

Abstract
Background
Ectodermal dysplasias are developmental disorders affecting tissues of ectodermal origin. To date, four different types of ectodermal dysplasia involving only hair and nails have been described. In an effort to understand the molecular bases of this form of ectodermal dysplasia, large Pakistani consanguineous kindred with multiple affected individuals has been ascertained from a remote region in Pakistan.
Objective
To identify the gene underlying the phenotype.
Methods
Microsatellite markers were genotyped in candidate regions and two point and multipoint parametric linkage analysis carried out.
Results
The disease locus was mapped to a 16.6 centimorgan region on chromosome 12q12–q14.1 (Zmax = 8.2), which harbours six type II hair keratin genes. DNA sequence analysis revealed a homozygous missense mutation in the hair matrix and cuticle keratin KRTHB5, leading to histidine substitution of a conserved arginine residue (R78H) located in the head domain.
Conclusions
This report provides the first direct evidence relating to the molecular pathogenesis of pure hair–nail ectodermal dysplasias.
Keywords: ectodermal dysplasias, hair, nail, hair keratins, missense mutation
Ectodermal dysplasia defines a family of disorders resulting in abnormal development of skin appendages (hair, nails, teeth, and sweat glands) during morphogenesis. Of the approximately 200 ectodermal dysplasia described so far, about 30 have been explained at the molecular level, with identification of the causative genes. Recently, Lamartine1 proposed that ectodermal dysplasias should be classified into four major subgroups according to the functions of the genes discovered: cell–cell signalling, cell adhesion, regulation of transcription, and development.
Ectodermal dysplasias that involve only hair and nails are uncommon. The majority of the hair and nail type have associated abnormalities, such as keratoderma or ichthyosis, skeletal problems, cardiac irregularities, mental or psychomotor retardation, cataracts, or haematological problems.2 Four different pure hair‐nail ectodermal dysplasias (MIM 602032) have been reported to date, the molecular bases of which are entirely unknown. Calzavara‐Pinton et al3 reported a family with autosomal recessive ectodermal dysplasia with clinical features of onychodysplasia and alopecia involving the scalp, beard, axillary and pubic hair, and absence of eyebrows and eyelashes. In a Brazilian family with autosomal dominant pure hair‐nail dysplasia, the affected individuals showed hypotrichosis of the whole body and short and fragile nails.4 Barbareschi et al5 described an autosomal dominant pure hair‐nail dysplasia in a mother and son. Both the affected individuals had hypotrichosis limited to the bilateral fronto‐temporal areas of the scalp. Micronychia and onychorrhexis were the main findings in the finger nails and toe nails. Harrison and Sinclair6 reported a hair‐nail ectodermal dysplasia in a three year old girl. Clinical features of this affected girl included generalised scalp hypotrichosis, absent eyebrows and eyelashes, and nail dystrophy in all digits.
To understand the molecular bases of pure hair‐nail ectodermal dysplasia, we have investigated a large Pakistani kindred with a unique hair‐nail disorder with no evidence of any other abnormality. Through parametric linkage analysis using microsatellite markers, we have localised the gene responsible for this autosomal recessive form of hair‐nail disorder to chromosome 12q12–q14.1, which contains type II hair keratin genes. Further, we have identified a causative mutation (R78H) in the hair matrix and cuticle keratin KRTHB5 gene.
Methods
Human subjects
A large inbred kindred of Pakistani origin was studied, in which four males and females each were affected with ectodermal dysplasia of hair and nail type (fig 1). Before the start of the study, approval was obtained from the Quaid‐i‐Azam University institutional review board. In addition, informed consent was obtained from all the family members who participated in the study. The family members rarely marry outside the community and consequently consanguineous unions are common. The pedigree (fig 1) provided convincing evidence of autosomal recessive mode of inheritance, and consanguineous loops accounted for all the affected persons being homozygous for the mutant allele. All of the affected individuals underwent examination at Department of Dermatology, Pakistan Institute of Medical Sciences (PIMS), Islamabad.

Figure 1 Drawing of the pedigree segregating ectodermal dysplasia of hair and nail type. Affected males and females are indicated by filled squares and circles, respectively. Double lines between figures are representative of consanguineous unions. Haplotypes for the most closely linked markers are shown below each symbol.
Extraction of genomic DNA and genotyping
Venous blood samples were obtained from 20 family members, including the eight affected individuals. Genomic DNA was extracted from whole blood following a standard protocol, quantified by spectrophotometric measurement of optical density at 260 nm and diluted to 40 ng/μl for amplification by polymerase chain reaction (PCR). PCR amplification of microsatellite markers was carried out according to a standard procedure in a total volume of 25 μl, containing: 40 ng genomic DNA, 20 pmol of each primer, 200 μM of each dNTP, 1 U of Taq DNA polymerase (MBI Fermentas, Sunderland, UK), and 1×PCR buffer (MBI‐Fermentas). PCR was carried out for 35 cycles, with the following thermal cycling conditions: 95°C for one minute, 57°C for one minute, 72°C for one minute, followed by final extension at 72°C for seven minutes in a thermal cycler 9600 (Perkin Elmer, Norwalk, Connecticut, USA). PCR products were resolved on 8% non‐denaturing polyacrylamide gel, along with the appropriate allelic ladder, and genotypes were assigned by visual inspection. The family was tested for linkage by using microsatellite markers tightly linked to candidate genes involved in related phenotypes. These included: type I keratin genes at 17q21.2 (D17S1299, D17S800, D17S930, D17S934, D17S791, D17S797); type II keratin genes at 12q13.13 (D12S297, D12S368, D12S398, D12S90); ectodermal dysplasia type 2 (ED2, MIM 129500) gene gap junction protein β‐6 (GJB6, MIM 604418) at 13q12.11 (D13S1316, D13S175, D13S633, D13S250, D13S787); ectodermal dysplasia type 3 (ED3, MIM 129490) gene ectodysplasin1 anhidrotic receptor (EDAR, MIM 604095) at 2q11–q13 (D2S1343, D2S2954, D2S340, D2S1889, D2S1893, D2S1891); ectodermal dysplasia type 4 (ED4, MIM 225060) gene poliovirus receptor‐like 1 (PVRL1, MIM 600644) at 11q23.3 (D11S1885, D11S4171, D11S4129, D11S924, D11S1299); and human hairless (HR, MIM 225060) gene at 8p12 (D8S298, D8S1786, D8S1048).
Linkage analysis
For two point linkage analysis we used the MLINK program of the FASTLINK computer package.7 The microsatellite markers were ordered according to their sequence based physical positions8 and the genetic map distances were estimated using MAP‐O‐MAT.9 Implementing the genetic map distances obtained from MAP‐O‐MAT, multipoint linkage analysis was carried out using ALLEGRO.10 Haplotypes were constructed using SIMWALK2.11,12 For the analysis, an autosomal recessive mode of inheritance with complete penetrance and a disease allele frequency of 0.001 were used.
Mutation analysis
The candidate region of the ectodermal dysplasia locus identified in the present study harbours type II hair keratin genes involved in the development of ectodermal appendages. To screen for mutations in the hair keratin genes KRTHB1 (MIM 602153), KRTHB2 (MIM 601078), KRTHB3 (MIM 602765), KRTHB4 (MIM 602766), KRTHB5 (MIM 602767), and KRTHB6 (MIM 601928), exons and splice junctions were PCR amplified from genomic DNA. PCR products were purified using the Marligen rapid PCR purification system (Marligen Biosciences Inc, Ijamsville, Maryland, USA) and were sequenced in ABI Prism 310 automated sequencer, using the Big Dye Terminator cycle sequencing kit (PE Applied Biosystems, Foster City, California, USA) following purification in Centri‐Sep spin columns (Applied Biosystems). Primer sequences to amplify exons and splice junctions of KRTHB1, KRTHB2, KRTHB3, KRTHB4, RKTHB5, and KRTHB6 genes are available on request.
Results
Clinical findings
Clinical information was obtained for all family members with particular attention to skin, dentition, sweating, nails, scalp, and body hair. All the affected individuals had total alopecia and nail dystrophy since birth. In affected individuals, hairs were absent from the scalp, face, chest, arms, and legs (fig 2); they were born completely devoid of eyebrows and eyelashes, and never developed axillary and pubic hair. In the affected individuals, nail abnormalities were present in all digits. Both finger nails and toe nails had a dystrophic appearance (fig 3). The patients were in good general health, sweated normally and were of normal intelligence. Eye abnormalities, ichthyosis, oral leucokeratosis, dental anomalies, palmoplantar keratosis and pigmentation were absent. Results of routine laboratory tests, including white blood cell count and granulocyte function, were normal. Heterozygous carrier individuals had normal hair and nails, and were clinically indistinguishable from genotypically normal individuals.
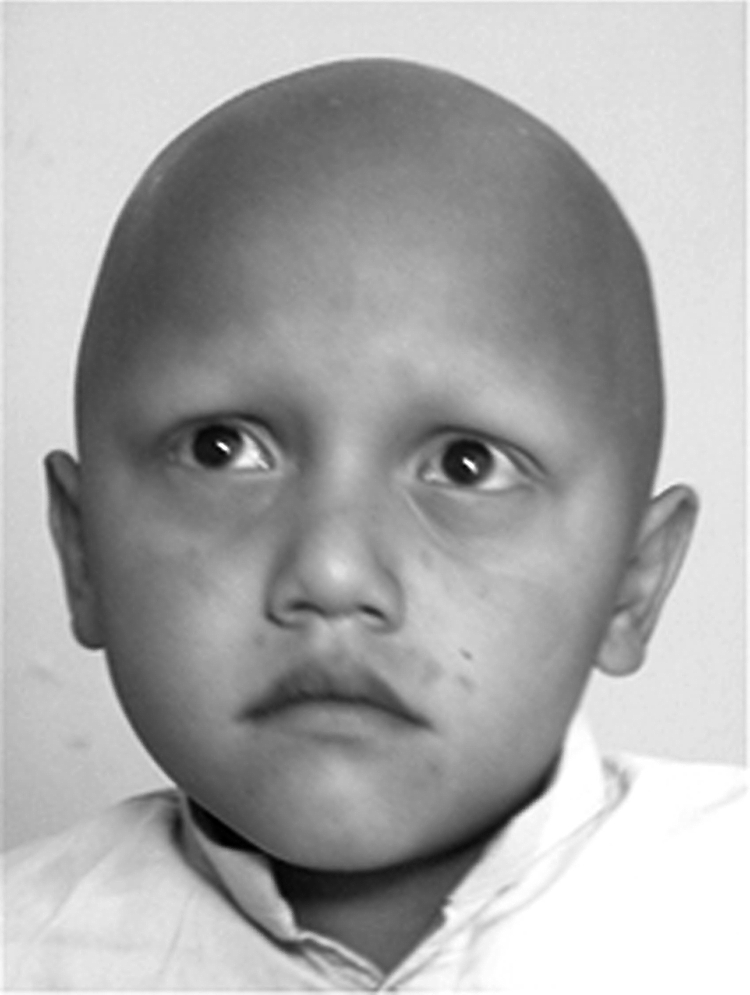
Figure 2 Clinical findings in ectodermal dysplasia of hair and nail type. Note the complete absence of hair on the scalp of an affected male (VI‐1). The eyebrows and eyelashes are completely missing. Written permission for the publication of this photograph was obtained from the child's parents.
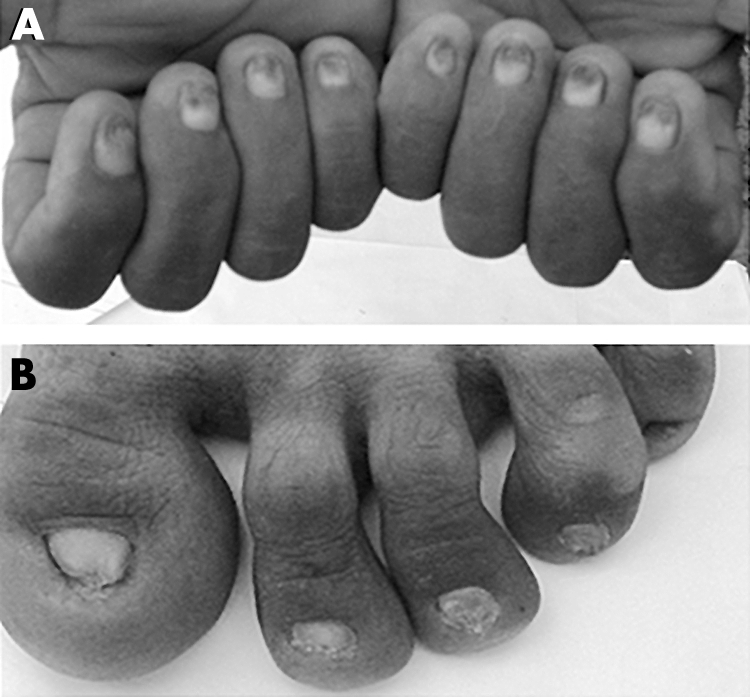
Figure 3 Clinical findings in ectodermal dysplasia of hair and nail type. (A) Phenotypic appearance of finger nails of an affected male (IV‐4). (B) Dystrophic toe nails of the same individual.
Localisation of hair‐nail ectodermal dysplasia to chromosome 12
To identify the gene underlying ectodermal dysplasia of hair and nail phenotype, a candidate gene approach was used. We carried out parametric linkage analysis with microsatellite markers mapping to candidate regions. These candidate regions were selected because genes within these regions are involved in related phenotypes. These included type I keratin genes at 17q21.2; type II keratin genes at 12q13.13; GJB6 gene at 13q12.11; EDAR gene at 2q11–q13; PVRL1 gene at 11q23.3; and HR gene at 8p12.
Linkage was excluded for all regions, with the exception of the type II keratin gene cluster on chromosome 12q13.13. A maximum two point logarithm of odds (LOD) ratio for linkage score (Zmax) of 4.54 at zero recombination was achieved with the marker D12S398 (table 1). Multipoint analysis also supported linkage to this region, with a maximum multipoint LOD score of 8.2 obtained at marker D12S398. The three unit support interval and region of homozygosity is flanked by markers D12S291 and D12S90, which is a 16.6 cM region, which corresponds to 15.0 Mb on the sequence based physical map.8
Table 1 Two point logarithm of odds (LOD) score results between the ectodermal dysplasia locus and chromosome 12 markers.
| Marker | cM* | Physical position† | LOD score at recombination fraction θ = | |||||
|---|---|---|---|---|---|---|---|---|
| 0.00 | 0.01 | 0.05 | 0.10 | 0.20 | 0.30 | |||
| D12S1034 | 0.0 | 26,139,414 | −4.34 | −1.87 | −0.61 | −0.20 | 0.03 | 0.05 |
| D12S1337 | 2.7 | 28,006,325 | −4.34 | −1.49 | −0.33 | −0.02 | 0.08 | 0.06 |
| D12S87 | 6.3 | 30,279,247 | 1.93 | 1.86 | 1.61 | 1.31 | 0.78 | 0.38 |
| D12S2080 | 7.7 | 33,305,760 | −4.34 | 2.52 | 2.74 | 2.40 | 1.52 | 0.69 |
| D12S291 | 9.6 | 41,688,530 | −4.34 | −2.49 | −1.15 | −0.64 | −0.23 | −0.08 |
| D12S85 | 15.6 | 45,622,990 | 2.36 | 2.29 | 2.02 | 1.69 | 1.08 | 0.56 |
| D12S297 | 20.1 | 50,899,321 | 3.21 | 3.12 | 2.77 | 2.34 | 1.53 | 0.80 |
| D12S368 | 20.1 | 50,917,731 | 2.77 | 2.69 | 2.36 | 1.95 | 1.19 | 0.56 |
| D12S398 | 24.7 | 51,483,354 | 4.54 | 4.41 | 3.92 | 3.30 | 2.11 | 1.06 |
| D12S90 | 26.2 | 56,710,412 | −4.34 | 2.28 | 2.49 | 2.20 | 1.37 | 0.62 |
| D12S1056 | 26.2 | 58,832,288 | 1.68 | 1.64 | 1.47 | 1.26 | 0.86 | 0.50 |
Mutational analysis
According to University of California Santa Cruz (UCSC) Genome Browser on Human (May 2004 assembly), type II hair keratin gene cluster lies in a 0.12 Mb region between markers D12S368 and D12S398 on chromosome 12q13.13. This region contains six type II hair keratin genes: KRTHB1, KRTHB2, KRTHB3, KRTHB4, KRTHB5, and KRTHB6. To search for a pathogenic mutation in our family, coding exons and splice sites of all of these genes were sequenced.
Sequence analysis of exon 1 of the KRTHB5 gene from all the affected individuals (IV‐4, V‐2, V‐3, V‐4, VI‐1, VI‐2, VI‐3, VI‐6) revealed a G to A transition at nucleotide position 233 (fig 4), resulting in arginine to histidine (R78H) amino acid substitution. Arginine at amino acid position 78 is highly conserved in the head domains of type II hair keratins in mouse, human, and sheep.13 This mutation was present in the heterozygous state in obligate carriers within the family (fig 4). To ensure that the mutation does not represent a neutral polymorphism in the Pakistani population, a panel of 100 unrelated unaffected individuals (200 chromosomes) was screened for mutation using PCR followed by sequencing, and the mutation was not identified outside the family (fig 4). Although it has been reported that hair keratin KRTHB2 lacks expression in nails14 and KRTHB4 shows expression only in the filiform papillae of the dorsal tongue,15 even these two genes were sequenced to exclude the possibility that mutation R78H is a rare polymorphism.
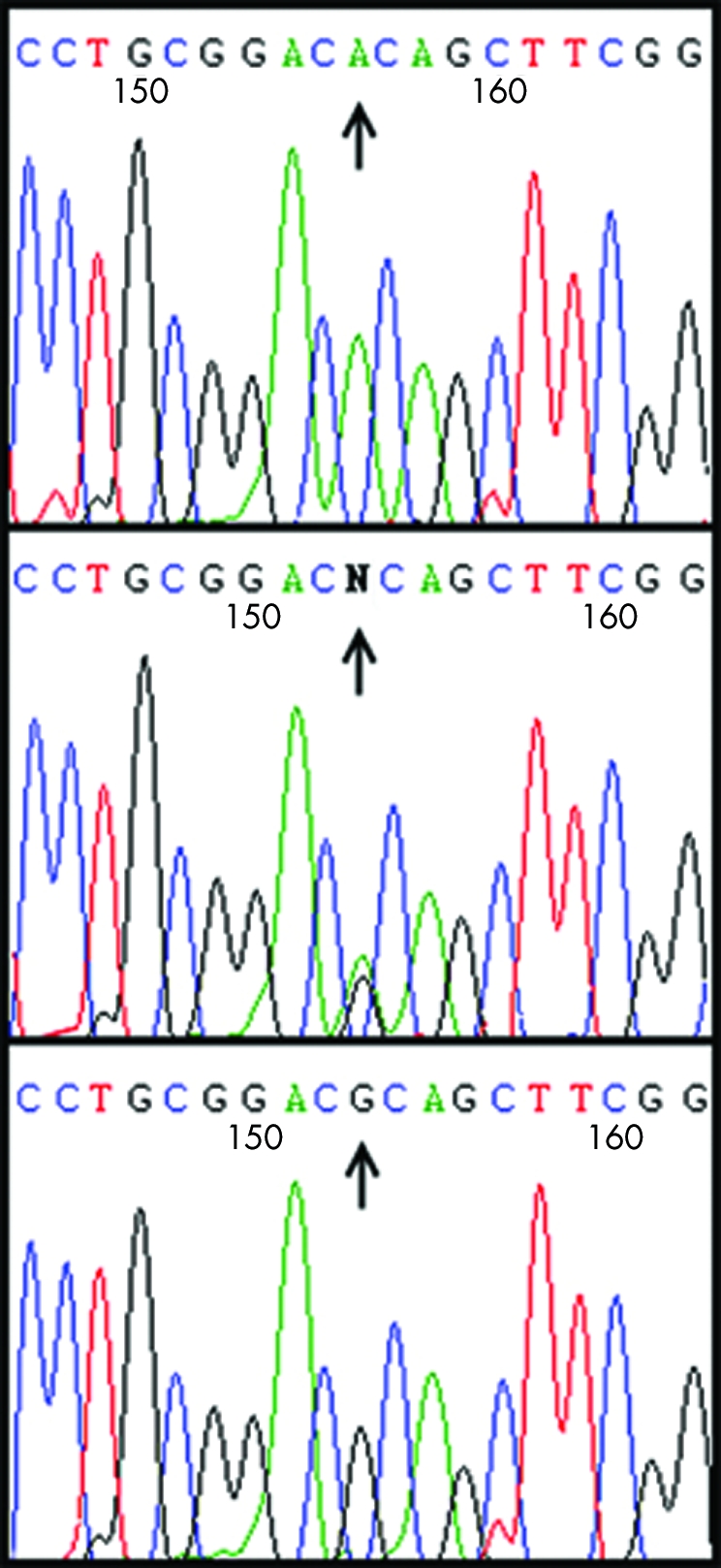
Figure 4 Mutation analysis of exon 1 of KRTHB5 gene. Sequence analysis of the gene from a homozygous affected individual (top panel), a heterozygous carrier (middle panel), and a control individual (bottom panel). The homozygous substitution of a nucleotide G→A at position 233 is shown in the mutant sequence (top panel).
Discussion
Ectodermal dysplasia with hair and nail abnormalities is a rare congenital disorder that is inherited as an autosomal recessive condition. Clinically, it is characterised by complete alopecia and dystrophy of finger nails and toe nails. In an effort to understand the molecular basis of this recessive form of ectodermal dysplasia, we studied a large Pakistani consanguineous family containing four affected males and four affected females. A candidate gene approach was used to localise the disease locus segregating in this family. Significant evidence of linkage was obtained for markers linked to the type II keratin gene cluster on chromosome 12q13.13. The candidate interval for recessive ectodermal dysplasia identified in the present family corresponds to a physical map distance of 15.0 Mb.8
Various hereditary diseases of skin, skin appendages, and oral mucosa have been shown to be caused by mutations in keratin genes.16 The keratin multigene family comprises two groups: the epithelial keratins, which are expressed in various types of epithelia, and the hair keratins, which are involved in the formation of hard keratinised structures such as hair and nail.15 Fifty four functional keratin genes (27 type I and 27 type II) have been identified in the human genome sequence database.17,18 The type II keratin gene domain, spanning 780 kb region on chromosome 12q13.13, contains 27 functional keratin genes including 21 epithelial and six hair keratin genes.15,17,18,19 Structurally, keratins have a central rod domain consisting of four highly conserved α‐helical subdomains, 1A, 1B, 2A, and 2B, separated by three non‐helical linkers, L1, L12, and L2. Non‐helical head and tail domains flank the rod domain and it is these domains that differ most between members of the keratin family. These end domains are made up of E1 (head) and E2 (tail) extreme end domains, the variable V1 (head) and V2 (tail) domains, and in type II keratins only, the homology H1 (head) and H2 (tail) domains. At the start and end of the rod domain are two short regions of highly conserved amino acids known as helix initiation motif and helix termination motif, respectively. Both type I and type II keratins form heterodimers, which assemble into tetramers and higher order structures that eventually form intermediate filaments of the cytoskeleton of mammalian cells. The pathology caused by mutation in a keratin gene is primarily determined by the tissue specific expression pattern of that keratin.20,21
The linkage interval of the hair‐nail ectodermal dysplasia locus on chromosome 12q13.13, identified in the present study harbours hair keratin genes KRTHB1, KRTHB2, KRTHB3, KRTHB4, KRTHB5, and KRTHB6. Considering the reported expression pattern of these keratins in hair and nails,14,15 KRTHB1, KRTHB2, KRTHB3, KRTHB4, KRTHB5, and KRTHB6 genes were selected for screening for pathogenic mutation. Sequence analysis of exon 1 of KRTHB5 keratin revealed a homozygous R78H mutation in all affected individuals. The codon for arginine 78 in KRTHB5 is CGC. The G to A transition leading to R78H amino acid substitution might be a result of methylated CpG deamination mutation of a 5‐methyl cytosine on the antisense strand, which leads to a CG to CA transition in the sense strand. Methylated CpG sequences are known to have a higher mutation rate than other dinucleotides.22 Arginine to histidine mutation has previously been reported in type I keratins K10 and K14, and is considered to be a hotspot mutation.23
Most of the reported pathogenic mutations in keratin genes are dominant missense mutations that alter amino acids in the helix initiation or termination motif. Mutations affecting these hot spot motifs of K5 or K14 are associated with a more severe epidermolysis bullosa simplex (EBS).20 However, mutations outside the hotspots have also been reported to affect protein stability and cause severe EBS phenotype.24,25,26,27,28 The homozygous missense mutation R78H in the V1 (head) domain of KRTHB5 identified here results in complete alopecia and nail dystrophy in all digits, thus further suggesting that mutation in the head domain, located outside the hot spot region, could lead to a severe phenotype.
Most of the dominant mutations found so far in keratins interfere with the assembly of intermediate filaments. R78H is a recessive mutation and produces a normal phenotype in its heterozygous state. This suggests that interference by the R78H mutation with assembly is probably not significant. However, the mutation may influence the availability of keratin for assembly—that is, it affects post‐translational processing or mRNA stability.
Parry et al13 have recently proposed a dynamic model to explain the role of the head domain of hair keratins in the organisation of the 1A helix of the rod domain. These investigators have identified several contiguous nonapeptide quasi‐repeats in the amino acid sequences of the head domains of type II hair keratins. These sequences strongly indicated the presence of alternating β‐strands and β‐turns to generate a four strand (or a five strand in KRTHB5) antiparallel β‐sheet in the head region. The model revealed that the twisted β‐sheet, postulated to be the key functional element in the head domain, wraps around and interacts strongly with the 1A segment in such a way that it could stabilise the coiled‐coil conformation for 1A. These predictions are consistent with the results presented by Strelkov et al29 indicating that segment 1A and the attached head domains of vimentin form dimers, whereas the equivalent peptide lacking the head domain forms only monomers. The R78H mutation affects a highly conserved residue located in one of the nonapeptide quasi‐repeats in the head domain of the keratin KRTHB5. This mutation could therefore affect the efficacy of wrap around of the head domain and hence affect its interaction with segment 1A. This could destabilise the coiled‐coil structure of 1A helix, which is important in intermediate filament assembly. Hence, it is speculated that the unavailability of KRTHB5 protein for assembly causes the abnormal hair and nail phenotype.
Langbein et al15 and Perrin et al14 have studied the expression pattern of 6 type‐II hair keratins in the hair follicle and nail, respectively. Immunohistochemical studies by Langbein et al have shown that type II hair keratins KRTHB5 and KRTHB2 define early stages of hair differentiation in the matrix (KRTHB5) and cuticle (KRTHB5 and KRTHB2), respectively.15 The hair keratins KRTHB1, KRTHB3, and KRTHB6 are expressed at an advanced stage of differentiation in the hair cortex cells while KRTHB4 is expressed in filiform papillae of the dorsal tongue rather than in the hair and nails. Perrin et al14 have reported that in the nail, hair keratin KRTHB5 is expressed in the epithelial tissue compartment comprised of apical and ventral matrix, and in the uppermost cell layers of the basal compartment of the ventral matrix and is absent from the compartment comprised of eponychium, nail bed epithelium, and hyponychium. Two other keratins, KRTHB1 and KRTHB6, similar to their sequential expression in the hair cortex, are consecutively expressed in the entire keratogenous zone of both the ventral and apical matrix of the nail.
The hair keratin KRTHB5 is expressed in matrix and precortex cells and in the entire hair cuticle.15 In the matrix and lowermost hair cuticle, two type I hair keratins KRTHA2 (MIM 602760) and KRTHA5 (MIM 602764) compete with KRTHB5 for filament formation.15 It is apparent that KRTHB5 mutation leads to unavailability of functional type II hair keratins to pair with its corresponding type I partners. Therefore, it is suggested that lack of functional KRTHB5 protein affects intermediate filament assembly, leading to the abnormal hair phenotype.
Electronic database information
Accession numbers and URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Bioinformatics Web Site, http://genome.ucsc.edu/
Marker based linkage mapping server, http://compgen.rutgers.edu/mapomat/
Acknowledgements
We thank the family members for their invaluable cooperation. We thank Professor Dr Anwar Ahmad from Department of Pathology; Dr Ikram ul Haque, and Dr Maqsood Anwar from Department of Dermatology, Pakistan Institute of Medical Sciences (PIMS), Islamabad, for their help with diagnosis of the disease. This study was supported by grant from Higher Education Commission (HEC), Islamabad, Pakistan.
Footnotes
Conflicts of interest: none declared
References
- 1.Lamartine J. Towards a new classification of ectodermal dysplasias. Clin Exp Dermatol 200328351–355. [DOI] [PubMed] [Google Scholar]
- 2.Pinheiro M, Freire‐Maia N. Ectodermal dysplasias: a clinical classification and a causal review. Am J Med Genet 199453153–162. [DOI] [PubMed] [Google Scholar]
- 3.Calzavara‐Pinton P, Carlino A, Benetti A, De Panfilis G. Pili torti and onychodysplasia: report of a previously undescribed hidrotic ectodermal dysplasia. Dermatologica 1991182184–187. [PubMed] [Google Scholar]
- 4.Pinheiro M, Freire‐Maia N. Hair‐nail dysplasia – a new pure autosomal dominant ectodermal dysplasia. Clin Genet 199241296–298. [DOI] [PubMed] [Google Scholar]
- 5.Barbareschi M, Cambiaghi S, Crupi A C, Tadini G. Family with “pure” hair‐nail ectodermal dysplasia. Am J Med Genet 19977291–93. [DOI] [PubMed] [Google Scholar]
- 6.Harrison S, Sinclair R. Hypotrichosis and nail dysplasia: a novel hidrotic ectodermal dysplasia. Aust J Dermatol 200445103–105. [DOI] [PubMed] [Google Scholar]
- 7.Cottingham R, Indury R M, Schaffer A A. Faster sequential genetic linkage computations. Am J Hum Genet 199353252–263. [PMC free article] [PubMed] [Google Scholar]
- 8.International Human Genome Sequencing Consortium Initial sequence and analysis of the human genome. Nature 2001409860–921. [DOI] [PubMed] [Google Scholar]
- 9.Kong X, Matise T C. MAP‐O‐MAT: internet‐based linkage mapping. Bioinformatics 200521557–559. [DOI] [PubMed] [Google Scholar]
- 10.Gudbjartsson D F, Jonasson K, Frigge M L, Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat Genet 20022512–13. [DOI] [PubMed] [Google Scholar]
- 11.Weeks D E, Sobel E, O'Connell J R, Lange K. Computer programs for multilocus haplotyping of general pedigrees. Am J Hum Genet 1995561506–1507. [PMC free article] [PubMed] [Google Scholar]
- 12.Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker‐sharing statistics. Am J Hum Genet 1996581323–1337. [PMC free article] [PubMed] [Google Scholar]
- 13.Parry D A D, Marekov L N, Steinert P M, Smith T A. A role for the 1A and L1 rod domain segments in head domain organization and function of intermediate filaments: structural analysis of trichocyte keratin. J Struct Biol 200213797–108. [DOI] [PubMed] [Google Scholar]
- 14.Perrin C, Langbein L, Schweizer J. Expression of hair keratins in the adult nail unit: an immunohistochemical analysis of the onychogenesis in the proximal nail fold, matrix and nail bed. Br J Dermatol 2004151362–371. [DOI] [PubMed] [Google Scholar]
- 15.Langbein L, Rogers M A, Winter H, Praetzel S, Schweizer J. The catalog of human hair keratins. II. Expression of the six type II members in the hair follicle and the combined catalog of human type I and II keratins. J Biol Chem 200127635123–35132. [DOI] [PubMed] [Google Scholar]
- 16.Rugg E L, Leigh I M. The keratins and their disorders. Am J Med Genet 2004131C4–11. [DOI] [PubMed] [Google Scholar]
- 17.Hesse M, Zimek A, Weberb K, Magin T M. Comprehensive analysis of keratin gene clusters in humans and rodents. Eur J Cell Biol 20048319–26. [DOI] [PubMed] [Google Scholar]
- 18.Rogers M A, Edler L, Winter H, Langbein L, Beckmann I, Schweizer J. Characterization of new members of the human Type II keratin gene family and a general evaluation of the keratin gene domain on chromosome 12q13.13. J Invest Dermatol 2005124536–544. [DOI] [PubMed] [Google Scholar]
- 19.Rogers M A, Winter H, Langbein L, Wolf C, Schweizer J. Characterization of a 300 kb region of human DNA containing the type II hair keratin gene locus. J Invest Dermatol 2000114464–472. [DOI] [PubMed] [Google Scholar]
- 20.Porter R M, Lane E B. Phenotypes, genotypes, and their contribution to understanding keratin function. Trends Genet 200319278–285. [DOI] [PubMed] [Google Scholar]
- 21.Coulombe P A, Omary M B. “Hard” and “soft” principles defining the structure, function and regulation of keratin intermediate filaments. Curr Opin Cell Biol 200214110–122. [DOI] [PubMed] [Google Scholar]
- 22.Cooper D N, Krawczak M. Cytosine methylation and the fate of CpG dinucleotides in vertebrate genomes. Hum Genet 198983181–188. [DOI] [PubMed] [Google Scholar]
- 23.Smith F J D. The molecular genetics of keratin disorders. Am J Clin Dermtol 20034347–364. [DOI] [PubMed] [Google Scholar]
- 24.Hovnanian A, Pollack E, Hilal L, Rochat A, Prost C, Barrandon Y, Goossens M. A missense mutation in the rod domain of keratin 14 associated with recessive epidermolysis bullosa simplex. Nat Genet 19933327–332. [DOI] [PubMed] [Google Scholar]
- 25.Humphries M M, Sheils D M, Farrar G J, Kumar‐Singh R, Kenna P F, Mansergh F C, Jordan S A, Young M, Humphries P. A mutation (Met to Arg) in the type I keratin (K14) gene responsible for autosomal dominant epidermolysis bullosa simplex. Hum Mutat 1993237–42. [DOI] [PubMed] [Google Scholar]
- 26.Humphries M M, Mansergh F C, Kiang A S, Jordan S A, Sheils D M, Martin M J, Farrar G J, Kenna P F, Young M M, Humphries P. Three keratin gene mutations account for the majority of dominant simplex epidermolysis bullosa cases within the population of Ireland. Hum Mutat 1996857–63. [DOI] [PubMed] [Google Scholar]
- 27.Galligan P, Listwan P, Siller G M, Rothnagel J A. A novel mutation in the L12 domain of keratin5 in the Köbner variant of epidermolysis bullosa simplex. J Invest Dermatol 1998111524–527. [DOI] [PubMed] [Google Scholar]
- 28.Liovic M, Stojan J, Bowden P E, Gibbs D, Vahlquist A, Lane E B, Komel R. A novel keratin 5 mutation (K5V186L) in a family with EBS‐K: a conservative substitution can lead to development of different disease phenotypes. J Invest Dermatol 2001116964–969. [DOI] [PubMed] [Google Scholar]
- 29.Strelkov S V, Herrmann H, Geisler N, Lustig A, Ivaninskii S, Zimbelmann R, Burkhard P, Aebi U. Divide‐and‐conquer crystallographic approach towards an atomic structure of intermediate filaments. J Mol Biol 2001306773–781. [DOI] [PubMed] [Google Scholar]