

Abstract
Xeroderma pigmentosum-variant (XP-V) patients have sun sensitivity and increased skin cancer risk. Their cells have normal nucleotide excision repair, but have defects in the POLH gene encoding an error-prone polymerase, DNA polymeraseη (polη). To survey the molecular basis of XP-V worldwide, we measured polη protein in skin fibroblasts from putative XP-V patients (aged 8–66 years) from 10 families in North America, Turkey, Israel, Germany, and Korea. Polη was undetectable in cells from patients in eight families, whereas two showed faint bands. DNA sequencing identified 10 different POLH mutations. There were two splicing, one nonsense, five frameshift (3 deletion and 2 insertion), and two missense mutations. Nine of these mutations involved the catalytic domain. Although affected siblings had similar clinical features, the relation between the clinical features and the mutations was not clear. POLH mRNA levels were normal or reduced by 50% in three cell strains with undetectable levels of polη protein, indicating that nonsense-mediated message decay was limited. We found a wide spectrum of mutations in the POLH gene among XP-V patients in different countries, suggesting that many of these mutations arose independently.
INTRODUCTION
To maintain DNA integrity, cells have multiple pathways to repair DNA damage caused by environmental agents. The failure of these systems leads to human disease. Xeroderma pigmentosum (XP) is an autosomal recessive disorder with an extremely high sensitivity to UV radiation, leading to a 1,000-fold increase in the incidence of sunlight-induced skin cancer, caused by defective DNA repair (Kraemer et al., 1987, 1994, 2007; Bootsma et al., 2002; Kraemer, 2003; Friedberg et al., 2006).
Genetic studies on cultured cells from XP patients revealed seven complementation groups, XP-A through XP-G, as well as a variant (XP-V) (OMIM no. 278750). The cells from XP-A through XP-G are defective in nucleotide-excision repair, which is responsible for the removal of UV-induced damage in genomic DNA (Van Steeg and Kraemer, 1999; Friedberg et al., 2006). Although different complementation groups exhibit diverse clinical features, these XP patients generally have early onset of skin cancers and about 20% (XP-A, XP-B, XP-G, and XP-D) exhibit neurological abnormalities (Rapin et al., 2000; Kraemer, 2003; Kraemer et al., 2007). In contrast, XP-V, which has been estimated to comprise approximately 20% of XP patients (Moriwaki and Kraemer, 2001), may have mild or severe clinical features, including late onset of skin cancers, but rarely have neurological abnormalities.
XP-V cells possess proficient nucleotide-excision repair (Cleaver, 1972; Robbins et al., 1974, 1975; Broughton et al., 2002; Tanioka et al., 2007), but display exaggerated delay in recovery of replicative DNA synthesis in association with defective post-replication repair (Lehmann et al., 1975; Itoh et al., 1996). XP-V cells are hypersensitive to killing by UV irradiation in the presence of caffeine (Arlett et al., 1975; Maher et al., 1976a; Broughton et al., 2002), are hypermutable after UV (Maher et al., 1976b; Wang et al., 1993), and have post-UV plasmid hypermutability in a host cell reactivation assay (Waters et al., 1993).
Extracts from XP-V cells are deficient in the ability to bypass a thymine–thymine cyclobutane pyrimidine dimer in a DNA replication assay (Svoboda et al., 1998). XP-V cells are unable to synthesize intact daughter DNA strands on UV-irradiated templates resulting from an inability to carry out translesion synthesis, the synthesis of DNA directly past damaged sites (Cordonnier et al., 1999). In 1999, the XPV gene was cloned and found to be POLH (OMIM *603968) (Johnson et al., 1999a; Masutani et al., 1999b), a human homolog of the yeast RAD30 gene (McDonald et al., 1997). This gene encodes DNA polymeraseη (polη) and is a member of the Y-family of DNA polymerases (Ohmori et al., 2001; Yang and Woodgate, 2007) related in structure to each other, but unrelated to classical DNA polymerases. The POLH gene is located on chromosome 6p21.1–6p12.3 spanning 40 kb and the calculated relative molecular mass (Mr) of POLH is 78.4 kDa. The messenger RNA size is of 3,464 bp (GenBank reference number NM_006502), but the size of the open reading frame is 2,139 bp as the first methionine is located in exon 2 (Johnson et al., 1999a; Masutani et al., 1999b; Yuasa et al., 2000). The function of polη in the error-free bypass of UV-induced DNA lesions has been well characterized (Johnson et al., 1999b; Masutani et al., 1999a, 2000). However, polη can also function as a highly mutagenic DNA polymerase when it replicates undamaged DNA, and it has an important role in generating immune diversity (Zeng et al., 2001; Yavuz et al., 2002).
Previous reports provided information about the molecular defects in XP-V patients from the United States, Europe, and Japan (Johnson et al., 1999a; Masutani et al., 1999b; Itoh et al., 2000a; Itoh and Linn, 2001; Broughton et al., 2002; Gratchev et al., 2003; Tanioka et al., 2007). The causative mutations reported in the POLH gene generally resulted in severe truncations of the protein and are effectively null alleles. We surveyed POLH mutations in XP-V patients from different parts of the world in order to assess the origin of this disorder and to attempt to correlate clinical features with molecular defects. We studied cells from 15 XP-V patients in 10 families from the United States, the Cayman Islands, Turkey, Israel, Germany, and Korea. We measured the polη protein levels and determined the causative mutations in the POLH gene.
RESULTS
Characteristics of XP-V patients and cells
We evaluated 15 putative XP-V patients in 10 families from the United States, the Cayman Islands, Turkey, Germany, Israel, and Korea (Table 1; Figure 1). XP31BE, a 60-year-old man in family A from the United States (Figure 1a), had multiple basal cell carcinomas (BCCs), squamous cell carcinomas (SCCs), and melanomas. As a teenager, the cancer-containing skin of his face was removed and replaced with sun-protected skin from his trunk. This grafted skin has remained cancer-free for more than 40 years to the present time. Unscheduled DNA synthesis (UDS) of his cells was normal. His fibroblasts had normal post-UV plasmid host cell reactivation, at least 10 times higher than XP-C cells (XP108DC) in this assay (Figure 2). These are typical features of XP-V cells (Arlett et al., 1975; Maher et al., 1976a; Waters et al., 1993; Itoh et al., 1996, 2000a; Broughton et al., 2002; Friedberg et al., 2006). XP1SE, a 66-year-old Korean woman in family B, had more than 10 BCCs and SCCs during the past 20 years (Kim and Chung, 2003). Her cells had normal host cell reactivation (data not shown).
Table 1.
Clinical features and cellular assays in XP-V patients
| Family | Cell line | Age/sex | Race or nationality | Cancer (type) | Cellular assay |
|---|---|---|---|---|---|
| A | XP31BE (GM13155) | 60 years M | Caucasian, USA | Yes (BCC, SCC, MM) | U, H |
| B | XP1SE | 66 years F | Korean | Yes (BCC, SCC) | H |
| C | XP71TMA*** (GM14875) | 8 years M | Turkish | Yes (SCC, BCC) | NT |
| C | XP70TMA*** (GM15697) | 17 years F | Turkish | Yes (SCC, BCC) | U, H |
| C | XP98TMA*** (GM15711) | 26 years F | Turkish | Yes (SCC) | NT |
| D | XP91TMA**** (GM15700) | 10 years M | Turkish | No | NT |
| D | XP92TMA**** (GM15713) | d 22years M | Turkish | Yes (SCC) | H |
| E | XP38BE* (AG02592) | 8 years M | Cayman Island | Yes (SCC conjunctiva) | U, H |
| E | XP39BE* (KR05822) | 18 years F | Cayman Island | Yes (BCC) | NT |
| F | XP161BE** | 46 years F | Caucasian, USA | Yes (BCC, MM) | C |
| F | XP164BE** | 52 years F | Caucasian, USA | Yes (BCC, SCC, MM) | C, H |
| G | XP3GO | 38 years M | German | Yes (BCC, SCC, MM) | C, H |
| H | XP139DC | 19 years M | USA | Yes (BCC) | U, C, H |
| I | XP224BE (GM16818) | 31 years F | Caucasian, USA | Yes (BCC, MM) | H |
| J | XP10TA | 53 years F | Iraqi–Israeli | Yes (MM) | H |
BCC, cutaneous basal cell carcinoma; C, post-UV hypersensitivity after caffeine treatment; d, age at death; H, normal host cell reactivation; MM, cutaneous melanoma; NT, not tested; SCC, cutaneous squamous cell carcinoma; U, normal post-UV unscheduled DNA synthesis; XP-V, xeroderma pigmentosum variant.
Siblings.
Siblings.
Siblings.
Siblings.
Figure 1. Clinical appearance of XP-V patients.

Family A: (a) XP31BE, 60 year Caucasian man had full-thickness skin removed from his face as a teenager because of multiple skin cancers. This skin was replaced with sun-protected skin from his abdomen and has been free of cancers ever since. He had BCC, SCC, and melanomas on non-grafted skin of his head and upper extremities. Family C: (b) C-1, XP71TMA, 8-year-old Turkish boy, had SCC. (c) C-2, XP70TMA, 17-year-old sister of XP71TMA, had SCC and BCC. (d) C-3, XP98TMA, 26-year-old sister of XP71TMA, had a large SCC on her left cheek. Family D: (e) D-1, XP91TMA, a 10-year-old Turkish man, had no skin cancers. (f) D-2, XP92TMA, brother of XP91TMA, died at age 22 years of metastasis of the SCC on his lip. Family F: (g) F-1, XP161BE, a 46-year-old Caucasian woman with multiple skin BCCs and melanomas. (h) F-2, XP164BE, 52-year-old sister of XP161BE, had multiple BCCs, SCCs, and melanomas. Family G: (i) XP3GO, a 30-year-old German man with multiple BCCs, SCCs, and melanomas. Family I: (j) XP224BE, a 31-year-old Caucasian woman, had multiple BCCs and melanomas.
Figure 2. Post-UV host cell reactivation assay showing normal DNA repair in XP-V cells.
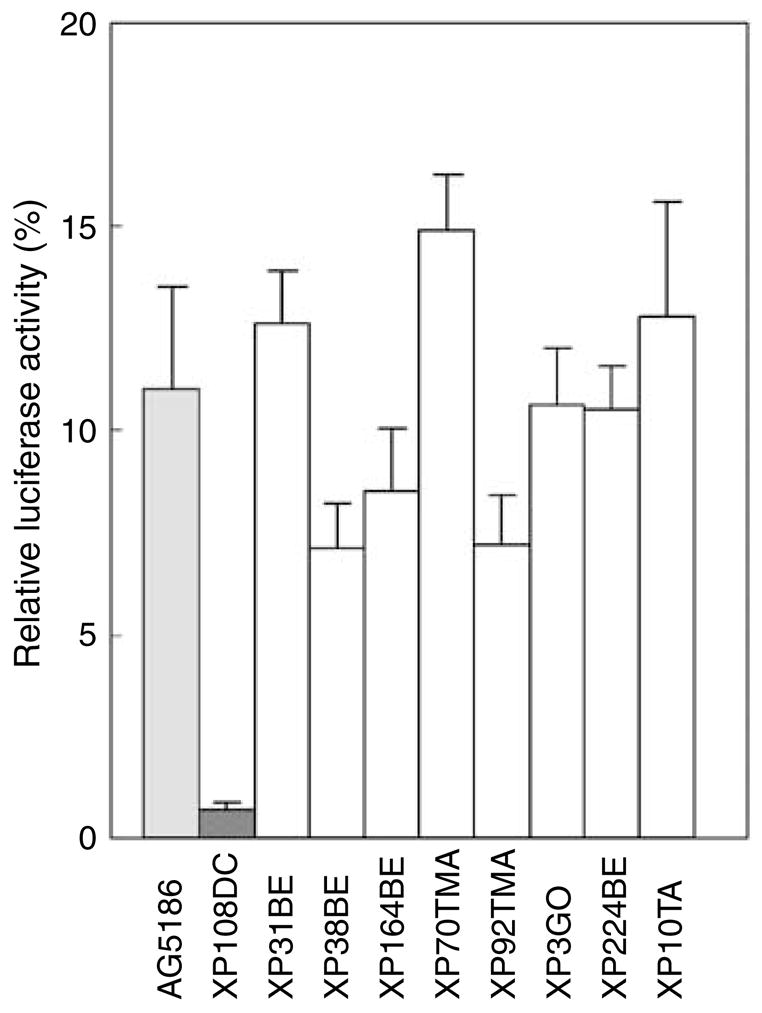
UV-treated or control unirradiated luciferase containing plasmid was transfected into cells from normal (AG5186) (light gray bar), XP-C (XP108DC) (dark gray bar), and putative XP-V donors (XP31BE, XP38BE, XP164BE, XP70TMA, XP92TMA, XP3GO, XP224BE, and XP10TA) (open bars). Two days later luciferase activity was measured. The relative luciferase activity of the UV treated to the control plasmid reflects repair activity of the cells. The XP-C cell had low activity and the putative XP-V cells had repair activity in the normal range. The mean ± SEM of triplicate experiments is shown.
Family C from Turkey had three affected siblings, XP71TMA, an 8-year-old boy with SCC and BCC (Figure 1b); XP70TMA, a 17-year-old girl with SCC and BCC (Figure 1c); and XP98TMA, a 26-year-old sister with SCC of her cheek (Figure 1d). Their parents are distant cousins. The cultured fibroblasts from patient XP70TMA showed normal post-UV UDS and post-UV plasmid host cell reactivation (Figure 2). XP91TMA, a 10-year-old boy in family D from Turkey was cancer free (Figure 1e). His elder brother, XP92TMA, died at 22 years of age from an SCC originating on his lower lip (Figure 1f). His cells had normal host cell reactivation (Figure 2). Their parents are known to be related to each other. However, families C and D lived in eastern Turkey, but were not known to be related.
XP38BE in family E from the Cayman Islands had a conjunctival SCC at the age of 8 years. UDS was 114% and host cell reactivation was normal (Figure 2). XP39BE, his affected sister, had seven BCCs by age 18. They are of mixed English/African heritage. In family F, two adult sisters, XP161BE (Figure 1g) and XP164BE (Figure 1h), from the United States, had similar clinical features. Both had BCC and melanoma, whereas XP164BE also had SCC. Their cultured cells showed normal-to-mild UV sensitivity, with post-UV hypersensitivity after caffeine treatment (Table 1). Cells from XP164BE had normal post-UV host cell reactivation (Figure 2).
XP3GO, a 38-year-old man in family G from Germany, had 12 primary melanoma, 3 SCCs, and 7 BCCs in the past 16 years (Figure 1i). His cultured skin fibroblasts had post-UV hypersensitivity after caffeine treatment (Table 1) and normal host cell reactivation (Figure 2). XP139DC, a 19-year-old man from the United States in family H, had a BCC. His cells showed normal UDS and normal host cell reactivation (data not shown). The cells had post-UV hypersensitivity to killing after caffeine treatment (Table 1).
XP224BE, a 31-year-old Caucasian woman in family I from the United States, had BCC and SCC (Figure 1j). Her cultured cells had normal host cell reactivation (Figure 2). XP10TA, a 53-year-old Iraqi–Israeli woman in family J, had two melanomas. Her parents were first cousins. Her cells had normal host cell reactivation (Figure 2).
The patients we examined in families A, B, C, D, F, G, I, and J had no XP-type neurological abnormalities (Rapin et al., 2000) and none were reported for the other patients by the referring doctors. These patients did not have loss of vision due to corneal clouding. They showed a range of cutaneous involvement and all showed freckling at an early age. The patients in families C and D had marked progression of pigmentary changes along with atrophy and telangiectasia. However, in the others these changes remained limited despite development of skin cancers. Their skin is similar in appearance to chronic sun damage in fair-skinned individuals in the general population.
Semi-quantification of polη protein expression
We used an immunoprecipitation (IP) procedure for detecting polη protein using monoclonal and polyclonal antibodies (Thakur et al., 2001; Laposa et al., 2003; Tanioka et al., 2007) in extracts from primary cells. As shown in Figure 3a, normal fibroblast whole-cell extracts (lanes 1, 2, 6, 7, 12, and 16) have a dark band of polη protein, with an estimated size, which is coincident with the expected size of 78.4 kDa. In contrast, cell lines XP224BE (lane 5) and XP10TA (lane 11) show faint bands, whereas the other XP-V cells (Figure 3a, lanes 3, 4, 8, 9, 10, 13, 15, 17, and 18) show undetectable levels of polη protein. Similarly, protein extracted from normal lymphoblastoid cells (AG10107 and AG10033) showed an intense band at the expected size and lymphoblastoid cells from XP31BE had an undetectable level of polη protein (data not shown). This assay demonstrates the large difference in levels of polη protein between normal and XP-V cells (Table 2).
Figure 3. Semi-quantitative estimation of polη and polι protein in XP and normal fibroblasts.

Normal and putative XP-V cells were assayed by IP-Western blotting of whole-cell extracts. (a) Polη protein and (b) polι protein.
Table 2.
Polη protein and mutations in the XP-V cell lines examined
| Polη mutations1 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele 1 | Allele 2 | |||||||||||
| Family | Cell line | IP-western | Genomic | cDNA | Type | Amino acid |
Size (aa)2 |
Genomic | cDNA | Type | Amino acid |
Size (aa)1 |
| A | XP31BE | Absent | g.24885 G>C
c.764+1G>C |
1. r.661_764 del104 (del exon 6)* | Splicing | p.Val221Profs*2 | 221 | Hemizygous | ||||
| 2. r.723_764 del 42 (partial del exon 6) | Splicing | p.Ser242_Ile255 del 14 | 699 | |||||||||
| B | XP1SE | Absent | g.11282 G4T (exon 4)***
c.490 G4T (exon 4)*** |
1.r.401_490 del90 (partial del exon 4) | Splicing | p.Gly134_Lys163 del30 | 683 | Homozygous | ||||
| 2.r.490+163(partial ins intron 4) | Splicing | p.Glu164Glyfs*29 | 191 | |||||||||
| C | XP70TMA | Absent | g.11246 C>T | c.454 C>T (exon 4) | Nonsense | p.Gln152* | 151 | Homozygous | ||||
| D | XP92TMA | Absent | g.11246 C>T | c.454 C>T (exon 4) | Nonsense | p.Gln152* | 151 | Homozygous | ||||
| E | XP38BE | Absent | g.34350_34351insG | c.1078dupG (exon 10)* | Frameshift | p.Asp360Gly fs*32 | 390 | Homozygous/hemizygous | ||||
| F | XP164BE | Absent | g.34350_34351insG | c.1078dupG (exon 10)* | Frameshift | p.Asp360Gly fs*32 | 390 | Homozygous | ||||
| G | XP3GO | Absent | g.24139_25203 del1065 | c.661_764 del104 (del exon 6)* | Frameshift | p.Val221Profs*2 | 221 | Homozygous | ||||
| H | XP139DC | Absent | c.1075_1244 del170 (del exon 10)** | Frameshift | p.Asn359Valfs*32 | 389 | g.37914_37915 del2 | c.1706_1707 del2(exon 11) | Frameshift | p.Thr569Argfs*10 | 577 | |
| I | XP224BE | Faint | g.6811_6812insT | c.149dupT (exon 3) | Frameshift | p.Ser51Glufs*3 | 52 | g.21656 A>G | c.658 A>G (exon 5) | Missense | p.Lys220Glu | 713 |
| J | XP10TA | Faint | g.21520 G>T | c.522 G>T (exon 5) | Missense | p.Trp174Cys | 713 | Homozygous | ||||
Polη, DNA polymerase-η; XP-V, xeroderma pigmentosum variant.
GenBank reference sequence NC_000006.1 for genomic sequence and NM_006502.1 for cDNA.
Predicted size.
Mutation reported in Johnson (1999) Science: 285; 263 at cDNA level.
Mutation reported in Broughton et al. (2002) at the cDNA level.
Mutation reported in Tanioka et al. (2007).
Mutation analysis
Sequencing of entire POLH coding region and exon/intron junctions were carried out for exon 2 through exon 11. The causative mutations were identified in all 10 cell lines. The mutations and the predicted proteins encoded by the mutant alleles are summarized in Table 2. Virtually all of the mutations were located in regions that are likely to affect the catalytic core of the enzyme. The exception was patient XP139DC, who had a mutation in one allele of exon 11 of POLH, encoding the C-terminus of polη (see below). Of the previously reported mutations in POLH that give rise to XP-V, only 6 out of 60 (including those described here) have been found in exon 11 (Johnson et al., 1999a; Masutani et al., 1999b; Itoh et al., 2000b; Itoh and Linn, 2001; Broughton et al., 2002; Gratchev et al., 2003; Tanioka et al., 2007), suggesting that mutations in this region may give rise to mild phenotypes that may be difficult to diagnose clinically as XP-V.
POLH splice donor site mutations in XP31BE and XP1SE
Sequencing XP31BE genomic DNA from family A revealed a mutation in the exon 6 splice donor site, G-to-C transversion (c.764+1G>C) (Figure 4a). The sequence at this exon–intron junction represents a strong donor site in the normal sequence and the single base substitution in XP31BE reduced its information content from 9.3 bits to −0.5 bits (Schneider, 1997a, b; Table S2). To confirm whether this splice donor site mutation results in alteration of POLH mRNA splicing, we performed reverse transcriptase-PCR (RT-PCR) surrounding exon 6, using two primers, a forward primer (c504F) in exon 5 and a reverse primer (c892R) in exon 8/7 junction using RNA from the XP31BE and a control (Figure 4b). RT-PCR showed that normal cells had a single band of the expected size. In contrast, XP31BE had two shorter bands. Sequencing of these two bands revealed two splice variants of POLH mRNA. Type I splice form, with the greater expression, has a deletion of the entire exon 6 (104 bp) (r.661_764del104) (Table 2). This out-of-frame deletion is expected to produce a 221-amino-acid truncated protein (p.Val221Profs*2). Type II splice form uses a cryptic splice donor site, 42 bp upstream from the end of exon 6, which has an information content of 3.8 bits (Table S2). The resulting in-frame deletion (r.723_764del42; p.ser242_Ile255del14) leads to shortening of the polη protein by 14 amino acids (Table 2). We examined inheritance of the mutation in family A by using PCR-restriction-fragment length polymorphism (RFLP) (Figure 4c). The G-to-C transversion creates a new DdeI restriction site, which cuts the mutant genomic DNA into two fragments. DdeI-treated DNA from the mother of XP31BE shows one normal band as well as the shorter bands, indicating that she is heterozygous for this mutation. However, DdeI-treated DNA from the XP31BE patient shows only the shorter bands. DdeI-treated DNA from the father does not show cutting by this enzyme, indicating that the G-to-C mutation is absent. But the genomic DNA sequence from the patient shows only the mutated base (C) without the correct base (G), indicating that he did not receive the correct base from either parent at this site (Figure 4a). Thus, we assume that this region of the DNA from his father is deleted. Hence, the XP31BE patient is hemizygous for this splicing mutation (Table 2).
Figure 4. Sequence analysis of family A.

(a) Genomic DNA of XP31BE shows change of G to C at exon 6 splice donor site (arrows). There is a single curve in the uncloned DNA, indicating homozygous or hemizygous mutation. (b) RT-PCR using RNA from XP31BE with forward primer C504F (in exon 5) and reverse primer C892R (spanning the exon 8/7 junction) showed a single 388-bp band in normal cells and two bands (arrows) at 284 and 346 bp in XP31BE cells. The diagram indicates deletion of the entire 104 bp of exon 6 in the type-I splice variant and partial deletion (42 bp) of exon 6 in the type-II splice variant. (c) PCR-RFLP assay for family A. The G-to-C mutation creates a new DdeI restriction site. The gel shows genomic DNA from the father, XP31BE, mother, and a normal donor. + Indicates DdeI treatment. The arrows indicate 63 and 54-bp bands of digested DNA. * Indicates the normal-size band.
XP1SE in family B had a homozygous splice donor mutation in exon 4 (c.490 G>T) (Table 2). This mutation reduced the information content of the splice donor site from 5.4 to 2.1 bits (Table S2). We found two alternatively spliced isoforms (r.401_490 del90 and r.490+163) resulting in proteins of 683 (p.Gly134_Lys163del30) and 191 amino acids (p.Glu164Glyfs*29), respectively, using cryptic donor sites within exon 4 (6.1 bits) and within intron 4 (6.5 bits) (Table S2). This same homozygous mutation was reported in six XP-V patients from Japan (Tanioka et al., 2007). Many Japanese XP-A patients have a splice mutation, which has been identified as a founder mutation carried by about 1% of the Japanese general population (Hirai et al., 2006). Possibly this POLH splice mutation in Japan and Korea is also a founder mutation.
Nonsense mutation in families C and D
XP70TMA in family C and XP92TMA in family D have the same mutation in exon 4. This G-to-C transversion at position 454 (Table 2) resulted in a nonsense mutation (p.Gln152*). The predicted protein size is 151 amino acids. This mutation creates a DdeI restriction site (Figure 5). The digested DNA from the mother of family C (XPH95TMA) shows three bands indicating that she was heterozygous for this mutation. The DNA from the affected siblings in both families showed complete digestion, indicating that the mutation is homozygous. This is consistent with the clinical history of consanguinity in each of these families.
Figure 5. Analysis of genomic DNA of families C and D.
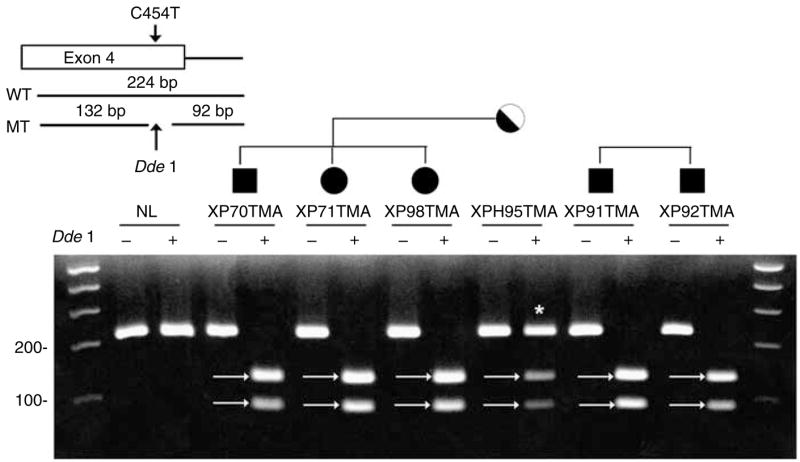
The C454T mutation in exon 4 creates a new DdeI restriction site. DNA from a normal donor, affected siblings XP70TMA, XP71TMA, XP98TMA, and their mother (XPH95TMA) in family C, and affected siblings XP91TMA and XP92TMA in family D were separated on 3% agarose gel. + Indicates DNA digested with DdeI. The digested DNA showed two bands of 92 and 132 bp (arrows). * Indicates the normal-size band.
There is no history of a common relative linking family C and family D. These families both live in eastern Turkey. Both of these XP-V families have multiple affected children (Figure 1, C-1, C-2, C-3, D-1, and D-2) and some of them have developed skin cancer at an early age. An earlier study of the inheritance of the XPC gene in a family from this region of Turkey examined microsatellite markers and found a genetic link to another XP-C family in Italy with a common ancestor about 300–500 years ago (Gozukara et al., 2001). Similar studies of microsatellites or single-nucleotide polymorphisms might indicate the extent of relationship between these families.
Truncation in the catalytic domain of POLH
The cells from patients in families E and F had the same mutation. Insertion of a single G at 1078 in exon 10 was identified in XP38BE (c.1078_1079dupG) (Figure 6a) and XP161BE (data not shown). This uncloned genomic DNA sequence does not show double bands, thus indicating that both alleles have the same insertion or that one allele has the mutation and the other allele is absent. This frameshift mutation codes for 31 unrelated amino acids before a termination codon at codon 391 (p.Asp360Glyfs*32) (Table 2). The mutation creates a new BslI site. Digestion of DNA from XPH253BE, the mother of family E (Figure 6b), produces three bands, indicating that she is heterozygous for this mutation. DNA from the affected siblings XP38BE and XP39BE is completely digested (Figure 6b), indicating that they are homozygous or hemizygous for this mutation, as suggested from the sequence tracing (Figure 6a). DNA from the father was not available for testing. PCR-RFLP analysis of DNA from family F (Figure 6c) shows complete digestion of the DNA from both affected sisters XP164BE and XP161BE. Digested DNA from both parents yields three bands, indicating that they are heterozygous for this mutation. Thus, we can conclude that the patients are homozygous (and not hemizygous) for this mutation. As expected, the children of patient XP161BE are heterozygous for this mutation (Figure 6c). Family E is from the Cayman Islands and family F from the United States. We have no history of consanguinity between these families. This mutation was also reported (Johnson et al., 1999a) in XPPHBE, a 30-year-old Caucasian man who has an affected brother, XP13BE (Robbins et al., 1974; Moshell et al., 1981).
Figure 6. Sequence analysis of families E and F.
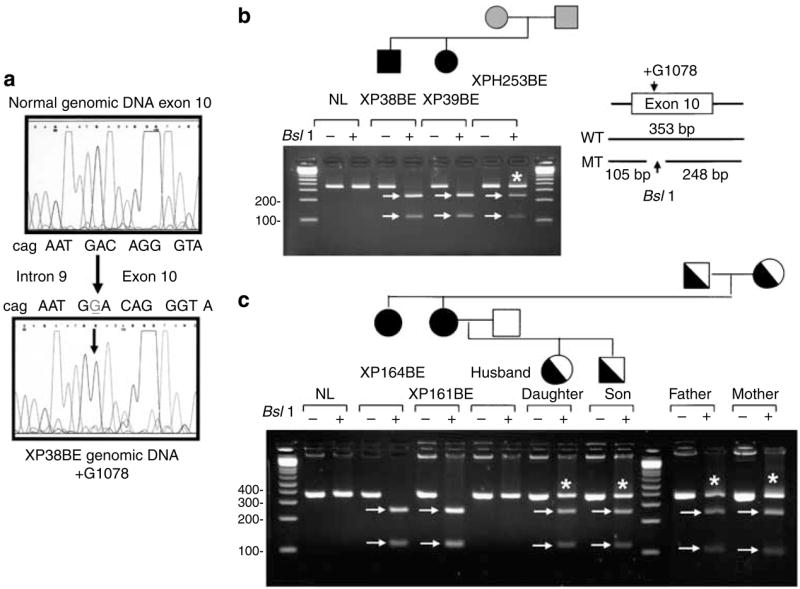
(a) Genomic DNA from XP38BE in family E shows an insertion of a G in exon 10. There is a single sequence following the insertion indicating that the DNA is homozygous or hemizygous for this frameshift mutation. (b) PCR-RFLP assay of family E. The insertion of G creates a new BsII site in the genomic DNA. Genomic DNA from a normal donor, patients XP38BE, XP39BE, and their mother, XPH253BE, was run on a gel. + Indicates BsII treatment. The 105 and 248-bp bands in the digested DNA are indicated by arrows. * Indicates the normal-size band. (c) PCR-RFLP analysis for family F. Genomic DNA from XP164BE, XP161BE, the husband, son, daughter, mother, and father of XP161BE was run on a gel. + Indicates BsII treatment. The 105 and 248-bp bands in the digested DNA are indicated by arrows. * Indicates the normal size band.
XP3GO cells in family G had a 1-kb deletion (g.24139_25203del1065) in the genomic DNA that includes exon 6 (Table 2). This homozygous deletion is predicted to result in a truncated protein of 221 amino acids (p.Val221-Profs*2). Mutated cDNA with this sequence was reported previously (Johnson et al., 1999a) in cell line XP5MA from an 86-year-old woman from Germany with “pigmented xerodermoid” (Hofmann et al., 1978). The mutation in the genomic DNA of XP5MA was not reported. Interestingly, the deletion of exon 6 was also seen in the type-I splice isoform in the cells from XP31BE who has a splice mutation (Table 2; Figure 4b).
XP139DC cells in family H had two different frameshift mutations (Table 2). Deletion of the 170 bases of exon 10 (c.1075_1244del170) resulted in an altered protein beginning at Asn359 and terminating after 389 amino acids (p.Asn359-Valfs*32). This same mutation was reported as homozygous at the cDNA level in XP6VI and XP75VI cells (Broughton et al., 2002). The other mutation in XP139DC cells was a 2 bp deletion in exon 11 (c.1706_1707del2). This would result in a truncated 577-amino-acid protein (p.Thr569Argfs*10).
XP224BE in family I is a compound heterozygote for a frameshift mutation and a missense mutation (Table 2). One allele has an insertion of a single T at nucleotide 149 in exon 3 (c.149_150dupT). This frameshift mutation creates a new termination signal 3 codons downstream, resulting in a truncated 52-amino-acid protein (p.ser51Glufs*3). This heterozygous mutation was identified in cells from her father (XPH227BE) (data not shown). The +T149 insertion inactivates a BtsI restriction site. A PCR-RFLP assay using BtsI confirmed that DNA from the patient, XP224BE, and her father, XPH227BE, were heterozygous for this mutation, and that DNA from her mother’s cells, XPH226BE, was not digested, indicating a normal sequence (data not shown).
Missense mutations
The second allele of XP224BE carried a transition mutation of A to G (c.658A>G) in exon 5 (Table 2). This missense mutation changed lysine 220 (AAG) to glutamate (GAG) (p.Lys220Glu), but preserved the size of the protein. This heterozygous mutation was also present in cells from her mother (XPH226BE) (data not shown). To determine if the POLH c.658A>G mutation was a common polymorphism, we screened DNA obtained from buccal swabs of 100 anonymous donors (Khan et al., 2000). DNA sequence analysis showed the normal A/A genotype for all 100 donors, indicating that the POLH c.658A>G mutation was not a common polymorphism (data not shown).
The mutation of XP10TA in family J was homozygous for a missense mutation in exon 5 based on the DNA sequence analysis and the family history of consanguinity (Table 2). This mutation (c.522 G>T), which changes tryptophan 174 to cystine (p.Trp174Cys), may affect protein conformation because of the conversion from a hydrophobic amino acid to a hydrophilic amino acid in the catalytic domain of POLH. We determined that c.522G>T was not a common polymorphism by screening 100 DNA samples from normal donors (Khan et al., 2000) using PCR-RFLP analysis with BsmAI. All of the donors’ samples had the restriction pattern of the normal G/G genotype (data not shown).
POLH mRNA in XP-V cells
Western blotting (Figure 3) and sequence analysis (Table 2) indicated that polη protein was not detectable in the mutant cells that yielded premature terminations, and a faint band could be seen in the mutants that had missense mutations. One mechanism for the observed large reduction in polη protein would be nonsense mediated message decay (Kuzmiak and Maquat, 2006). Nonsense-mediated message decay has been observed in cells from XPC patients who have mutations leading to premature stop codons (Khan et al., 2006). We measured POLH mRNA levels (Table 3; Table S1) in XP-V cell lines that had no detectible polη protein (Figure 3) resulting from a splice site mutation (XP31BE), a frameshift mutation (XP38BE), or a nonsense mutation (XP70TMA) (Table 2). Three normal fibroblast strains had a mean of 77.2 fg of full-length message (exon 1/exon 2 boundary primer), 81.9 fg (exon 2/exon 3 boundary primer), and 3 fg of POLH mRNA (exon 2-skipping primers) (Table 3). The cell strains with splicing and frameshift mutations had normal levels of POLH mRNA (Table 3), demonstrating absence of nonsense-mediated message decay in these cells. The cell strain with a nonsense mutation (XP70TMA) had POLH mRNA levels that were about half of normal (Table 3), demonstrating nonsense-mediated message decay in these cells. All of the cell strains had normal levels of skipping of exon 2 in POLH mRNA.
Table 3.
Polη mRNA levels in XP-V cell lines
| POLH WT (exons 1–2/2)1 | POLH WT (exon 2/3–2)1 | POLH-skipping exon 2 (exon 1/3–1)1 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Cell line | SQ | SQ mean | SQ | SQ mean | SQ | SQ mean | |||
| XP-V | |||||||||
| XP31BE | 81.5 | 80.8 | 2.5 | ||||||
| 76.0 | 78.6 | 102% | 103 | 92.9 | 113% | 3.6 | 3.3 | 109% | |
| 78.3 | 94.8 | 3.8 | |||||||
| XP38BE | 92.9 | 85.5 | 3.5 | ||||||
| 81.4 | 85.1 | 110% | 88.7 | 91.3 | 111% | 3.8 | 3.7 | 122% | |
| 80.9 | 99.6 | 3.7 | |||||||
| XP70TMA | 39.4 | 44.1 | 4.5 | ||||||
| 37.8 | 37.7 | 49% | 41.7 | 42.3 | 52% | 3.8 | 4.1 | 137% | |
| 36.0 | 41.2 | 4.1 | |||||||
| XP-V mean | 67.1 | XP-V mean | 75.5 | XP-V mean | 3.7 | ||||
| Normals | |||||||||
| AG5186 | 91.7 | 104 | 2.3 | ||||||
| 92.5 | 90.9 | 89.2 | 95.0 | 3.9 | 3.3 | ||||
| 88.6 | 91.7 | 3.6 | |||||||
| AG13153 | 77.4 | 93.3 | 2.9 | ||||||
| 103 | 89.7 | 94.7 | 92.0 | 4.9 | 3.8 | ||||
| 88.8 | 88.0 | 3.7 | |||||||
| AG13145 | 49.2 | 52.2 | 2.0 | ||||||
| 50.1 | 51.0 | 64.3 | 58.7 | 1.7 | 1.9 | ||||
| 53.6 | 59.6 | 2.1 | |||||||
| Normal mean | 77.2 | 100% | Normal mean | 81.9 | 100% | Normal mean | 3.0 | 100% | |
Polη, DNA polymeraseη; WT, wild type; XP-V, xeroderma pigmentosum variant.
Full-length (FL) and major alternative spliced (del ex 2) POLH mRNA levels (fg of plasmid DNA) in fibroblasts measured by quantitative real-time reverse transcriptase-PCR.
Polι in XP-V cells
DNA polymeraseι (polι) is another Y-family DNA polymerase (Woodgate, 1999; Ohmori et al., 2001; Kannouche et al., 2003) that was recently identified as playing a role in carcinogenesis in cells lacking polη (Wang et al., 2007). We measured the level of polι protein in XP-V cells with undetectable or with very low levels of polη (Figure 3b). All of the cells examined had levels of polι protein in the normal range (Figure 3b, and data not shown).
DISCUSSION
Laboratory identification of XP-V cells
XP-V classically has been defined by the combination of the clinical features of XP with normal post-UV cell survival that is reduced by caffeine, normal post-UV UDS and reduced post-UV DNA synthesis (Itoh et al., 1996). These conventional laboratory examinations separate XP-V from normal cells, but they cannot identify the molecular defect in XP-V cells. The identification of the POLH gene has greatly facilitated molecular diagnosis of XP-V. IP-western blotting was able to detect POLH protein in nuclear extracts from simian virus-transformed cells (Thakur et al., 2001; Laposa et al., 2003). In this procedure, POLH protein from SV-transformed normal cells showed a dark band at 78 kDa, whereas POLH protein in XP-V SV-transformed cells (XP30RO) was not detected. The monoclonal antibody for polη recognized the 300 amino acids at the C-terminal end of polη protein. XP30RO cells, with a 13-base deletion in exon 2, generate a severely truncated protein, which deletes the recognition site of the antibody. We have simplified this assay permitting use of whole-cell extracts, simplified IP, and measurement of protein concentration by a standard protocol. The simplified method detects polη levels in primary fibroblasts (Figure 3; Tanioka et al., 2007) and lymphoblastoid cells.
Undetectable polη protein: nonsense-mediated message decay and post-translational degradation
One mechanism for the observed large reduction in polη protein would be nonsense-mediated message decay (Kuzmiak and Maquat, 2006). For example, nonsense-mediated message decay has been observed in cells from XP-C patients who have mutations leading to premature stop codons (Khan et al., 2006). An XP-V cell strain (XP70TMA) with a nonsense mutation in the POLH gene (Table 2) had no detectable polη protein levels (Figure 3) and showed about 50% of normal POLH mRNA levels (Table 3). This reduction in mRNA level may be due to nonsense-mediated message decay; however, the level of reduction of mRNA is less than seen in XP-C cells with nonsense mutations (reduced to <25% of normal) (Khan et al., 2006). Northern blots from XP-V cells showed different levels of poly(A)+ RNA ranging from undetectable to normal levels in different cells with nonsense or frameshift mutations (Masutani et al., 1999b). We found normal levels of POLH mRNA in XP-V cell strains with no detectable polη protein and splice (XP31BE) or frameshift (XP38BE) mutations (Table 3). Thus, these cells have no evidence of nonsense-mediated message decay. The low polη protein levels may be caused by increased proteolysis mediated via ubiquitin binding, as demonstrated in studies of polη (Bienko et al., 2005) or its yeast homologue, RAD30 (Skoneczna et al., 2007).
Trace protein levels: missense mutations with alteration of RNA processing and protein degradation
XP10TA and XP224BE(2) have missense mutations in the catalytic domain, and western blotting showed faint bands of polη protein (Table 2; Figure 3a). XP10TA has a homozygous base substitution mutation in exon 5 that results in a missense mutation Trp174Cys. The c.522 G>T mutation increases the strength of a cryptic RNA splice acceptor (Schneider, 1997a, b) within exon 5 from 8.8 to 10.2 bits (data not shown). This mutation within an exon may result in altered RNA splicing as reported in cells from patients with isovaleric acidemia (Vockley et al., 2000), with accompanying low levels of protein. Similarly, the heterozygous c.658 A>G base substitution mutation in exon 5 in XP224BE reduces the information content of the donor from 7.8 to 6.8 bits (Table 3, and data not shown), possibly reducing the extent of normal splicing leading to low levels of polymeraseη protein.
Alternatively, the missense mutation may alter the stability of the protein as described for XPC (Yasuda et al., 2007) or for Escherichia coli umuC, the bacterial equivalent of polη (Woodgate et al., 1994). The two missense mutations, tryptophan (hydrophobic amino acid) 174 to cysteine (hydrophilic amino acid) in XP10TA and lysine (basic amino acid) 220 to glutamate (acidic amino acid) in XP224BE, may influence the polη protein conformation, resulting in instability or structure change at the epitope of antibody binding. One cell line, XP11BR, with a missense mutation in the catalytic domain, has been reported to be defective in translesion synthesis activity (Broughton et al., 2002). This glycine 263-to-valine alteration leads to the conversion from a hydrophilic amino acid to a hydrophobic amino acid.
Mutation spectrum and predicted POLH protein function
Following results of IP-western blotting, we sequenced eight cell lines, which have no band of polη protein and two cell lines with a faint band. We found 10 different mutations in 10 patients. These mutations were classified as splicing, frameshift, nonsense, and missense (Table 2). The functional structure of pol-η protein has been elucidated (Figure 7; Johnson et al., 1999a; Masutani et al., 1999b; Yamada et al., 2000; Kannouche et al., 2001, 2003; Ling et al., 2001, 2003; Broughton et al., 2002; Bomar et al., 2007; Yang and Woodgate, 2007). The N-terminal 400 amino acids are highly conserved in the Y-family polymerases (Boudsocq et al., 2002). The polymerase activities of polη reside entirely in the first 511 amino acids of the 713-amino-acid protein (Masutani et al., 1999a). The C-terminal 200-amino-acid region does not have polymerase function, but contains a bipartate nuclear localization signal between residues 682–698 and the C-terminal. The C-terminal 120 amino acids of polη are sufficient for nuclear localization, and for localization into foci (Kannouche et al., 2001). Cell-free extracts from XP-V with severe truncation in the catalytic domain were shown to be defective in translesion synthesis activity. This suggests that mutant alleles with truncations between 52 and 390 amino acids in our patients’ cells (XP31BE, XP3GO, XP38BE, XP161BE, XP70TMA, XP92TMA, XP224BE(1), and XP1SE variant II) would have defective polymerase activity.
Figure 7. Mutation spectrum and predicted proteins in XP-V cells.

The top line shows the 11 exons of POLH genomic sequence on chromosome 6 as filled rectangles. The location of frameshift, nonsense, and missense mutations in each allele for the cells studied is shown above the line and the splice site mutations are shown below the line. The second line shows the mRNA with the ATG initiation codon in exon 2 and the TAG stop codon in exon 11. The 713-amino-acid pol-η protein is shown in the third line. The N-terminal 400 amino acids are highly conserved in Y-family polymerases, and contain the catalytic domain of the polymerase. There is a nuclear localization signal located at amino acids 682–698 (NLS). The C-terminal region from amino acids 628–662 contains a C2H2 zinc finger that is involved in DNA-binding ubiquitin. A PCNA-binding site is located at the extreme C-terminus of the protein. The bottom portion of the figure shows the predicted size of the protein from each allele or splice variant form from the XP-V patients and the description of the mutation at the cDNA and protein level. PCNA, proliferating-cell nuclear antigen.
Splice variant II of XP31BE has a 13-amino-acid in-frame deletion in exon 6. XP3DU with in-frame deletion in exon 3 also has defective bypass ability in a translesion synthesis assay (Broughton et al., 2002). This suggests that mutations found in the patients’ cell lines with undetectable or faint bands of polη protein in western blotting are causative for XP-V.
Splicing abnormalities
Information theory is an important tool to rank normal and mutant splice junctions, and most splice junctions that are fully functional have an information content of at least 2.4 bits (Schneider, 1997a, 1997b; Rogan et al., 1998). Splice-site mutations may result in exon skipping, activation of cryptic splice sites, creation of a pseudo-exon within intron, or intron retention. The splice donor at exon 6 in normal sequence has an information content of 9.3 bits (Table S2). The exon 6 splice donor site mutation in XP31BE inactivates the normal splice donor, resulting in two splice variants, one of which results from skipping of exon 6 and the other from activation of a cryptic splice site in exon 6 (Figures 4 and 7). Another XP-V cell (XP1RO) had a splice acceptor site mutation in exon 2 (Masutani et al., 1999b). This mutation also reduced information content at the splice acceptor from 3.4 to <0 bits, and led to skipping of exon 2. XP1SE has a mutation within exon 4, which reduces the splice donor site information from 5.4 bits to 2.1 bits.
Origin, inheritance, and clinical features of XP-V mutations in different parts of the world
Some DNA-repair genes also function as basal transcription factors and are thus essential for survival. Complete inactivation of both alleles of these genes is not compatible with life. Thus, mutations in affected patients must preserve some genetic activity and hence only a limited set of mutations would be observed in at least one allele. An example of this is the XPD gene where unrelated patients from many different countries have mutations at the same site (R683W) (Taylor et al., 1997; Lehmann, 2003; Friedberg et al., 2006). This mutation has been shown to retain some activity (Dubaele et al., 2003). In contrast, the XPC gene is not essential for life and can be completely inactivated with undetectable levels of XPC protein (Khan et al., 2006). In the XPC gene, many different mutations have been found without an accumulation at one site (Chavanne et al., 2000; Gozukara et al., 2001; Friedberg et al., 2006; Khan et al., 2006).
The XP-V gene appears to be closer to the model of a nonessential gene, with no one site having a preponderance of mutations (Johnson et al., 1999a; Broughton et al., 2002; Tanioka et al., 2007) and undetectable levels of polη protein (Figure 3). We found 10 mutations in POLH in 10 families. Two families from Turkey (families C and D) had the same homozygous nonsense mutation, whereas two families from North America (families E and F) had the same homozygous frameshift mutation. A patient from Korea had the same probable founder mutation as that in Japanese XP-V patients (Tanioka et al., 2007). The other five families had seven different mutations. These probably arose independently.
The 15 XP-V patients we studied had a range of clinical features (Table 1). None had XP related neurological abnormalities (Rapin et al., 2000; Kraemer et al., 2007) or loss of vision due to corneal clouding. They all developed freckles at an early age. In some of the patients, this did not progress (for example, XP224BE and XP10TA) and their skin looked similar to that of chronic sun damage in fair-skinned individuals in the general population. While in others, for example, in patients from Turkey (families C and D) (Figure 1), the skin changes resulted in extensive cutaneous pigmentation and atrophy. All of the patients, except for two of the youngest, had skin cancer and six had melanoma. This is similar to the average age of reported skin cancer in more than 800 XP patients of less than 10 years (Kraemer et al., 1987, 1994). The group includes four (27%) XP-V patients older than 45 years (Table 1). This is a relatively old age for XP patients compared with the 5% of 785 reported XP patients who were older than 45 years (Kraemer et al., 1987), and may reflect a tendency of XP-V patients to live longer than XP patients with defects in nucleotide-excision repair genes. However, one XP-V patient, XP92TMA (Table 1; Figure 1, D-2) died of metastatic SCC of his lip at 22 years of age. Our first XP-V patient, XP4BE, had ocular involvement, extensive cutaneous changes, more than 100 skin cancers, and died at 27 years of metastatic melanoma (Robbins et al., 1974, 1975).
A correlation between the clinical features and the different POLH mutations is not clear (see also Tanioka et al., 2007). Cells from the two patients with missense mutations (XP24BE and XP10TA) showed trace polη protein on the western blots (Figure 3), and were from adults without marked progression of their pigmentary abnormalities. The patients with extensive cutaneous pigmentation and atrophy (families C and D) had a homozygous nonsense mutation that resulted in no detectible polη protein (Figure 3). However, other patients with absent polη protein did not show these extensive cutaneous changes.
XP and polι protein
We also examined the level of polη protein in 16 cell strains from other suspected UV-sensitive, cancer-prone XP-V patients who were referred to the authors. We found them to have normal post-UV cell survival, host cell reactivation, or DNA repair (data not shown). However, unlike true XP-V cells, these cells had normal levels of polη protein and we were unable to detect any mutations in POLH (data not shown). We therefore considered the possibility that these clinical phenotypes may have arisen because of mutations in another polymerase involved in the translesion synthesis of UV photoproducts. A good candidate enzyme appeared to be polι, which is a paralog of polη (Ohmori et al., 2001). Polι has also been shown to physically interact with polη, and both polymerases colocalize to sites of UV-induced DNA damage (Kannouche et al., 2002). Furthermore, polι can bypass cyclobutane dimers in vitro (Vaisman et al., 2003; Frank and Woodgate, 2007) and mice lacking polι exhibit mild sensitivity to UV-light (Dumstorf et al., 2006). We therefore measured the level of polι protein in these cells, as well as sequenced the entire POLI gene. However, all cells exhibited normal levels of polι protein and did not contain any mutations in POLI (data not shown). The molecular defects causing the phenotype resembling XP-V in these cells therefore remain to be determined.
MATERIALS AND METHODS
Patients and cells
Patients were examined in accordance with the Institutional Review Boards at their institutions (National Institutes of Health, Bethesda, MD; Yüzüncü Yil University Medical School, Van, Turkey; Wuerzburg University, Wurzburg, Germany; Armed Forces Institute of Pathology, Washington, DC; Yonsei University College of Medicine, Seoul, Korea; and Tel Aviv University, Tel Aviv, Israel) or their cells were obtained from the Human Genetic Mutant Cell Repository, Camden, NJ. The studies followed the Declaration of Helsinki Principles and patients gave informed consent for these studies. The cell lines examined in this study are listed in Table 1. Patients’ skin primary fibroblasts (XP31BE (GM13155), XP38BE (AG02592), XP70TMA (GM14873), XP92TMA (GM15713), XP224BE (GM16818), and lymphoblastoid cell lines (XP31BE (GM13154) and normals (AG10107) and (XP23BE - XPC (AG10033)) were obtained from the Human Genetic Mutant Cell Repository, Camden, NJ. XP10TA cells were established at Tel Aviv University. XP161BE, XP164BE, XP139DC, and the XP-C strain, XP108DC, were supplied by the Department of Environmental and Toxicologic Pathology, Armed Forces Institute of Pathology, Washington, DC. XP3GO primary fibroblasts were established at the Department of Dermatology, Georg-August-University, Goettingen, Germany. XP1SE was obtained from Department of Dermatology and Cutaneous Biology Research Institute, Yonsei University College of Medicine, Seoul, Korea. Normal skin primary fibroblasts (AG4659, AG5186, AG13153, and AG13145) were obtained from the Human Aging Cell Repository, Camden, NJ. Fibroblast cell lines were grown in DMEM (Invitrogen, Grand Island, NY) containing 4mM glutamine and 10% fetal calf serum, as previously described (Khan et al., 2004).
Host cell reactivation assay
To screen the UV sensitivity of the cells, we performed post-UV host cell reactivation assay as described previously (Emmert et al., 2002). The pCMVLuc reporter gene plasmid (a generous gift from M Hedayati and L Grossman, John Hopkins University, Baltimore, MD) was used to measure post-UV host cell reactivation. A 200-ng weight of CsCl-purified pCMVLuc, either 1,000 Jm−2 UV-irradiated or unirradiated, was transfected into 1.5×105 fibroblast cells per well using 4 μl of Lipofectamine (Invitrogen) in a total 1ml of OPTI-MEM medium (Invitrogen) for 5 hours. After 48 hours of incubation, luciferase activity was measured with a luminometer (Monolight 2010; Analytical Luminescence Laboratory, San Diego, CA) using luciferase assay reagent (Promega, Madison, WI) according to the manufacturer’s protocol. Relative luciferase activities are presented as a percentage of activities obtained with UV-irradiated versus unirradiated control plasmids.
Semi-quantitative estimation of polη and polι protein expression
Polη and polι protein levels were assessed by IP followed by western blotting as described previously (Thakur et al., 2001; Kannouche et al., 2003; Laposa et al., 2003; Tanioka et al., 2007). Polι protein was detected using a rabbit polyclonal antibody directed against a keyhole limpet hemocyanin-conjugated peptide corresponding to the extreme C-terminal 15 amino-acid residues (AEWKRTGSDFHIGHK) of poli (Kannouche et al., 2002).
PCR amplification, sequencing of POLH and POLI, and measurement of POLH mRNA
DNA was isolated using DNAzol reagent as per vendor’s protocol (Invitrogen). Total cytoplasmic RNA was isolated from cells by the RNA aqueous small-scale phenol-free total RNA isolation kit (Ambion, Austin, TX) according to the vendor’s protocol. To sequence the entire coding region and exon–intron junction sites of POLH and POLI genes, each exon and the entire coding region were amplified using intronic and exonic primers (Table S1). The primers were designed on the basis of the GenBank reference sequences (accession no. NC_000006 for POLH genomic sequence; NM_006502 for POLH cDNA sequence; and NC_000018 for POLI genomic sequence). RT-PCR amplification of the entire coding region of POLH was performed using primers S0 (GATCCCTTCTCGGTTTCTCC) and AS24 (ATCCTACAGGCAAGCCTGAG) (Masutani et al., 1999b), with two PCR involving denaturing at 94 °C for 1minute, 35 cycles of denaturing at 94 °C for 20 seconds, and annealing/extension at 70 °C for 3minutes, followed by extension at 70 °C for 3 minutes. PCR products were sequenced by using the Thermo Sequenase II dye-terminator cycle sequencing Premix Kit (Amersham Pharmacia, Piscataway, NJ). POLI sequence analysis was performed similarly as described for POLH. Real-time quantitative RT-PCR for measurement of POLH mRNA was performed with pairs of primers, where one primer spanned an exon–exon boundary as described by Khan et al. (2002) (2006) (Table S1).
Mutations were described according to the recommendations of the Human Genome Variation Society (den Dunnen and Antonarakis, 2000) (http://www.hgvs.org/mutnomen/) and the nomenclature was checked using the mutalyzer website http://www.lovd.nl/mutalyzer/1.0.1/.
DNA sequence information analysis
The sequences of normal splice donor and acceptor sites of each exon and all of the sequences with mutations we identified were scanned with the donor and acceptor individual information weight matrices (Schneider, 1997a, b). This analysis can be performed on a web server, https://splice.cmh.edu/. The normal sequences of splice donor and acceptor sites of each exon and information contents are calculated and listed in Table S2.
Supplementary Material
Table S1. Primer sequences used.
Table S2. Human pol-η gene: sequence and information content of normal and mutant splice sites.
Acknowledgments
This study was partially supported by the intramural research program of the National Cancer Institute, the National Institute of Child Health and Human Development, and the National Institutes of Health, Bethesda, MD. SE was supported by the Deutsche Forschungsgemeinschaft DFG (EM 63/3-1 and GRK 1034). HS and EA were supported by the Israel Cancer Association. We thank Tala Shalavi BS and Vanessa Muniz-Medina BS for technical assistance.
Abbreviations
- BCC
basal cell carcinoma
- RT-PCR
reverse transcriptase- PCR
- SCC
squamous cell carcinoma
- UDS
unscheduled DNA synthesis
- XP
xeroderma pigmentosum
- XP-V
xeroderma pigmentosum variant
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Arlett CF, Harcourt SA, Broughton BC. The influence of caffeine on cell survival in excision-proficient and excision-deficient xeroderma pigmentosum and normal human cell strains following ultraviolet-light irradiation. Mutat Res. 1975;33:341–6. doi: 10.1016/0027-5107(75)90209-2. [DOI] [PubMed] [Google Scholar]
- Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–4. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- Bomar MG, Pai MT, Tzeng SR, Li SS, Zhou P. Structure of the ubiquitin-binding zinc finger domain of human DNA Y-polymerase eta. EMBO Rep. 2007;8:247–51. doi: 10.1038/sj.embor.7400901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootsma D, Kraemer KH, Cleaver JE, Hoeijmakers JHJ. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler KW, editors. The Genetic Basis of Human Cancer. 2. New York: McGraw-Hill; 2002. pp. 211–37. [Google Scholar]
- Boudsocq F, Ling H, Yang W, Woodgate R. Structure-based interpretation of missense mutations in Y-family DNA polymerases and their implications for polymerase function and lesion bypass. DNA Repair (Amst) 2002;1:343–58. doi: 10.1016/s1568-7864(02)00019-8. [DOI] [PubMed] [Google Scholar]
- Broughton BC, Cordonnier A, Kleijer WJ, Jaspers NG, Fawcett H, Raams A, et al. Molecular analysis of mutations in DNA polymerase eta in xeroderma pigmentosum-variant patients. Proc Natl Acad Sci USA. 2002;99:815–20. doi: 10.1073/pnas.022473899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavanne F, Broughton BC, Pietra D, Nardo T, Browitt A, Lehmann AR, et al. Mutations in the XPC gene in families with xeroderma pigmentosum and consequences at the cell, protein, and transcript levels. Cancer Res. 2000;60:1974–82. [PubMed] [Google Scholar]
- Cleaver JE. Xeroderma pigmentosum: variants with normal DNA repair and normal sensitivity to ultraviolet light. J Invest Dermatol. 1972;58:124–8. doi: 10.1111/1523-1747.ep12538913. [DOI] [PubMed] [Google Scholar]
- Cordonnier AM, Lehmann AR, Fuchs RP. Impaired translesion synthesis in xeroderma pigmentosum variant extracts. Mol Cell Biol. 1999;19:2206–11. doi: 10.1128/mcb.19.3.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. HumMutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Dubaele S, Proietti De Santis L, Bienstock RJ, Keriel A, Stefanini M, Van Houten B, et al. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol Cell. 2003;11:1635–46. doi: 10.1016/s1097-2765(03)00182-5. [DOI] [PubMed] [Google Scholar]
- Dumstorf CA, Clark AB, Lin Q, Kissling GE, Yuan T, Kucherlapati R, et al. Participation of mouse DNA polymerase iota in strand-biased mutagenic bypass of UV photoproducts and suppression of skin cancer. Proc Natl Acad Sci USA. 2006;103:18083–8. doi: 10.1073/pnas.0605247103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmert S, Slor H, Busch DB, Batko S, Albert RB, Coleman D, et al. Relationship of neurologic degeneration to genotype in three xeroderma pigmentosum group G patients. J Invest Dermatol. 2002;118:972–82. doi: 10.1046/j.1523-1747.2002.01782.x. [DOI] [PubMed] [Google Scholar]
- Frank EG, Woodgate R. Increased catalytic activity and altered fidelity of human DNA polymerase iota in the presence of manganese. J Biol Chem. 2007;282:24689–96. doi: 10.1074/jbc.M702159200. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. Washington, DC: ASM Press; 2006. [Google Scholar]
- Gozukara EM, Khan SG, Metin A, Emmert S, Busch DB, Shahlavi T, et al. A stop codon in xeroderma pigmentosum group C families in Turkey and Italy: molecular genetic evidence for a common ancestor. J Invest Dermatol. 2001;117:197–204. doi: 10.1046/j.1523-1747.2001.01424.x. [DOI] [PubMed] [Google Scholar]
- Gratchev A, Strein P, Utikal J, Sergij G. Molecular genetics of xeroderma pigmentosum variant. Exp Dermatol. 2003;12:529–36. doi: 10.1034/j.1600-0625.2003.00124.x. [DOI] [PubMed] [Google Scholar]
- Hirai Y, Kodama Y, Moriwaki S, Noda A, Cullings HM, MacPhee DG, et al. Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res. 2006;601:171–8. doi: 10.1016/j.mrfmmm.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Hofmann H, Jung EG, Schnyder UW. Pigmented xerodermoid: first report of a family. Bull Cancer (Paris) 1978;65:347–50. [PubMed] [Google Scholar]
- Itoh T, Linn S. XP43TO, previously classified as xeroderma pigmentosum group E, should be reclassified as xeroderma pigmentosum variant. J Invest Dermatol. 2001;117:1672–4. doi: 10.1046/j.0022-202x.2001.01619.x. [DOI] [PubMed] [Google Scholar]
- Itoh T, Linn S, Kamide R, Tokushige H, Katori N, Hosaka Y, et al. Xeroderma pigmentosum variant heterozygotes show reduced levels of recovery of replicative DNA synthesis in the presence of caffeine after ultraviolet irradiation. J Invest Dermatol. 2000a;115:981–5. doi: 10.1046/j.1523-1747.2000.00154.x. [DOI] [PubMed] [Google Scholar]
- Itoh T, Linn S, Ono T, Yamaizumi M. Reinvestigation of the classification of five cell strains of xeroderma pigmentosum group E with reclassification of three of them. J Invest Dermatol. 2000b;114:1022–9. doi: 10.1046/j.1523-1747.2000.00952.x. [DOI] [PubMed] [Google Scholar]
- Itoh T, Ono T, Yamaizumi M. A simple method for diagnosing xeroderma pigmentosum variant. J Invest Dermatol. 1996;107:349–53. doi: 10.1111/1523-1747.ep12363303. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999a;285:263–5. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Prakash S, Prakash L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Poleta. Science. 1999b;283:1001–4. doi: 10.1126/science.283.5404.1001. [DOI] [PubMed] [Google Scholar]
- Kannouche P, Broughton BC, Volker M, Hanaoka F, Mullenders LH, Lehmann AR. Domain structure, localization, and function of DNA polymerase eta, defective in xeroderma pigmentosum variant cells. Genes Dev. 2001;15:158–72. doi: 10.1101/gad.187501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannouche P, Fernandez De Henestrosa AR, Coull B, Vidal AE, Gray C, Zicha D, et al. Localization of DNA polymerases eta and iota to the replication machinery is tightly coordinated in human cells. EMBO J. 2002;21:6246–56. doi: 10.1093/emboj/cdf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannouche P, Fernandez De Henestrosa AR, Coull B, Vidal AE, Gray C, Zicha D, et al. Localization of DNA polymerases eta and iota to the replication machinery is tightly coordinated in human cells. EMBO J. 2003;22:1223–33. doi: 10.1093/emboj/cdf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SG, Metin A, Gozukara E, Inui H, Shahlavi T, Muniz-Medina V, et al. Two essential splice lariat branchpoint sequences in one intron in a xeroderma pigmentosum DNA repair gene: mutations result in reduced XPC mRNA levels that correlate with cancer risk. Hum Mol Genet. 2004;13:343–52. doi: 10.1093/hmg/ddh026. [DOI] [PubMed] [Google Scholar]
- Khan SG, Metter EJ, Tarone RE, Bohr VA, Grossman L, Hedayati M, et al. A new xeroderma pigmentosum group C poly(AT) insertion/deletion polymorphism. Carcinogenesis. 2000;21:1821–5. doi: 10.1093/carcin/21.10.1821. [DOI] [PubMed] [Google Scholar]
- Khan SG, Muniz-Medina V, Shahlavi T, Baker CC, Inui H, Ueda T, et al. The human XPC DNA repair gene: arrangement, splice site information content and influence of a single nucleotide polymorphism in a splice acceptor site on alternative splicing and function. Nucleic Acids Res. 2002;30:3624–31. doi: 10.1093/nar/gkf469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SG, Oh KS, Shahlavi T, Ueda T, Busch DB, Inui H, et al. Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis. 2006;27:84–94. doi: 10.1093/carcin/bgi204. [DOI] [PubMed] [Google Scholar]
- Kim J, Chung KY. Removal by Mohs micrographic surgery and reconstruction using combined local flaps. Korean J Dermatol. 2003;41:1354–8. [Google Scholar]
- Kraemer KH. Heritable diseases with increased sensitivity to cellular injury. In: Freedberg IM, et al., editors. Fitzpatrick’s Dermatology in General Medicine. New York: McGraw-Hill; 2003. pp. 1508–21. [Google Scholar]
- Kraemer KH, Lee M-M, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer: the xeroderma pigmentosum paradigm. Arch Dermatol. 1994;130:1018–21. [PubMed] [Google Scholar]
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–50. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience. 2007;145:1388–96. doi: 10.1016/j.neuroscience.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmiak HA, Maquat LE. Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med. 2006;12:306–16. doi: 10.1016/j.molmed.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Laposa RR, Feeney L, Cleaver JE. Recapitulation of the cellular xeroderma pigmentosum-variant phenotypes using short interfering RNA for DNA polymerase H. Cancer Res. 2003;63:3909–12. [PubMed] [Google Scholar]
- Lehmann AR. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie. 2003;85:1101–11. doi: 10.1016/j.biochi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Lehmann AR, Kirk-Bell S, Arlett CF, Paterson MC, Lohman PH, De Weerd-Kastelein EA, et al. Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV-irradiation. Proc Natl Acad Sci USA. 1975;72:219–23. doi: 10.1073/pnas.72.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Boudsocq F, Plosky BS, Woodgate R, Yang W. Replication of a cis–syn thymine dimer at atomic resolution. Nature. 2003;424:1083–7. doi: 10.1038/nature01919. [DOI] [PubMed] [Google Scholar]
- Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- Maher VM, Ouellette LM, Curren RD, McCormick JJ. Caffeine enhancement of the cytotoxic and mutagenic effect of ultraviolet irradiation in a xeroderma pigmentosum variant strain of human cells. Biochem Biophys Res Commun. 1976a;71:228–34. doi: 10.1016/0006-291x(76)90272-2. [DOI] [PubMed] [Google Scholar]
- Maher VM, Ouellette LM, Curren RD, McCormick JJ. Frequency of ultraviolet light-induced mutations is higher in xeroderma pigmentosum variant cells than in normal human cells. Nature. 1976b;261:593–5. doi: 10.1038/261593a0. [DOI] [PubMed] [Google Scholar]
- Masutani C, Araki M, Yamada A, Kusumoto R, Nogimori T, Maekawa T, et al. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999a;18:3491–501. doi: 10.1093/emboj/18.12.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C, Kusumoto R, Iwai S, Hanaoka F. Mechanisms of accurate translesion synthesis by human DNA polymerase eta. EMBO J. 2000;19:3100–9. doi: 10.1093/emboj/19.12.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999b;399:700–4. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- McDonald JP, Levine AS, Woodgate R. The Saccharomyces cerevisiae RAD30 gene, a homologue of Escherichia coli dinB and umuC, is DNA damage inducible and functions in a novel error-free postreplication repair mechanism. Genetics. 1997;147:1557–68. doi: 10.1093/genetics/147.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki S, Kraemer KH. Xeroderma pigmentosum—bridging a gap between clinic and laboratory. Photodermatol Photoimmunol Photomed. 2001;17:47–54. doi: 10.1034/j.1600-0781.2001.017002047.x. [DOI] [PubMed] [Google Scholar]
- Moshell AN, Tarone RE, Newfield SA, Andrews AD, Robbins JH. A simple and rapid method for evaluating the survival of xeroderma pigmentosum lymphoid lines after irradiation with ultraviolet light. In Vitro. 1981;17:299–307. doi: 10.1007/BF02618141. [DOI] [PubMed] [Google Scholar]
- Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, et al. The Y-family of DNA polymerases. Mol Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000;55:1442–9. doi: 10.1212/wnl.55.10.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins JH, Kraemer KH, Flaxman BA. DNA repair in tumor cells from the variant form of xeroderma pigmentosum. J Invest Dermatol. 1975;64:150–5. doi: 10.1111/1523-1747.ep12533310. [DOI] [PubMed] [Google Scholar]
- Robbins JH, Kraemer KH, Lutzner MA, Festoff BW, Coon HG. Xeroderma pigmentosum. An inherited disease with sun sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Ann Intern Med. 1974;80:221–48. doi: 10.7326/0003-4819-80-2-221. [DOI] [PubMed] [Google Scholar]
- Rogan PK, Faux BM, Schneider TD. Information analysis of human splice site mutations. Hum Mutat. 1998;12:153–71. doi: 10.1002/(SICI)1098-1004(1998)12:3<153::AID-HUMU3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Schneider TD. Information content of individual genetic sequences. J Theor Biol. 1997a;189:427–41. doi: 10.1006/jtbi.1997.0540. [DOI] [PubMed] [Google Scholar]
- Schneider TD. Sequence walkers: a graphical method to display how binding proteins interact with DNA or RNA sequences. Nucleic Acids Res. 1997b;25:4408–15. doi: 10.1093/nar/25.21.4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoneczna A, McIntyre J, Skoneczny M, Policinska Z, Sledziewska-Gojska E. Polymerase eta is a short-lived, proteasomally degraded protein that is temporarily stabilized following UV irradiation in Saccharomyces cerevisiae. J Mol Biol. 2007;366:1074–86. doi: 10.1016/j.jmb.2006.11.093. [DOI] [PubMed] [Google Scholar]
- Svoboda DL, Briley LP, Vos JM. Defective bypass replication of a leading strand cyclobutane thymine dimer in xeroderma pigmentosum variant cell extracts. Cancer Res. 1998;58:2445–8. [PubMed] [Google Scholar]
- Tanioka M, Masaki T, Ono R, Nagano T, Otoshi-Honda E, Matsumura Y, et al. Molecular analysis of DNA polymerase eta gene in Japanese patients diagnosed as xeroderma pigmentosum variant type. J Invest Dermatol. 2007;127:1745–51. doi: 10.1038/sj.jid.5700759. [DOI] [PubMed] [Google Scholar]
- Taylor EM, Broughton BC, Botta E, Stefanini M, Sarasin A, Jaspers NG, et al. Xeroderma pigmentosum and trichothiodystrophy are associated with different mutations in the XPD (ERCC2) repair/transcription gene. Proc Natl Acad Sci USA. 1997;94:8658–63. doi: 10.1073/pnas.94.16.8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur M, Wernick M, Collins C, Limoli CL, Crowley E, Cleaver JE. DNA polymerase eta undergoes alternative splicing, protects against UV sensitivity and apoptosis, and suppresses Mre11-dependent recombination. Genes Chromosomes Cancer. 2001;32:222–35. doi: 10.1002/gcc.1186. [DOI] [PubMed] [Google Scholar]
- Vaisman A, Frank EG, Iwai S, Ohashi E, Ohmori H, Hanaoka F, et al. Sequence context-dependent replication of DNA templates containing UV-induced lesions by human DNA polymerase iota. DNA Repair (Amst) 2003;2:991–1006. doi: 10.1016/s1568-7864(03)00094-6. [DOI] [PubMed] [Google Scholar]
- Van Steeg H, Kraemer KH. Xeroderma pigmentosum and the role of UV-induced DNA damage in skin cancer. Mol Med Today. 1999;5:86–94. doi: 10.1016/s1357-4310(98)01394-x. [DOI] [PubMed] [Google Scholar]
- Vockley J, Rogan PK, Anderson BD, Willard J, Seelan RS, Smith DI, et al. Exon skipping in IVD RNA processing in isovaleric acidemia caused by point mutations in the coding region of the IVD gene. Am J Hum Genet. 2000;66:356–67. doi: 10.1086/302751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Woodgate R, McManus TP, Mead S, McCormick JJ, Maher VM. Evidence that in xeroderma pigmentosum variant cells, which lack DNA polymerase eta, DNA polymerase iota causes the very high frequency and unique spectrum of UV-induced mutations. Cancer Res. 2007;67:3018–26. doi: 10.1158/0008-5472.CAN-06-3073. [DOI] [PubMed] [Google Scholar]
- Wang YC, Maher VM, Mitchell DL, McCormick JJ. Evidence from mutation spectra that the UV hypermutability of xeroderma pigmentosum variant cells reflects abnormal, error-prone replication on a template containing photoproducts. Mol Cell Biol. 1993;13:4276–83. doi: 10.1128/mcb.13.7.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters HL, Seetharam S, Seidman MM, Kraemer KH. Ultraviolet hypermutability of a shuttle vector propagated in xeroderma pigmentosum variant cells. J Invest Dermatol. 1993;101:744–8. doi: 10.1111/1523-1747.ep12371686. [DOI] [PubMed] [Google Scholar]
- Woodgate R. A plethora of lesion-replicating DNA polymerases. Genes Dev. 1999;13:2191–5. doi: 10.1101/gad.13.17.2191. [DOI] [PubMed] [Google Scholar]
- Woodgate R, Singh M, Kulaeva OI, Frank EG, Levine AS, Koch WH. Isolation and characterization of novel plasmid-encoded umuC mutants. J Bacteriol. 1994;176:5011–21. doi: 10.1128/jb.176.16.5011-5021.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada A, Masutani C, Iwai S, Hanaoka F. Complementation of defective translesion synthesis and UV light sensitivity in xeroderma pigmentosum variant cells by human and mouse DNA polymerase eta (In Process Citation) Nucleic Acids Res. 2000;28:2473–80. doi: 10.1093/nar/28.13.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc Natl Acad Sci USA. 2007;104:15591–8. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda G, Nishi R, Watanabe E, Mori T, Iwai S, Orioli D, et al. In vivo destabilization and functional defects of the xeroderma pigmentosum C protein caused by a pathogenic missense mutation. Mol Cell Biol. 2007;27:6606–14. doi: 10.1128/MCB.02166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavuz S, Yavuz AS, Kraemer KH, Lipsky PE. The role of polymerase eta in somatic hypermutation determined by analysis of mutations in a patient with xeroderma pigmentosum variant. J Immunol. 2002;169:3825–30. doi: 10.4049/jimmunol.169.7.3825. [DOI] [PubMed] [Google Scholar]
- Yuasa M, Masutani C, Eki T, Hanaoka F. Genomic structure, chromosomal localization and identification of mutations in the xeroderma pigmentosum variant (XPV) gene. Oncogene. 2000;19:4721–8. doi: 10.1038/sj.onc.1203842. [DOI] [PubMed] [Google Scholar]
- Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat Immunol. 2001;2:537–41. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences used.
Table S2. Human pol-η gene: sequence and information content of normal and mutant splice sites.