kg |
Standards rodent diet was provided by Harlan Teklad (Cat: 8640, 22/5 rodent diet), the formula is also enriched with vitamins, amino acids and minerals.
2.2. Preparation of 13-HPODE
13-Hydroperoxyoctadecadienoic acid (13-HPODE) was prepared as we previously described [2]. Briefly, approximately 10 g of linoleic acid (3.6 mmol/L) were oxidized with 3 million units of soybean lipoxidase per gram of fatty acid (3 h at 37 °C, pH 11). Oxidation process was periodically checked and additional lipoxidase was added until a complete oxidation was attained. An increase in absorption at 234 nm was used for monitoring the reaction progress in spectrophotometer (Uvikon XL, Biotech Instuments, CA, USA). The amount of conjugated diene formed was determined employing the molar extinction coefficient 23 mM−1 C−1 at 234 nm. Lipid peroxide generated was further measured using leuco methylene blue assay. 13-HPODE was then extracted with diethyl ether and dried under liquid nitrogen.
2.3. Protein extractions, measurement and Western blot
Liver lysates were prepared in 250 mM sucrose, 10 mM Tris–HCl, pH 7.4, 1 mM EDTA as previously described [12]. Protein concentration in laysates was measured using Bradford reagent. Twenty micrograms of whole-cell lysate was subjected to 12% SDS-PAGE followed by transfer to nitrocellulose membranes. Immunoblots were developed using primary antibodies against PPARα and anti rabbit IgG secondary antibodies conjugated with horseradish peroxidase.
2.4. Total RNA preparation and analysis
Mouse liver was homogenized in Trizol reagent. The total hepatic RNA was isolated using Trizol reagent according to manufacturer's directions. RNA extracted from liver was quantified using The Qubit™ Quantitation Fluorometer and Quant-iT™ Reagents made by Invitrogen.
RNA products were analyzed on a 1% agarose gel electrophoresis and DNA was visualized by ethidium bromide staining using a UV-light box. The intensities of the bands on the images, the purity and the integrity of the RNA were determined by UVP BioSpectrum® Imaging System, Upland, CA, USA.
2.5. Quantitative RT PCR
The sequences of the primers, the protocol and validation of the RT-competitive PCR assays were performed prior to analysis. cDNA synthesis was performed in a total volume of 20 μL of 2XRT mix, RT enzyme, DEPC-treated water (Invitrogen kit) containing 1 μL of RNA using GT-Storm PCR system, Gene Technologies Ltd., Essex, UK. cDNA synthesized after the mixture containing RNA was incubated in micro-tubes at 25 °C for 10 min for denaturation, and annealed at 50 °C for 30 min after which the reaction was terminated at 85 °C for 5 min and later chilled down. Bio-Rad iQ5TM, Hercules, CA, USA RT-PCR system was used for the quantitative real-time PCR studies. Total volume of 20 μL SYBR, DEPC-treated water contains 1 μL each of cDNA template. Forward and reverse primers (10 μM) for each gene was separately pipetted in a 96 well plate. The samples underwent a number of cycles of 2 min at 50, 95 and 60 °C for total of 40 cycles. The following primers were designed and used in the study: mouse GAPDH (forward: CCTGCACCACCAACTGCTTA, reverse: TCATGAGCCCTTCCACAATG. mouse PPARα (forward: AAG AGG GCT GAG CGT AGG T, reverse: GGC CGG TTA AGA CCA GAC T. mouse APOA5 (forward: GAA CGC TTG GTGACTGGA AT, reverse: TCG CCT TAC GTG TGA GTTTG. mouse APOClll (forward: GTG TTG CAG ATG TGC CTG TT, reverse: GGA GGG GTG AAG ACA TGAGA. mouse hepatic lipase (forward: GAC TGG ATC TCC CTG GCA TA, reverse: AGG TGA ACT TTG CTC CGA GA. mouse acetyl-CoA oxidase (forward: CCACATATGACCCCAAGACC, reverse: AGGCATGTAACCCGTAGCAC.
Results for the expression of specific mRNAs were always presented relative to the expression of the control gene (GAPDH).
2.6. Plasma lipids determination
Plasma total cholesterol, high-density lipoproteins and triglycerides were measured by enzymatic methods. The total cholesterol and triglycerides were analyzed using ready to use reagents obtained from the Beckman Coulter Clinical Diagnostics Division, USA. LDL and HDL were measured using kits purchased from Equal Diagnostics, (Exton PA USA). The analysis was performed in a Beckman Coulter CX4 auto analyzer.
2.7. Statistical analysis
All data was expressed as mean ± S.D. Statistical analysis was performed using ANOVA for group comparison and Student's t-test for pairs. P ≤ 0.05 was considered significant.
3. Results and discussion
The animals were divided into two groups. The control group was fed normal chow mixed with oleic acid (18 mg/day) and the experimental group was fed normal chow mixed with 13-HPODE (18 mg/day). The standard mouse chow used in this study contains 3.32% linoleic acid. This amount by itself is sufficient to serve as control for the experimental group which was in addition supplemented with oxidized linoleic acid in form of 13-HPODE. It is well understood that fatty acids may be auto oxidized resulting in different oxygenated groups, mainly hydroxy, keto, and epoxy, as well as short-chain fatty acetyl groups as the main products. The rates of auto-oxidation of fatty acids are known to increase with the increases in the number of double bonds. Although the control and experimental diets were prepared and stored at a condition that is less favorable for auto-oxidation, and due to the fact that linoleic acid is more rapidly oxidized compared to oleic acid, we designed the study to further supplement the control group with oleic acid to provide for a less oxidized fatty acid and serve as control.
After 15 days of feeding, the mice on the 13-HPODE containing diet showed a significant reduction (41.14%, P < 0.02) of TG levels in their plasma compared to controls (Table 2). The decrease in TG was accompanied by a decrease in total cholesterol (15.43%) and elevation in HDL (1.43%). However, changes in total cholesterol and HDL were not significant. Evidently, the reduction in TG was a result of the administration of oxidized fatty acids and not due to feeding of polyunsaturated fatty acids, as both diets contains considerable amount of linoleic acid, however changes in TG levels was only observed in group supplemented with 13-HPODE. Also repeated studies from our laboratory comparing different strains of mice fed diet rich in polyunsaturated fatty acid or monounsaturated fatty acids did not show any difference in the level of TG (unpublished data). The increase in plasma TG-rich lipoproteins may have resulted from either increased production from the liver and intestine, by means of upregulated synthetic and secretory pathways, or through decreased peripheral catabolism, mainly from reduced lipoprotein lipase activity [13,14]. The decreased plasma TG levels of 13-HPODE fed mice in this study is apparent due to an active catabolic process and seems to be influenced by dietary intake. The diet and body weight of mice on both groups were closely monitored. The mice in both groups had similar eating patterns and gained similar weight over the 2 weeks study (data not shown).
Table 2.
Mice plasma lipids
| Group | Control n = 9 | 2 to 13-HPODE n = 9 | P value |
|---|---|---|---|
| Cholesterol mg/dl | 62.53 ± 9.19 | 52.88 ± 21.04 | NS |
| HDL (mg/dl) | 38.94 ± 14.87 | 39.5 ± 17.83 | NS |
| TG (mg/dl) | 103.74 ± 38.6 | 61.061 ± 44.5 | P < 0.02 |
Plasma lipids were measured using commercially available kits, the difference in TG between both groups was significant (P ≤ 0.02). The decrease in TC and the slight increase in HDL was not significant (NS). Values are means ± S.D.
PPARα regulates HDL-cholesterol levels via transcriptional induction of the synthesis of apolipoproteins A (ApoA-I, and ApoA-II). It also mediates action on hepatic APOClll production and increases lipoprotein lipase through lipolysis. PPARα agonists in turn, stimulate cellular fatty acid uptake conversion to acyl-CoA derivatives and catabolism by the β-oxidation pathways. This, combined with a reduction in fatty acid and TG synthesis, results in a decrease in VLDL production [5,15]. We therefore decided to examine whether the decrease in TG is mediated through PPARα. Liver protein was extracted and PPARα Western blotting was performed, and e also quantified PPARα mRNA in liver cells. Our results indicated that there was no PPARα gene induction and the changes in TG levels did not seem to be mediated through the activation of PPARα gene expression or protein synthesis (Fig. 1). There was no significant difference in the Western blot results or gene expression data between control mice and those on 13-HPODE mixed diet. Interestingly, hepatic lipase was significantly (P < 0.007) down regulated among mice fed oxidized fatty acid (Fig. 2). The role of hepatic lipase as a lipolytic enzyme mediating the hydrolysis of TG and phospholipids is well established [16]. The fact that hepatic lipase is down regulated among experimental mice fed oxidized fatty acids in this study clearly indicates that the decreases in TG levels among experimental group is not caused by the action of hepatic lipase. It also indicates that hydrolysis may not be the mechanism that contributed towards the lowering of TG levels. Although it appears that feeding oxidized fatty acids decreases hepatic lipase gene expression, the mechanisms of action of oxidized fatty acids on lipase is not clear.
Fig. 1.
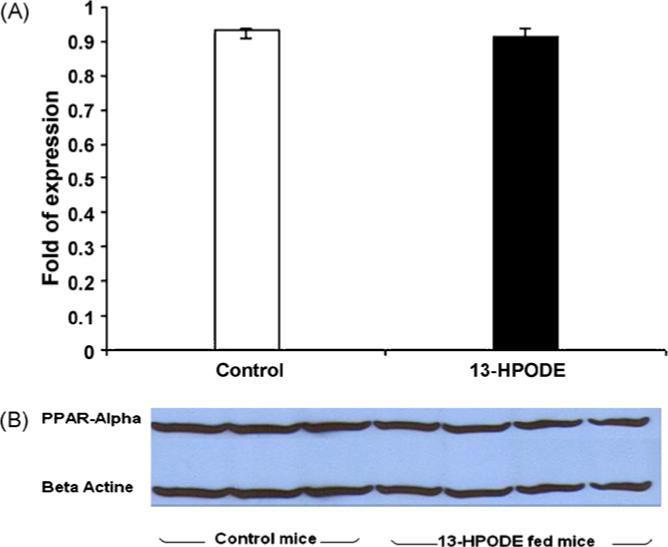
(A) PPARα gene expression. Mice PPARα gene expression have not shown significant difference between the control and 13-HPODE fed mice. Values are means ± S.D. (B) Western blot analysis of PPARα in the liver is shown. Gels were immunoblotted by using anti-PPARα and anti-β-actin (used as an internal control) antibodies. Control mice on oleic acid mixed chow (n = 9) were compared to experimental mice on 13-HPODE mixed chow (n = 9), results did not show significant difference between the two groups; data is represented by three mice from control group, and four from experimental mice.
Fig. 2.
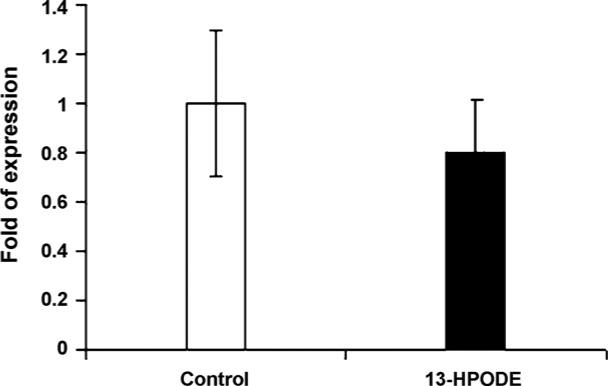
Mice hepatic lipase gene expression. The hepatic lipase gene expression was significantly down regulated among the experimental mice fed 13-HPODE when compared to control (P ≤ 0.007). Values are means ± S.D.
We further examined whether acetyl-CoA oxidase gene expression is affected by the dietary supplementation of oxidized fatty acid. To do this, we measured the quantity of mRNA for acetyl-CoA oxidase (Fig. 3). Acetyl-CoA oxi- dase was significantly (P < 0.02) increased among mice fed oxidized fatty acids compared to control. The increase of acetyl-CoA oxidase brings an interesting piece of evidence. It shows that the decreases in triglycerides may have resulted through involvement of acetyl-CoA oxidase due to a PPARα ligand binding activation. It has been reported that the addition of 0.5% of trans-10, cis-12 conjugated linoleic acid isomer to the diet increases liver fatty acid oxidation leading to decreased hepatic and serum triacylglycerols [17]. Stimulation of acetyl-CoA oxidase by alpha-linolenic acid-rich perilla oil was reported to lower plasma triacylglycerol level in rats [18]. However, Staels et al. have shown that fibrates down regulate APOClll expression independent of induction of peroxisomal acyl coenzyme A oxidase [19]. We further investigate the involvement of APOClll and APOA5, as seen in Fig. 4, APOA5 gene expression has significantly increased among mice on OxFA diet and APOClll gene expression has decreased significantly in this group compared to controls. Several reports from animal and clinical studies have documented the involvement of APOClll in TG metabolism [7,20–22]. APOClll is a component of TG-rich lipoproteins synthesized mainly in the liver and to some extent in the intestine. The major physiological role of APOClll appears to be as an inhibitor of lipoprotein lipase. Therefore, plasma APOClll concentrations are positively associated with triglyceride levels. Consistent with this role, over expression of the human APOClll gene in mice resulted in dramatically increased plasma TG concentrations. Contrary to APOClll, transgenic mice expressing the hAPOA5 gene have significantly decreased TG concentrations whereas the APOA5 knockout mouse has significantly increased plasma TG concentrations compared with wild-type mice [10,11,23]. It is not very clear how APOA5 may modulate the decreases in TG levels. However, data from in vitro studies have suggested that APOA5 may act by increasing lipoprotein lipase (LPL) activity in a fashion similar to that of apolipoprotein C-II, the physiological LPL activator. This hypothesis is supported by some in-vitro studies showing that recombinant APOA5 stimulated LPL activity in the presence of apolipoprotein C-II [24]. However, to our knowledge, there is no published data on in vitro studies that describe the mechanism(s) by which APOA5 is involved in lowering levels of TG. The physicochemical properties of APOA5 protein moiety, has lipid-binding activity as demonstrated by the ability to form lipid–protein complexes. Also, APOA5 displays high affinity, low elasticity and slow binding kinetics at hydrophobic interfaces and leads to the postulation that it might retard TG-rich particle assembly in the liver and inhibit the secretion of VLDL [25]. The other argument is that APOA5 is an activator of intravascular TG hydrolysis by LPL. However, in the current study, LPL does not seem to be involved in this process. In mice, adenoviral over expression of murine APOA5 resulted in a decreased production rate of VLDL triglycerides. Most importantly, as determined by APOB kinetic studies using stable isotopes Marcais et al. have shown that the VLDL production rate was normal but the fractional catabolic rate of VLDL-APOB was decreased more than 20-fold in patients lacking normal APOA5 [26]. These and previously mentioned evidence demonstrate that APOA5 and ApoClll may have modulated the OxFA induced TG decreases.
Fig. 3.
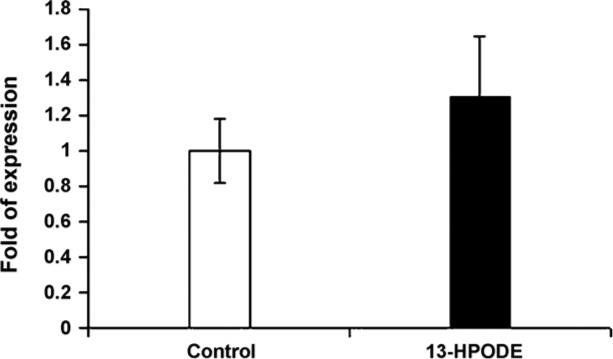
Mice acetyl-CoA oxidase gene expressions. Acetyl-CoA oxidase gene expression was significantly upregulated among mice supplemented with diet mixed with 13-HPODE (P ≤ 0.02). Values are means ± S.D.
Fig. 4.
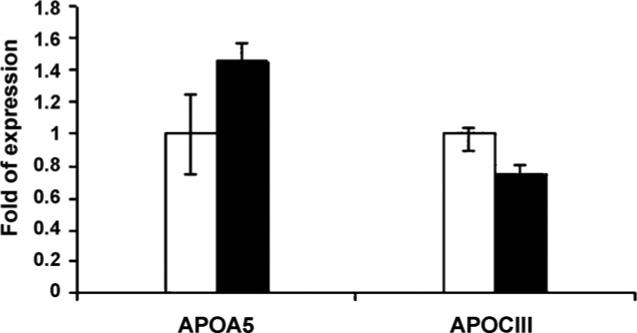
Mice APOA5 and APOClll gene expression. APOA5 was significantly upregulated (P ≤ 0.001) among mice supplemented with diet containing 13-HPODE; however APOClll was significantly (P 0.04) down regulated in this group compared to the control mice fed diet containing oleic acid. Data expressed as means ± S.D. Open bars represent control group whereas the black bars represent the experimental mice on 13-HPODE supplemented diet.
The results suggest that oxidized fatty acids might act through activation of a transcriptional mechanism independent of PPARα gene expression or protein synthesis possibly involving PPARα ligand binding [3] as demonstrated by the upregulation of acetyl-CoA oxidase. This mechanism might affect lipoprotein metabolism and contribute towards lowering of triglycerides levels. APOA5/APOClll seem to modulate the decreases in triglycerides. However, the mechanism is not clearly understood.
In conclusion, the results presented in the study suggest that dietary oxidized fat might be beneficial in the absence of cholesterol. Oxidized lipids have been shown to induce catalase [27], MnSOD [28], heme oxygenase, nitric oxide synthase [29], apo A1 synthesis [30], and glutathione synthase [28]. However, these conclusions are drawn from in vitro cell culture studies. Considering that a plethora of studies have also shown toxic, pro-inflammatory and pathological effects in similar systems, one has to conclude the potential beneficial effects are mere responses to harmful effects. In fact, at high concentrations, dietary oxidized fatty acids appear to induce inflammatory changes in the liver (unpublished results). Thus, the current results may simply mean that these compounds are biologically active and could induce mechanisms in the system that could be important under controlled conditions.
Acknowledgments
This work was supported by NIH grants HL69038 and DK056353. The help of John Canon, Larissa Brophy, Sainath Babu and Priscilla Jaichander during the course of the study is greatly appreciated.
References
- 1.Staprans I, Pan X-M, Rapp JH, Feingold KR. The role of dietary oxidized cholesterol and oxidized fatty acids in the development of atherosclerosis. Mol Nutr Food Res. 2005;49(11):1075–82. doi: 10.1002/mnfr.200500063. [DOI] [PubMed] [Google Scholar]
- 2.Khan-Merchant N, Penumetcha M, Meilhac O, Parthasarathy S. Oxidized fatty acids promote atherosclerosis only in the presence of dietary cholesterol in low-density lipoprotein receptor knockout mice. J Nutr. 2002;132(11):3256–62. doi: 10.1093/jn/132.11.3256. [DOI] [PubMed] [Google Scholar]
- 3.Delerive P, Furman C, Teissier E, et al. Oxidized phospholipids activate PPARalpha in a phospholipase A2-dependent manner. FEBS Lett. 2000;471(1):34–8. doi: 10.1016/s0014-5793(00)01364-8. [DOI] [PubMed] [Google Scholar]
- 4.Ringseis R, Muschick A, Eder K. Dietary oxidized fat prevents ethanol-induced triacylglycerol accumulation and increases expression of PPARalpha target genes in rat liver. J Nutr. 2007;137(1):77–83. doi: 10.1093/jn/137.1.77. [DOI] [PubMed] [Google Scholar]
- 5.Auwerx J, Schoonjans K, Fruchart JC, Staels B. Regulation of triglyceride metabolism by PPARs: fibrates and thiazolidinediones have distinct effects. J Atheroscler Thromb. 1996;3(2):81–9. doi: 10.5551/jat1994.3.81. [DOI] [PubMed] [Google Scholar]
- 6.Fruchart JC, Duriez P. HDL and triglyceride as therapeutic targets. Curr Opin Lipidol. 2002;13(6):605–16. doi: 10.1097/00041433-200212000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Breuer HW. Hypertriglyceridemia: a review of clinical relevance and treatment options: focus on cerivastatin. Curr Med Res Opin. 2001;17(1):60–73. [PubMed] [Google Scholar]
- 8.Verges B. Cardiovascular risk and dyslipidemias. Ann Endocrinol. 1998;59(4):335–43. [PubMed] [Google Scholar]
- 9.Wierzbicki AS. Fibrates after the FIELD study: some answers, more questions. Diab Vasc Dis Res. 2006;3(3):166–71. doi: 10.3132/dvdr.2006.025. [DOI] [PubMed] [Google Scholar]
- 10.van der Vliet HN, Sammels MG, Leegwater AC, et al. Apolipoprotein A-V: a novel apolipoprotein associated with an early phase of liver regeneration. J Biol Chem. 2001;276(48):44512–20. doi: 10.1074/jbc.M106888200. [DOI] [PubMed] [Google Scholar]
- 11.Pennacchio LA, Olivier M, Hubacek JA, et al. Two independent apolipoprotein A5 haplotypes influence human plasma triglyceride levels. Hum Mol Genet. 2002;11(24):3031–8. doi: 10.1093/hmg/11.24.3031. [DOI] [PubMed] [Google Scholar]
- 12.Wilcke M, Alexson SE. Characterization of acyl-CoA thioesterase activity in isolated rat liver peroxisomes. Partial purification and characterization of a long-chain acyl-CoA thioesterase. Eur J Biochem. 1994;222(3):803–11. doi: 10.1111/j.1432-1033.1994.tb18927.x. [DOI] [PubMed] [Google Scholar]
- 13.Lopez A. Serum lipid transport systems: recent advances. Lipids. 1971;6(6):369–77. doi: 10.1007/BF02531373. [DOI] [PubMed] [Google Scholar]
- 14.Keys A. Blood lipids in man—a brief review. J Am Diet Assoc. 1967;51(6):508–16. [PubMed] [Google Scholar]
- 15.Fruchart J-C, Duriez P. Mode of action of fibrates in the regulation of triglyceride and HDL-cholesterol metabolism. Drugs Today (Barc) 2006;42(1):39–64. doi: 10.1358/dot.2006.42.1.963528. [DOI] [PubMed] [Google Scholar]
- 16.Santamarina-Fojo S, Changting H, Amar M. The role of hepatic lipase in lipoprotein metabolism and atherosclerosis. Curr Opin Lipidol. 1998;9:211–9. doi: 10.1097/00041433-199806000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Macarulla T, Fernandez-Quintela A, Zabala A, et al. Effects of conjugated linoleic acid on liver composition and fatty acid oxidation are isomer-dependent in hamster. Nutrition. 2005;21(4):512–9. doi: 10.1016/j.nut.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 18.Kim H-K, Choi H. Stimulation of acyl-CoA oxidase by alpha-linolenic acid-rich perilla oil lowers plasma triacylglycerol level in rats. Life Sci. 2005;77(12):1293–306. doi: 10.1016/j.lfs.2004.10.082. [DOI] [PubMed] [Google Scholar]
- 19.Staels B, Vu-Dac N, Kosykh VA, et al. Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme A oxidase. A potential mechanism for the hypolipidemic action of fibrates. J Clin Invest. 1995;95(2):705–12. doi: 10.1172/JCI117717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klein Richard L, McHenry M Brent, Lok Kerry H, et al. DCCT/EDIC Research Group, Apolipoprotein C-III protein concentrations and gene polymorphisms in type 1 diabetes: associations with microvascular disease complications in the DCCT/EDIC cohort. J Diabetes Complications. 2005;19(1):18–25. doi: 10.1016/j.jdiacomp.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 21.van Dijk KW, Rensen PCN, Voshol PJ, Havekes LM. The role and mode of action of apolipoproteins CIII and AV: synergistic actors in triglyceride metabolism? Curr Opin Lipidol. 2004;15(3):239–46. doi: 10.1097/00041433-200406000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Zeng Q, Dammerman M, Takada Y, et al. An apolipoprotein CIII marker associated with hypertriglyceridemia in Caucasians also confers increased risk in a west Japanese population. Hum Genet. 1995;95(4):371–5. doi: 10.1007/BF00208957. [DOI] [PubMed] [Google Scholar]
- 23.Talmud PJ, Hawe E, Martin S, et al. Relative contribution of variation within the APOC3/A4/A5 gene cluster in determining plasma triglycerides. Hum Mol Genet. 2002;11(24):3039–46. doi: 10.1093/hmg/11.24.3039. [DOI] [PubMed] [Google Scholar]
- 24.Schaap FG, Rensen PCN, Voshol PJ, et al. ApoAV Reduces Plasma Triglycerides by Inhibiting Very Low Density Lipoprotein-Triglyceride (VLDL-TG) Production and Stimulating Lipoprotein Lipase-mediated VLDL-TG Hydrolysis. J Biol Chem. 2004;279(27):27941–7. doi: 10.1074/jbc.M403240200. [DOI] [PubMed] [Google Scholar]
- 25.Calandra S, Oliva CP, Tarugi P, Bertolini S. APOA5 and triglyceride metabolism, lesson from human APOA5 deficiency. Curr Opin Lipidol. 2006;17(2):122–7. doi: 10.1097/01.mol.0000217892.00618.54. [DOI] [PubMed] [Google Scholar]
- 26.Marcais C, Verges B, Charriere S, et al. Apoa5 Q139X truncation pre-disposes to late-onset hyperchylomicronemia due to lipoprotein lipase impairment. J Clin Invest. 2005;115(10):2862–9. doi: 10.1172/JCI24471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meilhaca O, Zhoua M, Santanam N, Parthasarathy S. Lipid peroxides induce expression of catalase in cultured vascular cells. J Lipid Res. 2000;41:1205–13. [PubMed] [Google Scholar]
- 28.Parthasarathy S, Santanam N, Ramachandran S, Meilhac O. Oxidants and antioxidants in atherogenesis. An appraisal. J Lipid Res. 1999;40(12):2143–57. [PubMed] [Google Scholar]
- 29.Ramasamy S, Parthasarathy S, Harrison DG. Regulation of endothelial nitric oxide synthase gene expression by oxidized linoleic acid. J Lipid Res. 1998;39(2):268–76. [PubMed] [Google Scholar]
- 30.Rong R, Ramachandran S, Penumetcha M, et al. Dietary oxidized fatty acids may enhance intestinal apolipoprotein A-I production. J Lipid Res. 2002;43(4):557–64. [PubMed] [Google Scholar]