

Abstract
An excessive amount of matrix metalloproteinase-9 (MMP-9) has been well documented in inflammatory diseases, including chronic wounds and cancers. Secreted as a zymogen, proMMP-9 can be irreversibly converted to a mature form through cleavage of the N-terminal propeptide domain. Although the converting enzyme for proMMP-9 in human tissues is unknown, we previously found that tumor necrosis factor-α (TNF-α) promotes activation of proMMP-9 in human skin, and characterized the converting activities as tissue-associated chymotrypsin-like proteinases. On the other hand, the pathophysiologic inhibitor to prevent proMMP-9 maturation also remains elusive. In this regard, we observed the presence of the inhibitory property in burn blister fluid that abrogates the skin extract–mediated activation of proMMP-9. Then we determined that α-1-antichymotrypsin (α-ACT), an acute-phase factor abundantly present in the blister, effectively inhibited proMMP-9 activation in human and rodent skin. In contrast, the aminophenylmercuric acetate-induced ‘‘cysteine switch’’ and activation of proMMP-9 were not affected by α-ACT. TNF-α-induced activation of proMMP-9 by the explants of human skin was inhibited by α-ACT but not by related α-1-antitrypsin. α-ACT specifically attenuated maturation of proMMP-9 but not proMMP-2 or proMMP-13. Furthermore, short peptides that mimic the reactive center loop (RCL) of α-ACT were sufficient to inhibit the conversion. Mutation analysis demonstrated that a conserved leucine within the RCL was critical for α-ACT-exerted inhibition. In chronic wounds, a large amount of mature MMP-9 was associated with fragmentation and inactivation of α-ACT. Taken together, these results demonstrate that, to the best of our knowledge, α-ACT is a previously unreported pathophysiologic inhibitor that controls proMMP-9 activation in skin tissue.
INTRODUCTION
Large-scale remodeling of the extracellular matrix occurs in embryonic development and recedes to a minimal level in adult tissues to maintain tissue homeostasis (Werb, 1997). Tissue remodeling is regulated by proteinases and their inhibitors at the level of expression, deposition, activation, and inhibition (Birkedal-Hansen, 1995; Parks, 1999). In many inflammation-associated diseases, such as chronic wounds and cancers, proteinases are predominantly expressed, which activates quiescent cells, promotes cell migration, and triggers cell differentiation (Han, 2006). Matrix metallo-proteinase-9 (MMP-9), previously called gelatinase B, is believed to function in the remodeling of the basement membrane zone because several extracellular matrix proteins in the basement membrane zone, such as type IV collagen and BP180, have been identified as the substrates for this proteinase (Seltzer et al., 1989; Stahle-Backdahl et al., 1994). Nascent proMMP-9 (92 kDa in human) is a zymogen that remains latent through the interaction between a conserved cysteine within the N-terminal propeptide domain and a zinc ion within the catalytic domain. The activation of proMMP by disruption of the cysteine–zinc bond is executed mainly by cleavage of the propeptide domain of ~100 amino-acid residues, which consequently leads to reorganization of the cysteine interactions and conformation changes, a process called ‘‘cysteine switch’’ (Springman et al., 1990). In early development and tissue injury, proMMP-9 is transiently expressed and then declines as tissue remodeling proceeds (Salo et al., 1994; Young and Grinnell, 1994; Moses et al., 1996; Tarlton et al., 1997). However, in prolonged inflammation, proMMP-9 is readily converted into the active or mature form (82 kDa) (Wysocki et al., 1993; Weckroth et al., 1996; Yager et al., 1996; Han et al., 2001b; Ladwig et al., 2002). Excessive and prolonged expression and activation of MMPs are etiologic causes of chronic diseases due, at least in part, to excessive tissue destruction (Trengove et al., 1999; Warner et al., 2001).
The mechanism of proMMP-9 activation in human diseases is currently unknown. Isolated human skin cells such as keratinocytes or dermal fibroblasts can secrete proMMP-9 in response to defined cytokine stimulation; however, the activation of proMMP-9 is rarely reconstituted in pure cell cultures (Oikarinen et al., 1993; Han et al., 2001b). The failure of proMMP-9 activation in cell cultures is likely due to the lack of ‘‘tissue milieu,’’ the interaction among cells, cytokines, and extracellular matrix. In this regard, we successfully reconstituted the activation of proMMP-9 through organ culture of human skin treated with specific cytokines (Han et al., 2001a). Through this approach, we have found that transforming growth factor-β (TGF-β) promotes expression of proMMP-9, whereas tumor necrosis factor-α (TNF-α) exerts maturation of the zymogen by explants of human skin. Furthermore, we characterized the converting factor in human skin as a tissue-associated chymotrypsin-like proteinase (Han et al., 2002). Based on such findings, we searched for physiologic or pathologic inhibitors that control proMMP-9 activation in human skin. First, we found that the TNF-α-mediated activation of proMMP-9 in human skin is sufficiently regulated through downregulation of tissue inhibitor of metalloproteinase 1 (TIMP-1), a tissue inhibitor binding to proMMP-9 and preventing the zymogen from access to the converting enzyme. Then we embarked on a search for the pathophysiologic inhibitor antagonizing the conversion per se. Initially, we noticed the presence of inhibitory factor(s) in thermal burn blister fluid, which prevents the proteolytic processing of proMMP-9. This finding indicates that the inhibitor may be an acute-phase factor produced in response to trauma. Given the inhibition of proMMP-9 activation by N-tosyl-phenylalanyl-chloromethyl ketone (TPCK) and chymostatin, two specific inhibitors of chymotrypsin (Han et al., 2002, 2005), we speculated on the presence of an equivalent pathophysiologic inhibitor. We then examined whether the acute-phase factor(s) had antichymotrypsin activity. Among them, we noticed α-1-antichymotrypsin (α-ACT), an acute-phase factor produced in the liver in response to trauma. This led us to examine α-ACT for its inhibition of proMMP-9 conversion in human and rodent skin. Indeed, α-ACT potently inhibits the TNF-α-induced conversion of proMMP-9 in explants of human skin. Moreover, we found that α-ACT, but not MMP inhibitor, also inhibits proMMP-9 activation in rat skin; conversely, IL-1-induced activation of proMMP-9 in rat hepatic stellate cells is refractory to α-ACT but is antagonized by MMP inhibitor (Han et al., 2007). Thus, activation of proMMP-9 appears to be controlled by tissue-specific activators and inhibitors in an evolutionally conserved manner. To understand the mechanism of inhibition, we defined a critical role of the P1 leucine in the reactive center loop (RCL). Further, synthetic peptides derived from the RCL are sufficient for the inhibitory function. The implication of α-ACT in control proMMP-9 activation is further supported by our clinical findings: (1) in the early stage of trauma, the absence of proMMP-9 activation is associated with a large amount of intact α-ACT; and (2) with the progression of wound healing, the maturation of proMMP-9 is associated with degradation and inactivation of α-ACT. Taken together, we demonstrated, for the first time, to the best of our knowledge, that α-ACT is a pathophysiologic inhibitor that controls proMMP-9 activation in skin tissue.
RESULTS
TNF-α-induced activation of proMMP-9 in human skin is inhibited by α-ACT but not by α-1-antitrypsin
To maximally produce mature MMP-9, the normal human skin was costimulated by TNF-α (10 ng ml−1) and TGF-β (1 ng ml−1). Purified α-ACT and α-1-antitrypsin (α-AT) were added directly to the culture at the indicated concentration. After 3 days of culture, the conditioned medium was resolved by zymography. As shown in Figure 1a, in the absence of cytokine treatment, the 72-kDa proMMP-2 and a minimal amount of 92-kDa proMMP-9 were produced in the conditioned medium. In response to the cytokines, 92-kDa proMMP-9 was substantially induced and converted into 82-kDa mature form. α-ACT at 10 μg ml−1 totally blocked the maturation of proMMP-9 but had no effect on the expression of the zymogen. In contrast, α-AT was found to have no significant effect on proMMP-9 activation, indicating the fine specificity of the converting enzyme as chymotrypsin-like serine proteinase. Importantly, this effective concentration of α-ACT (0.01 mg ml−1) is only about 1/50 of the maximal plasma concentration in the acute response to injury (0.5 mg ml−1), implying the pathophysiologic relevance of the inhibitor in restraining proMMP-9 activation (Travis et al., 1990). To determine whether such inhibition can be extended to other species, we stimulated the rat skin explants with TNF-α together with α-ACT. As expected, TNF-α-induced maturation of proMMP-9 was completely inhibited by exogenous α-ACT (data not shown).
Figure 1. Cytokine-induced activation of proMMP-9 in human skin is inhibited by α-ACT but not by α-AT.
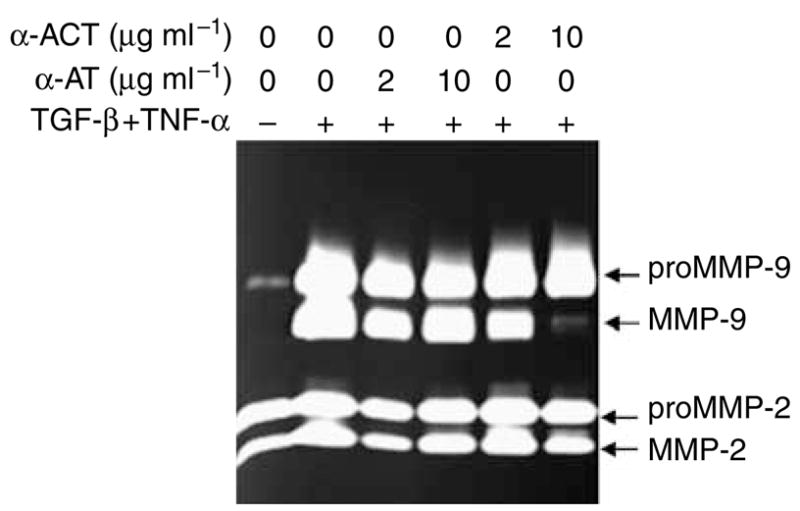
Explants of full thickness normal human skin donors were cultured in DMEM and treated with TGF-β and TNF-α. Purified human α-ACT and α-AT were added to the skin culture at the indicated concentration. After culture for 3 days, the conditioned medium was resolved by gelatinolytic zymography. The identity of MMP-9 and MMP-2 has been previously confirmed by western blot.
In vitro inhibition of proMMP-9 activation by α-ACT
α-ACT was added to the reactions containing purified proMMP-9 and human skin extract that contained the converting enzyme as described previously (Han et al., 2002). As shown in Figure 2a, α-ACT at 0.5 μg ml−1 exhibited partial inhibition; at 5 μg ml−1, the inhibitor nearly blocked the conversion as indicated by the accumulation of proMMP-9. The α-ACT protein was confirmed by western blot analysis (Figure 2b). Importantly, α-ACT exerted its inhibition at 5 μg ml−1, which was 1% of the amount in the plasma in the acute phase (500 μg ml−1), suggesting a pathophysiologic relevance of the inhibitor in acute-phase reaction by attenuation of proMMP-9 activation.
Figure 2. In vitro inhibition of proMMP-9 activation by α-ACT.
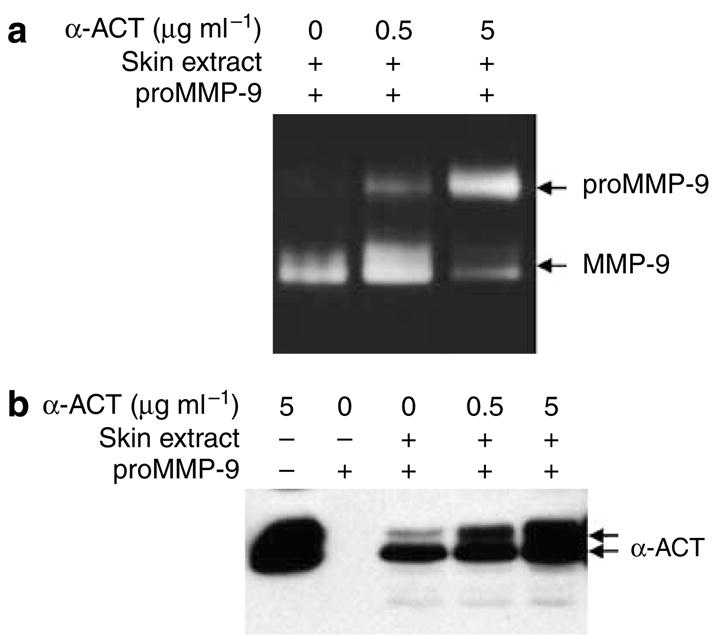
To directly assess the inhibitory function of α-ACT, the skin extract was incubated with proMMP-9 and recombinant α-ACT. (a) After a 16-hour reaction, the products were resolved by gelatinolytic zymography. (b) The α-ACT protein was measured by western blot.
The P1 leucine in the RCL of α-ACT is necessary for the inhibition of proMMP-9 activation
α-ACT belongs to a family of serine protease inhibitors called serpins and exerts its proteolytic inhibition through a ‘‘suicide’’ mechanism by which the RCL of the inhibitor is inserted into the catalytic domain of the proteases (Travis et al., 1990). In this regard, we changed the critical leucine (P1) to serine, designated P1m, and the adjacent serine (P1′) to alanine, called P1′m (Figure 3a). Six histidines were added as a tag to the C terminus of the α-ACT variants to facilitate purification. The wild-type α-ACT and two mutant variants were expressed by lentivirus in immortalized human keratinocytes (IKCs). α-ACT variants from the conditioned medium were purified using Ni-NTA chromatography to near homogeneity, as shown by protein staining and western blot (Figure 3b and c). We performed the converting assay by incubating proMMP-9 with human skin extract and α-ACT variants. As shown in the zymography, the wild-type α-ACT at 50 μg ml−1 clearly blocked the maturation process, whereas the leucine mutation (P1m) abolished its inhibition (Figure 3d). In contrast, the P′1 serine mutant (P1′m) still retains its inhibition, showing efficiency similar to that of the wild type (Figures 2 and 3e). Thus, the P1 leucine in the RCL is critical for α-ACT to inhibit proMMP-9 conversion.
Figure 3. A conserved leucine (P1) in the RCL of α-ACT is essential for inhibition of proMMP-9 activation.

(a) Sequence information of the RCL of human α-ACT indicates the P1 leucine and the variants. (b and c) The α-ACT variants were expressed by IKCs transduced by lentivirus. A polyhistidine tag was added to the C terminus of the α-ACT variants. The α-ACT variants were purified from the conditioned medium by Ni-NTA column and examined by Coomassie blue and western blot analysis. (d and e) Inhibition by the α-ACT variants was assayed by incubation with proMMP-9 and skin extract, and the reactions were revealed by zymography.
The RCL of α-ACT is sufficient to inhibit proMMP-9 activation
We further addressed the question of whether the RCL of α-ACT is sufficient to exert the inhibition of proMMP-9 activation. To this end, we synthesized a linear peptide of 25 residues corresponding to the RCL from amino-acid residues 153 to 177 of human α-ACT (Figure 4a). A cyclic peptide of the RCL was also created by introducing two cysteines followed by oxidation. First, we performed an in vitro assay by incubating the peptides or α-ACT protein with human skin extract together with proMMP-9. Both the linear and cyclic peptides potently inhibited the conversion (Figure 4b). We then tested the inhibition of the RCL peptides by organ culture of human skin. As shown in Figure 4c, the cytokine-induced conversion of proMMP-9 in human skin was completely inhibited by the peptides at 140 nM. As an internal control, the expression of proMMP-2 and proMMP-9 as well as the conversion of proMMP-2 were not affected by the peptides, which underlines the specificity of α-ACT on proMMP-9 maturation. Thus, the RCL of α-ACT is necessary and sufficient to attenuate conversion of proMMP-9 in human skin.
Figure 4. Synthetic peptides derived from the RCL are sufficient to inhibit proMMP-9 but not proMMP-2 activation.
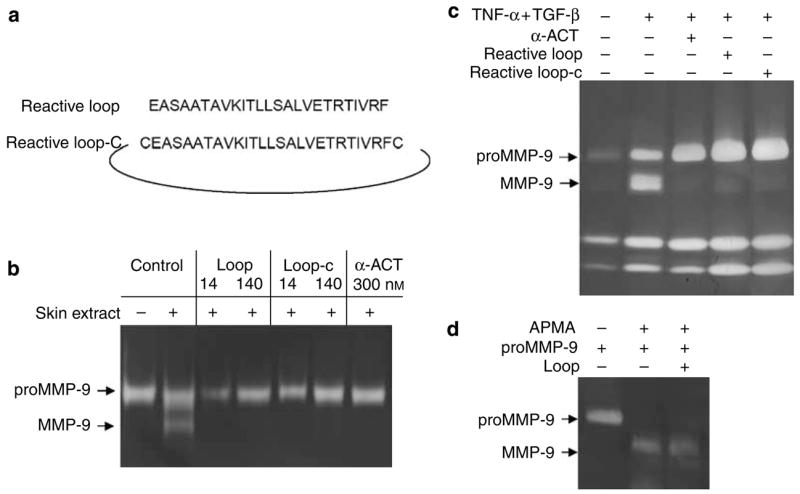
(a) The sequence of the peptides derived from the RCL. (b) For in vitro inhibition, peptides or α-ACT protein were incubated with proMMP-9 and the skin extract, and reactions were resolved by zymography. (c) Ex vivo inhibition was carried out by culture of human skin explants with TGF-β and TNF-α together with the peptides or α-ACT protein. (d) Activation of proMMP-9 was induced by APMA, and the effect of an RCL peptide was measured in the system. APMA, 4-aminophenylmercuric acetate.
In addition to proteolytic cleavage, latent MMPs can also be activated by organomercurial compounds such as 4-aminophenylmercuric acetate through disruption of the bond between the conserved cysteine in the prodomain and the zinc atom in the catalytic domain, leading the consequent autocleavage, a process called ‘‘cysteine switch’’ (Springman et al., 1990). As shown in Figure 4d, 4-aminophenylmercuric acetate (0.7 mg ml−1) efficiently provoked maturation of proMMP-9, converting from 92 to 82 kDa. The peptide of the RCL had no effect on the conversion, but at the same concentration it effectively attenuated the skin proteinase-mediated activation of the zymogen. As expected, the organomercurial compound–induced activation of proMMP-9, which bypasses the initial step of proteolytic cleavage, is refractory to the serine protease inhibitor (data not shown).
Burn blister fluid contains a large amount of α-ACT, which prevents proMMP-9 activation
We initially speculated that the latent form of proMMP-9 observed in the acute phase of injury is due to the presence of inhibitory components in wound fluid. To test this hypothesis, we directly measured the possible inhibitory function of thermal burn blister fluid. To reduce the background of endogenous gelatinases, the thermal burn blisters were cleaned with gelatin-conjugated Sepharose 4B. The resulting blister fluid was added to the assay system containing proMMP-9 and human skin extracts. As shown in Figure 5a, the formation of a 82-kDa band was completely prevented by the blister fluid from all three patients. We thus demonstrated the presence of an inhibitory factor(s) in the acute phase of injured tissues that controls the activation of proMMP-9. We then measured the protein level of α-ACT in the burn blister fluid by western blot analysis. As shown in Figure 5b, a 65-kDa band from the burn blister was recognized by the antibodies and comigrated with the purified human α-ACT. Based on the standard curve of purified α-ACT, we estimated the concentration of the inhibitor in the blister at 0.2 mg ml−1, which is close to the pathophysiologic level of the acute-phase factor in plasma (0.5 mg ml−1) and about 20 times higher than the effective concentration (0.01 mg ml−1; Figure 1). If α-ACT indeed prevents activation of proMMP-9 in the acute phase of injury, the MMP-9 should be in latent form in the blister fluid. Gelatinases in the blister fluid were pulled down by gelatin-conjugated Sepharose 4B and resolved by zymography. As shown in Figure 5c, the majority of MMP-9 from the patients was proMMP-9 (92 kDa) without the mature 82-kDa active form.
Figure 5. Inhibition of proMMP-9 activation by burn blister fluid is associated with a large amount of α-ACT.
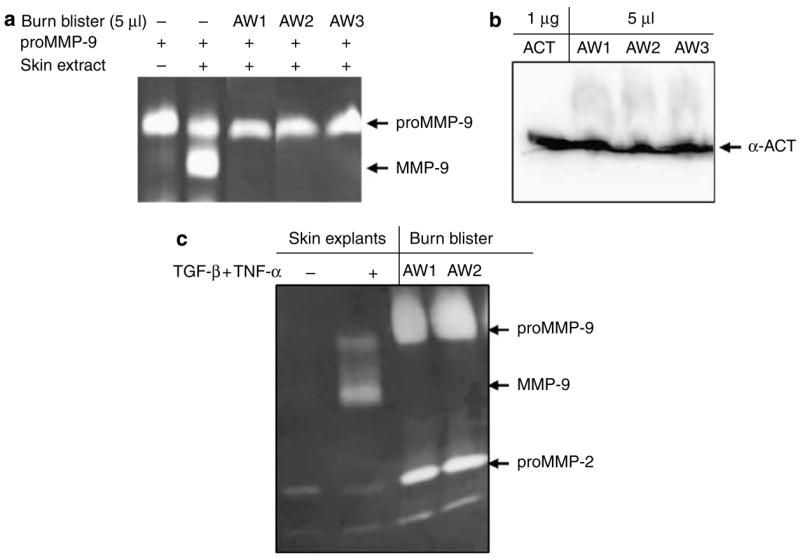
(a) Blister fluid from thermal burn patients (n = 6) was incubated with proMMP-9 and skin extract. After a 16-hour incubation, the reactions were resolved by zymography. (b) The same blister fluid was examined for α-ACT by western blot analysis. (c) Gelatinases from the blister were pulled down by gelatin-conjugated Sepharose 4B and resolved by zymography. The proMMP-9 and MMP-9 were indicated by resolving the conditioned medium from human skin culture stimulated by TGF-β and TNF-α.
The α-ACT in chronic wounds is degraded and non-functional, which is associated with maturation of proMMP-9
If α-ACT plays a pathophysiologic role by inhibiting maturation of proMMP-9 in the acute phase, as shown in Figure 5, the activation of the zymogen observed during the progression of wound healing, on the other hand, should rely on the breakdown or inactivation of α-ACT. To examine this notion, we surveyed α-ACT and MMP-9 from wound tissues for such a relationship. Biopsies from chronic wounds (more than 30 days without closure) of thermal burn patients and healed skin were examined for gelatinases and α-ACT. As shown in Figure 6a, no MMP-9 was found in healed skin tissue, whereas both active and latent MMP-9 was observed in the non-healing tissues. In contrast, proMMP-2 was constitutively expressed by both healed and non-healing skin tissue. Western blot analysis showed the presence of 65-kDa α-ACT as well as the fragmented and conjugated products in the non-healing skin tissue, which may be associated with the inactivation of the inhibitor in human skin (Figure 6b and c). A large-scale survey of inactivation of α-ACT in non-healing wounds has been conducted (Reiss et al., in preparation). In addition, we tested inactivation and fragmentation of α-ACT by incubating the purified inhibitor with human skin explants. Exogenous α-ACT was truncated by incubation with human skin. Thus, the massive activation of proMMP-9 that occurs in wounds is linked to the inactivation of α-ACT.
Figure 6. Activation of proMMP-9 in non-healing wounds is associated with degradation of α-ACT.
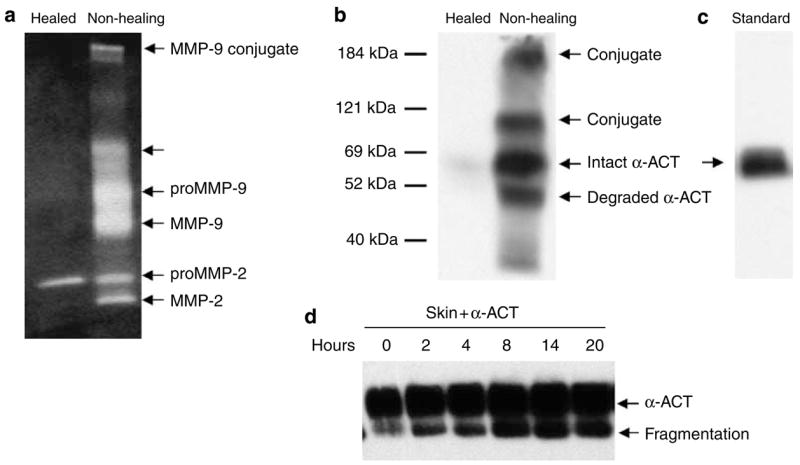
(a) The biopsies of healed and non-healing wounds (n = 6) were resolved by zymography. (b and c) The presence of α-ACT in the wound fluid (n = 6) was examined by western blot analysis. Intact, fragmented, and conjugated α-ACT proteins were indicated. (d) Degradation of α-ACT in human skin was assayed by incubating the recombinant protein with human skin explants for the indicated time and then resolved by western blot analysis.
Cathepsin G–induced activation of proMMP-9 is inhibited by α-ACT
To further strengthen the notion of chymotrypsin-mediated activation of proMMP-9, we directly tested conversion of proMMP-9 by a known chymotrypsin-like proteinase, cathepsin G, derived from neutrophils in secreted azurophil granules. As shown in Figure 7a, incubation of purified proMMP-9 with cathepsin G led the conversion of proMMP-9 into its 82-kDa form. Conversely, incubation with α-ACT resulted in inhibition of the maturation of the zymogen. Indeed, α-ACT at 1 μg ml−1 showed initial inhibition, and at 10 μg ml−1 it totally prevented the cathepsin G–mediated conversion of proMMP-9. Such concentration of inhibition matches well with the inhibitor against the human skin-mediated conversion of proMMP-9, as shown in Figures 1 and 2. Further, we tested the possible conjugation of α-ACT to the converting enzyme. As shown, α-ACT was found to be conjugated to cathepsin G but not to elastase (Figure 7b).
Figure 7. A model showing proMMP-9 activation in human skin is mediated by chymotrypsin-like proteinase and inhibited by TIMP-1 and α-ACT.
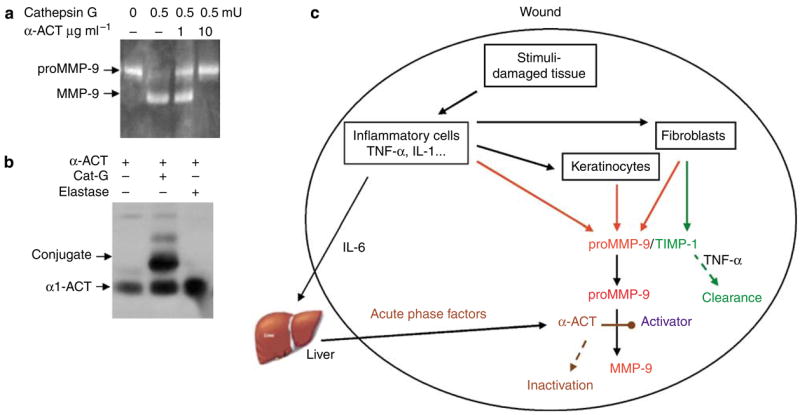
(a) To verify the concept that α-ACT-exerted inhibition of proMMP-9 activation occurs via the targeting of a chymotrypsin-like enzyme in human skin, we tested α-ACT for its inhibition of cathepsin G–mediated maturation of proMMP-9. The α-ACT was incubated with proMMP-9 together with cathepsin G, and the reaction was resolved by zymography. (b) Conjugation of cathepsin G, but not elastase, with α-ACT was measured by western blot analysis for α-ACT. (c) A proposed model for induction, activation, and inhibition of proMMP-9 in human skin during tissue injury and healing. Two inhibitors—TIMP-1, which binds to proMMP-9, and α-ACT, which directly targets the converting enzyme—participate in controlling the maturation of proMMP-9. Downregulation of TIMP-1 and inactivation of α-ACT are prerequisites for conversion of proMMP-9.
DISCUSSION
The serine proteinase inhibitors (serpins) are a superfamily of proteins with diverse physiological functions, including the control of blood coagulation, complement activation, programmed cell death, development, and wound healing (Travis et al., 1988; Baumann and Gauldie, 1994). This family of inhibitors has a molecular weight of about 350–500 amino acids and folds into a conserved structure that uses a unique suicide substrate-like inhibitory mechanism. Serpins are of pathophysiologic importance because the defective inhibitors have been found in blood clotting disorders, emphysema, cirrhosis, and dementia (Carrell et al., 1989; Travis et al., 1990). In this study, we identified a previously unreported function for a particular serpin in wound healing. Specifically, we found a biochemical function of α-ACT that controls the proteolytic activation of proMMP-9. At the molecular level, we demonstrated that the RCL and the P1 leucine within the loop are critical for the inhibition. Eventually, the peptides derived from the RCL of α-ACT are sufficient to hamper the activation of proMMP-9 in skin. Based on the evidence of a large amount of intact α-ACT and lack of activation of the zymogen in the acute phase, we suggest that α-ACT is a pathophysiologic inhibitor for proMMP-9 activation. Conversely, the loss of α-ACT during the progression of wound healing is associated with the proteolytic activation of proMMP-9.
In response to injury, proMMP-9 and α-ACT are expressed almost simultaneously, and the level of acute-phase factors including α-ACT peaks at approximately 8 hours (Batstone et al., 1983; Salo et al., 1994; Moses et al., 1996; Tarlton et al., 1997). Such a temporal relationship could explain the latency of proMMP-9 found in acute wounds or after surgical trauma. Despite such correlation, MMP-9 and α-ACT are seemingly regulated in a different manner. MMP-9 is expressed mostly by inflammatory cells as well as many stromal cells in response to cytokine stimulation. For instance, we noticed that both epidermal keratinocytes and dermal fibro-blasts can express proMMP-9 in response to proinflammatory cytokines (Han et al., 2001a). On the other hand, α-ACT is produced mostly by hepatocytes in response to innate immunity, including IL-6 and IL-1 stimulation (Castell et al., 1989; Fey et al., 1989). In fact, IL-6, originally called hepatocyte-stimulatory factor, was regarded as a major wound signal for liver cells to secrete acute-phase factors (Baumann et al., 1986). Thus, it is possible that wound signals, such as extracellular calcium and ATP within the damaged tissue, activate the keratinocytes and inflammatory cells to release IL-6, which in turn circulates to the liver and turns on the acute-phase response. Consequently, a large amount of α-ACT reaches the wound bed and controls MMP-9 activation.
On the other hand, TIMP-1 conducts its inhibition of MMP-9 in a different manner, by direct binding to proMMP-9, which may physically block the access of the zymogen to converting enzymes (O’Connell et al., 1994). Thus, we propose a model of two inhibitors—TIMP-1, which exerts its action on proMMP-9 (at the substrate level), and α-ACT, which directly targets the converting enzyme—to achieve the control of proMMP-9 activation in tissue injury (Figure 7d). Conversely, downregulation or inactivation of the two inhibitors is a prerequisite for the conversion of proMMP-9 in degenerative diseases, including cancer metastasis and wound healing. Additional evidence to support this two-inhibitor hypothesis has been provided by our previous work, which showed that the TNF-α-exerted activation of proMMP-9 in human skin is mediated by downregulation of TIMP-1 (Han et al., 2002). Our results are consistent with clinical observations that in acute wounds both TIMP-1 and α-ACT are upregulated, in line with the latency of proMMP-9 (Batstone et al., 1983; Ladwig et al., 2002), whereas in chronic wounds TIMP-1 was found to be downregulated (Saarialho-Kere, 1998). In this report, we show the degradation of α-ACT in the non-healed wounds. α-ACT protein in the wound bed is probably inactivated through proteolytic cleavage of α-ACT, a general mechanism of clearance of serpins (Kress, 1983; Potempa et al., 1991; Rao et al., 1995). Proteolytic inactivation of α-ACT and α-AT by neutrophils was observed in arthritic joints (Abbink et al., 1993). This observation is in line with our data showing cathepsin G–mediated activation of proMMP-9 and inhibition by α-ACT (Figure 6). Similarly, inactivation of α-ACT in skin tissue is likely also mediated by mast cell chymase (Schechter et al., 1989). Despite these points, it is not known which proteinase(s) is involved in the inactivation of α-ACT in chronic wounds. Thus, the inactivation of α-ACT and the activation of proMMP-9 are tightly associated in pathologic development in degenerative diseases. Our model may also shed light on the observed sequential production and activation of MMP-9 with breast cancer progression (Rha et al., 1997). The findings reported here may also be relevant to other degenerative diseases in which there is dysregulated MMP-9 activation. For example, it has been found that α-ACT is closely associated with the β-amyloid deposits in the brains of individuals with Alzheimer’s disease, implying a role for this inhibitor in amyloid deposition (Abraham and Potter, 1989). This suggests that α-ACT can promote the formation of the neurotoxic amyloid deposits that are a pathologic hallmark of the disease (Potter et al., 2001). Others have found that MMP-9 in the human hippocampus retains its latent form, whereas active MMP-9 is capable of degrading the β-amyloid peptide (Backstrom et al., 1996; Lim et al., 1997). Accumulation of amyloid could be due to a lack of degradation. Thus, it is plausible that the overdeposition of β-amyloid in Alzheimer’s may be due, in part, to accumulation of α-ACT, which consequently blocks maturation of proMMP-9, leading to retarded matrix clearance.
MATERIALS AND METHODS
Materials and reagents
Cytokines were purchased from R&D Systems (Minneapolis, MN). Antibodies to α-ACT were purchased from Neomarkers (Runcorn, Cheshire, UK). Anti-MMP-9 (C-terminus) was from Chemicon (Billerica, MA). The enhanced chemiluminescence and gelatin Sepharose 4B were purchased from Amersham (Little Chalfont, Buckinghamshire, UK). α-AT, α-ACT, and cathepsin G were purchased from Calbiochem (Gibbstown, NJ). DNA ladders and protein markers were supplied through the courtesy of Transgen (Beijing, China). Peptides derived from the RCL of α-ACT were synthesized by JPT Peptide Technologies GmbH (Volmerstrasse, Berlin, Germany).
Biopsies and animals
Clinical biopsies, including those of normal skin and chronic wounds, were collected according to the protocol approved by the Internal Review Board (IRB) at the University of Southern California, and consent was obtained from patients. The study was performed according to the Declaration of Helsinki Principles. The fluid that accumulated within burn blisters was collected by sterile aspiration within 48 hours after injury and stored at −80 °C for batch analysis. Chronic wounds were defined as those that did not close for more than 30 days while similar wounds in the same patient had healed. The 6-mm punch biopsies were placed in 2-ml DMEM with antibiotics (200 U ml−1 penicillin G sodium, 200 U ml−1 streptomycin sulfate, and 0.5 μg ml−1 amphotericin B). To accumulate the secreted factors, the biopsies were incubated in the medium for 6 hours with the supply of 5% CO2 at 37 °C. The conditioned medium and blister fluid were cleared of debris by centrifugation at 5,000 g before zymography or western blot analysis. Animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Southern California. The pieces of rat skin were cultured in DMEM with antibiotics and cytokines for 3 days.
Organ culture and cytokine stimulation of human skin
Normal human skin was obtained, in an IRB-approved manner, from patients undergoing reconstructive or aesthetic surgery. Fat and connective tissue were removed. The full-thickness skin was decontaminated by incubation in DMEM containing an antibiotic at 4 °C overnight. The skin was cut into 0.25-cm2 pieces and incubated in DMEM at 37 °C with 5% CO2 for 8 hours. To reduce the effects of endogenous soluble factors in the skin induced by the harvesting process, the medium was changed three times during the 8-hour incubation. Finally, the pieces of skin were floated in 2 ml DMEM with or without TGF-β(1 ng ml−1) and TNF-α (10 ng ml−1). For the inhibition experiment, purified α-ACT or α-AT was added to the skin organ culture. The cultures were maintained at 37 °C with 5% CO2. The conditioned media were sampled at the intervals indicated in the text for gelatinolytic zymography and western blot analysis as mentioned below.
Plasmid construction, lentivirus, and preparation of proMMP-9 and α-ACT variants
The cDNA coding human proMMP-9 (clone ID: 4054882) was from Open Biosystems (Huntsville, AL). The full-length cDNA was cloned into a lentiviral vector, and a replication-defective virus was prepared by packing in HEK 293T. IKCs were transduced by the virus coding either MMP-9 or green fluorescent protein as a control. Transduced IKCs were grown in keratinocyte grown medium (Invitrogen, Carlsbad, CA), and conditioned medium was collected. The conditioned medium was centrifuged at 4,000 g for 20 minutes and the supernatant was loaded into a gelatin-conjugated Sepharose 4B column. The column was washed with buffer containing 1% Triton X-100 in 100 mM NaCl and 50 mM Tris, pH 7.5, and MMP-9 was eluted by 6 M urea. The eluted MMP-9 was dialyzed by NT buffer (100 mM NaCl and 50 mM Tris, pH 7.5). MMP-9 activity was measured by zymography, identity was confirmed by western blot analysis, purity was measured by SDS-PAGE and silver staining, and concentration was measured by A280 absorption. Human α-ACT cDNA in pCMV-SPORT6 was from Open Biosystems. A tag of six histidines was added to the C terminus before the stop codon. Two variants in the active loop of α-ACT were generated by Quick-Change mutagenesis (Stratagene, La Jolla, CA). A conserved leucine residue (P1) in the active loop of wild type (-ctc ctt tct gca-) was mutated to serine (-ctc tct tct gca-), designated as P1m (Figure 3). The adjacent serine (P1′) in the active loop was changed to alanine (-ctc tct gct gca-), designated as P1′m. The three α-ACT variants were cloned into the lentiviral vector. IKC was transduced by the lentivirus. The α-ACT in the conditioned medium was purified by Ni-NTA kit from Qiagen (Valencia, CA). Purified protein was examined by western blot analysis, SDS-PAGE, and protein staining.
Acknowledgments
This work was supported by grants from the National Institutes of Health (AR051558 to YPH and GM 050967 to WLG) and Robert May Wright Foundation (YPH). We thank Ling Zhou and Lan Qin for technical support and Wes Grimm for proofreading.
Abbreviations
- α-ACT
α-1-antichymotrypsin
- α-AT
α-1-antitrypsin
- IKC
immortalized human keratinocyte
- MMP
matrix metalloproteinase
- RCL
reactive center loop
- TGF-β
transforming growth factor-β
- TNF-α
tumor necrosis factor-α
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Abbink JJ, Kamp AM, Nuijens JH, Swaak TJ, Hack CE. Proteolytic inactivation of alpha 1-antitrypsin and alpha 1-antichymotrypsin by neutrophils in arthritic joints. Arthritis Rheum. 1993;36:168–80. doi: 10.1002/art.1780360206. [DOI] [PubMed] [Google Scholar]
- Abraham CR, Potter H. Alpha 1-antichymotrypsin in brain aging and disease. Prog Clin Biol Res. 1989;317:1037–48. [PubMed] [Google Scholar]
- Backstrom JR, Lim GP, Cullen MJ, Tokes ZA. Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-beta peptide (1–40) J Neurosci. 1996;16:7910–9. doi: 10.1523/JNEUROSCI.16-24-07910.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batstone GF, Levick PL, Spurr E, Shakespeare PG, George SL, Ward CM. Changes in acute phase reactants and disturbances in metabolism after burn injury. Burns Incl Therm Inj. 1983;9:234–9. doi: 10.1016/0305-4179(83)90052-9. [DOI] [PubMed] [Google Scholar]
- Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15:74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- Baumann H, Hill RE, Sauder DN, Jahreis GP. Regulation of major acute-phase plasma proteins by hepatocyte-stimulating factors of human squamous carcinoma cells. J Cell Biol. 1986;102:370–83. doi: 10.1083/jcb.102.2.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkedal-Hansen H. Proteolytic remodeling of extracellular matrix. Curr Opin Cell Biol. 1995;7:728–35. doi: 10.1016/0955-0674(95)80116-2. [DOI] [PubMed] [Google Scholar]
- Carrell RW, Aulak KS, Owen MC. The molecular pathology of the serpins. Mol Biol Med. 1989;6:35–42. [PubMed] [Google Scholar]
- Castell JV, Gomez-Lechon MJ, David M, Andus T, Geiger T, Trullenque R, et al. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett. 1989;242:237–9. doi: 10.1016/0014-5793(89)80476-4. [DOI] [PubMed] [Google Scholar]
- Fey GH, Hattori M, Northemann W, Abraham LJ, Baumann M, Braciak TA, et al. Regulation of rat liver acute phase genes by interleukin-6 and production of hepatocyte stimulating factors by rat hepatoma cells. Ann N Y Acad Sci. 1989;557:317–29. doi: 10.1111/j.1749-6632.1989.tb24024.x. discussion 329–31. [DOI] [PubMed] [Google Scholar]
- Han YP. Matrix metalloproteinases, the pros and cons, in liver fibrosis. J Gastroenterol Hepatol. 2006;21(Suppl 3):S88–91. doi: 10.1111/j.1440-1746.2006.04586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YP, Downey S, Garner WL. Interleukin-1alpha-induced proteolytic activation of metalloproteinase-9 by human skin. Surgery. 2005;138:932–9. doi: 10.1016/j.surg.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YP, Nien YD, Garner WL. Tumor necrosis factor-alpha-induced proteolytic activation of pro-matrix metalloproteinase-9 by human skin is controlled by down-regulating tissue inhibitor of metalloproteinase-1 and mediated by tissue-associated chymotrypsin-like proteinase. J Biol Chem. 2002;277:27319–27. doi: 10.1074/jbc.M202842200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YP, Tuan TL, Hughes M, Wu H, Garner WL. Transforming growth factor-beta- and tumor necrosis factor-alpha-mediated induction and proteolytic activation of MMP-9 in human skin. J Biol Chem. 2001a;276:22341–50. doi: 10.1074/jbc.M010839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YP, Tuan TL, Wu H, Hughes M, Garner WL. TNF-alpha stimulates activation of pro-MMP2 in human skin through NF-(kappa)B mediated induction of MT1-MMP. J Cell Sci. 2001b;114:131–9. doi: 10.1242/jcs.114.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YP, Yan C, Zhou L, Qin L, Tsukamoto H. A matrix metallo-proteinase-9 activation cascade by hepatic stellate cells in trans-differentiation in the three-dimensional extracellular matrix. J Biol Chem. 2007;282:12928–39. doi: 10.1074/jbc.M700554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress LF. Inactivation of human plasma alpha 1-antichymotrypsin by microbial proteinases. Acta Biochim Pol. 1983;30:159–64. [PubMed] [Google Scholar]
- Ladwig GP, Robson MC, Liu R, Kuhn MA, Muir DF, Schultz GS. Ratios of activated matrix metalloproteinase-9 to tissue inhibitor of matrix metalloproteinase-1 in wound fluids are inversely correlated with healing of pressure ulcers. Wound Repair Regen. 2002;10:26–37. doi: 10.1046/j.1524-475x.2002.10903.x. [DOI] [PubMed] [Google Scholar]
- Lim GP, Russell MJ, Cullen MJ, Tokes ZA. Matrix metalloproteinases in dog brains exhibiting Alzheimer-like characteristics. J Neurochem. 1997;68:1606–11. doi: 10.1046/j.1471-4159.1997.68041606.x. [DOI] [PubMed] [Google Scholar]
- Moses MA, Marikovsky M, Harper JW, Vogt P, Eriksson E, Klagsbrun M, et al. Temporal study of the activity of matrix metalloproteinases and their endogenous inhibitors during wound healing. J Cell Biochem. 1996;60:379–86. doi: 10.1002/(SICI)1097-4644(19960301)60:3%3C379::AID-JCB9%3E3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- O’Connell JP, Willenbrock F, Docherty AJ, Eaton D, Murphy G. Analysis of the role of the COOH-terminal domain in the activation, proteolytic activity, and tissue inhibitor of metalloproteinase interactions of gelatinase B. J Biol Chem. 1994;269:14967–73. [PubMed] [Google Scholar]
- Oikarinen A, Kylmaniemi M, Autio-Harmainen H, Autio P, Salo T. Demonstration of 72-kDa and 92-kDa forms of type IV collagenase in human skin: variable expression in various blistering diseases, induction during re-epithelialization, and decrease by topical glucocorticoids. J Invest Dermatol. 1993;101:205–10. doi: 10.1111/1523-1747.ep12363823. [DOI] [PubMed] [Google Scholar]
- Parks WC. Matrix metalloproteinases in repair. Wound Repair Regen. 1999;7:423–32. doi: 10.1046/j.1524-475x.1999.00423.x. [DOI] [PubMed] [Google Scholar]
- Potempa J, Fedak D, Dubin A, Mast A, Travis J. Proteolytic inactivation of alpha-1-anti-chymotrypsin. Sites of cleavage and generation of chemotactic activity. J Biol Chem. 1991;266:21482–7. [PubMed] [Google Scholar]
- Potter H, Wefes IM, Nilsson LN. The inflammation-induced pathological chaperones ACT and apo-E are necessary catalysts of Alzheimer amyloid formation. Neurobiol Aging. 2001;22:923–30. doi: 10.1016/s0197-4580(01)00308-6. [DOI] [PubMed] [Google Scholar]
- Rao CN, Ladin DA, Liu YY, Chilukuri K, Hou ZZ, Woodley DT. Alpha 1 -antitrypsin is degraded and non-functional in chronic wounds but intact and functional in acute wounds: the inhibitor protects fibronectin from degradation by chronic wound fluid enzymes. J Invest Dermatol. 1995;105:572–8. doi: 10.1111/1523-1747.ep12323503. [DOI] [PubMed] [Google Scholar]
- Rha SY, Kim JH, Roh JK, Lee KS, Min JS, Kim BS, et al. Sequential production and activation of matrix-metalloproteinase-9 (MMP-9) with breast cancer progression. Breast Cancer Res Treat. 1997;43:175–81. doi: 10.1023/a:1005701231871. [DOI] [PubMed] [Google Scholar]
- Saarialho-Kere UK. Patterns of matrix metalloproteinase and TIMP expression in chronic ulcers. Arch Dermatol Res. 1998;290(Suppl):S47–54. doi: 10.1007/pl00007453. [DOI] [PubMed] [Google Scholar]
- Salo T, Makela M, Kylmaniemi M, Autio-Harmainen H, Larjava H. Expression of matrix metalloproteinase-2 and -9 during early human wound healing. Lab Invest. 1994;70:176–82. [PubMed] [Google Scholar]
- Schechter NM, Sprows JL, Schoenberger OL, Lazarus GS, Cooperman BS, Rubin H. Reaction of human skin chymotrypsin-like proteinase chymase with plasma proteinase inhibitors. J Biol Chem. 1989;264:21308–15. [PubMed] [Google Scholar]
- Seltzer JL, Weingarten H, Akers KT, Eschbach ML, Grant GA, Eisen AZ. Cleavage specificity of type IV collagenase (gelatinase) from human skin. Use of synthetic peptides as model substrates. J Biol Chem. 1989;264:19583–6. [PubMed] [Google Scholar]
- Springman EB, Angleton EL, Birkedal-Hansen H, Van Wart HE. Multiple modes of activation of latent human fibroblast collagenase: evidence for the role of a Cys73 active-site zinc complex in latency and a ‘‘cysteine switch’’ mechanism for activation. Proc Natl Acad Sci USA. 1990;87:364–8. doi: 10.1073/pnas.87.1.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahle-Backdahl M, Inoue M, Guidice GJ, Parks WC. 92-kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest. 1994;93:2022–30. doi: 10.1172/JCI117196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlton JF, Vickery CJ, Leaper DJ, Bailey AJ. Postsurgical wound progression monitored by temporal changes in the expression of matrix metalloproteinase-9. Br J Dermatol. 1997;137:506–16. doi: 10.1111/j.1365-2133.1997.tb03779.x. [DOI] [PubMed] [Google Scholar]
- Travis J, Guzdek A, Potempa J, Watorek W. Serpins: structure and mechanism of action. Biol Chem Hoppe Seyler. 1990;371(Suppl):3–11. [PubMed] [Google Scholar]
- Travis J, Shieh BH, Potempa J. The functional role of acute phase plasma proteinase inhibitors. Tokai J Exp Clin Med. 1988;13:313–20. [PubMed] [Google Scholar]
- Trengove NJ, Stacey MC, MacAuley S, Bennett N, Gibson J, Burslem F, et al. Analysis of the acute and chronic wound environments: the role of proteases and their inhibitors. Wound Repair Regen. 1999;7:442–52. doi: 10.1046/j.1524-475x.1999.00442.x. [DOI] [PubMed] [Google Scholar]
- Warner RL, Lewis CS, Beltran L, Younkin EM, Varani J, Johnson KJ. The role of metalloelastase in immune complex-induced acute lung injury. Am J Pathol. 2001;158:2139–44. doi: 10.1016/S0002-9440(10)64685-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckroth M, Vaheri A, Lauharanta J, Sorsa T, Konttinen YT. Matrix metalloproteinases, gelatinase and collagenase, in chronic leg ulcers. J Invest Dermatol. 1996;106:1119–24. doi: 10.1111/1523-1747.ep12340167. [DOI] [PubMed] [Google Scholar]
- Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439–42. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- Wysocki AB, Staiano-Coico L, Grinnell F. Wound fluid from chronic leg ulcers contains elevated levels of metalloproteinases MMP-2 and MMP-9. J Invest Dermatol. 1993;101:64–8. doi: 10.1111/1523-1747.ep12359590. [DOI] [PubMed] [Google Scholar]
- Yager DR, Zhang LY, Liang HX, Diegelmann RF, Cohen IK. Wound fluids from human pressure ulcers contain elevated matrix metalloproteinase levels and activity compared to surgical wound fluids. J Invest Dermatol. 1996;107:743–8. doi: 10.1111/1523-1747.ep12365637. [DOI] [PubMed] [Google Scholar]
- Young PK, Grinnell F. Metalloproteinase activation cascade after burn injury: a longitudinal analysis of the human wound environment. J Invest Dermatol. 1994;103:660–4. doi: 10.1111/1523-1747.ep12398424. [DOI] [PubMed] [Google Scholar]