

Abstract

The disilylation of α-amino acids 1 to provide 2 (72–87%) was achieved without racemization. An unprecedented borane-mediated semi-reduction strategy was devised to convert 2 to stable, isolable oxazaborolidines 3 (100%) which were hydrolyzed to provide 5 (49–60%) as pure, stable compounds. Analysis of the Mosher amides (8) of the γ-amino esters 7 reveals that ≤2% racemization occurs in the 1→8 conversions.
The synthetically versatile α-amino aldehydes have found numerous applications in the construction of important pharmaceuticals, natural products and modified proteins.1,2 These polyfunctional compounds also can function as transition state analogue inhibitors for various proteases.3 Because the free amino aldehydes undergo rapid self-condensation, numerous N-protected derivatives have been reported with the most common being N-alkoxycarbonyl (e.g., Boc, Fmoc, benzyloxycarbonyl (Z)), N-triarylmethyl (e.g., trityl, 9-phenylfluoren-9-yl (PhFl)) or N,N-dibenzyl.1 Semi-reductive strategies have been devised for the conversion of esters or amide derivatives of α-amino acids to these aldehydes.1a,4a However, these procedures can give impure products which contain both esters and alcohols.4 Thus, they are more commonly produced from the reduction of α-amino acid derivatives to the corresponding alcohols followed by their selective oxidation to the desired aldehydes.1
Racemization is a major issue when electron-withdrawing groups are employed in the N-substituted amino aldehydes.1,5 Moreover, these intermediates exhibit both thermal and chromatographic instability. Thus, they are generally isolated and either used immediately or stored in the cold.4b In fact, this is a general problem in even N,N-dibenzyl derivatives which decompose with chromatographic purification and storage.1b In this case, the removal of the benzyl groups through catalytic hydrogenation can also restrict the potential synthetic applications of these intermediates.1b,6 Rapoport’s use of the N-PhFl protection largely solved these racemization and purification problems.7 However, the installation of this group employs stoichiometric quantities of the environmentally unfriendly Pb(NO3)2, and its removal requires harsh conditions (e.g., TFA, 8 h, 80 °C or Li/NH3 (l)). Thus, the appropriate precursors to these aldehydes are not commercially available and these groups are rarely used.
For some time, we have had an interest in new chemistry orchestrated through the triisopropylsilyl (TIPS) substitution.8 The highly compact and effective steric bulk of the triisopropyl substitution on silicon not only retards reactions at the silyl center, but also at adjacent centers. We felt that the TIPS group should combine ease of installation and removal with effective steric bulk which impedes the racemization of enolizable amino aldehydes (i.e., 5). Consideration of these properties and the above limitations for the known α-amino aldehydes led us to examine the potential of the bulky, electron-donating TIPS group to provide thermally stable and isolable N-protected α-amino aldehydes (5) in enantiomerically and chemically pure form. We envisaged the N,O-bis-(triisopropylsilyl)-α-amino acids 2 as convenient precursors to 5 (Scheme 1). While both TIPS amines and esters are known,8a the only reported N,O-disilylated α-amino acids are the hydrolytically unstable trimethylsilyl derivatives.9 We expected this process to be straightforward. However, considerable effort was required to find conditions which provide an efficient route to 2 without its partial racemization. Fortunately, through the slow addition of TIPSOTf to 1/(i-Pr)2NEt in refluxing THF, the clean 1→2 conversion was achieved (Table 1). The unwanted formation of polytetrahydrofuran was completely avoided through this protocol. More significantly, the inclusion of the bulky Hünig’s base in the procedure provided the key to avoiding any loss of optical purity in the product 2 even for challenging systems such as O-benzylserine (2f) or phenylglycine (2g). This is a major issue for these examples when DBU is used as the base.
Scheme 1.

Table 1.
N-TIPS-α-Amino Aldehydes 5 from 1
| R | series | 2 (%)a | 3b (%) (c/t)c | 5 (%)a |
|---|---|---|---|---|
| Me | a | 74 | 100 (31/69) | 56 |
| Prd | b | 72 | 100 (18/82) | 56 |
| (CH2)2SMe | c | 72 | 100 (13/87) | 56 |
| i-Bu | d | 82 | 100 (14/86) | 54 |
| Bn | e | 87 | 100 (20/80) | 52 |
| CH2OBn | f | 82 | 100 (20/80) | 50 |
| Ph | g | 85 | 100 (25/75) | 49 |
Yields of isolated analytically pure material.
The yields for 3 from this process were quantitative (100 ± 2%).
The cis isomer exhibits J(H4–H5) = 5–6 Hz while the trans isomer has a negligible 3-bond H-H coupling J(H4–H5) ~ 0 Hz.
The D-amino acid was also silylated to give D-2b (83%) and reduced to D-3b (100%, c/t = 15/85) and hydrolyzed to D-5b (60%).
With the highly soluble and stable silyl derivatives 2 in hand, we chose to examine their semi-reduction with borane-dimethyl sulfide complex (DMSB). While DIBAL-H is commonly employed for related processes with amino esters,1a,4a to our knowledge, the analogous borane-based process is unprecedented. TIPS esters specifically are fully reduced to the corresponding alcohol when heated neat with 1 mol equiv of DMSB. In fact, when a 1:1:1 mixture of 2e, PhCH2COOTIPS and DMSB is heated, only the PhCH2COOTIPS is reduced (to PhCH2CH2OH) with 2e being unaffected. With the bulky 2°-amine present in 2, after the initial “hydroboration” of the carbonyl, we view the BH2 moiety as reversibly complexing this amine, ultimately reacting further to produce hydrogen and the oxazaborolidine 3. This reaction diminishes both the Lewis acidity and the mobility of the boron atom. Thus, 3 is stable, showing no tendency to undergo β-elimination of a BOTIPS moiety to generate an aldehydic carbonyl group which would be further reduced. The oxazaborolidines 3 are produced quantitatively as cis/trans mixtures (Table 1). These isomers exhibit a number of clearly resolved signals in both the 1H- and 13C NMR spectra which can be used to evaluate this ratio in each case (see Supporting Information). Interestingly, while these BH oxazaborolidines evidently exist as monomers (ν(B-H) = 2550–2580 cm−1, 11B NMR δ 31–33), we were unable to observe a doublet in the 1H-coupled 11B NMR spectra of 3. This phenomenon has been observed by Corey in related systems.10 Remarkably, despite containing the mixed acetal moiety, 3 is stable and can be stored under nitrogen for months without significant decomposition!
Very mild reaction conditions were developed to effect the clean hydrolysis of 3 through 4 to provide the pure α-amino aldehydes 5. This was accomplished through the rate-controlled addition of a solution of water (3 equiv, 1.5 M in THF) to 3 (0.50 M in THF) with its mixed acetal, B—N and B—H functional groups. Dilution with ethyl acetate and rapid filtration through activated (I) dry neutral alumina removes the B(OH)3, giving a mixture of 5, TIPSOH and an intermediate hemiacetal 4 which we identified through NMR experiments.11 After 4–5 h, 4 collapses cleanly to 5 and TIPSOH. Subsequent concentration and rapid distillation gives a mixture of TIPSOH and the aldehyde.12 Careful distillation to remove the TIPSOH (bp 40–42 °C, 0.4 mm Hg) gives pure 5 in good yields (49–60%). Notably, the N-TIPS protection faithfully serves its function to preserve the chemical and optical purity of 5 even when it is filtered through alumina at ambient temperatures. Moreover, these aldehydes can be stored in the cold (−20 °C) under a nitrogen atmosphere for weeks without significant decomposition. Skeletal rearrangements have not been observed in 5 even during filtrations through alumina, distillations or the other manipulations employed in their isolation and storage. It must be emphasized that 5 is the only known α-amino aldehyde which can be distilled without decomposition or racemization.13 The bulky, electron-donating N-TIPS group14 provides particularly effective protection for amino aldehydes.
Computer-generated models reveal the effectiveness of the N-TIPS group in not only blocking access to the amino nitrogen atom, but also, in limiting access to the α-hydrogen and processes which lead to the racemization of 5 (Figure 1).15
Figure 1.

Space-filling MM model of phenylglycinal 5g.
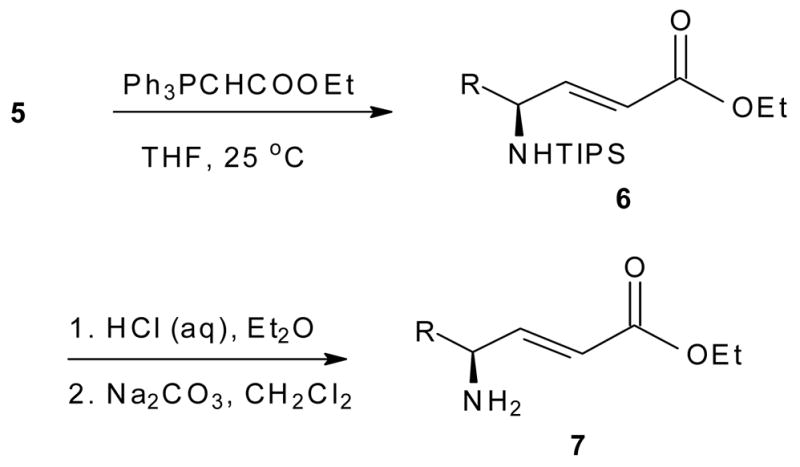
Traditionally, the optical purities of N-protected α-amino aldehydes have been obtained through the analysis of the corresponding amino alcohols.1 However, we chose to combine this determination with a demonstration of the synthetic value of 5 through their conversion to the corresponding γ-amino-α,β-unsaturated esters 6 through a simple Wittig olefination.1,2,5b
While rare in nature, unsaturated derivatives of amino acids provide intriguing building blocks for making significant structural changes to the backbone of peptides.16 This conversion also facilitates a new entry to geometrically and optically pure cysteine and serine protease inhibitors.17 The removal of the N-TIPS protection takes place rapidly under very mild conditions (0.16 N HCl, 0 °C). Moreover, employing a simple basic workup permits 7 to be isolated in analytically pure form as the free base with preservation of the trans C=C geometry (Table 2).
Table 2.
γ-Amino-α,β-unsaturated Amino Esters from 5
| series | 6 (%)a | 7 (%) | ee (%)a |
|---|---|---|---|
| A | 73 | 62 | 99 |
| B | 80 | 64 | 99c |
| C | 84 | 64 | 98 |
| D | 85 | 62 | 98 |
| e | 84 | 71 | 98 |
| f | 85 | 66 | 98 |
| g | 90 | 70 | 99 |
Yields correspond to the product obtained from the pure aldehyde 5.
Product ee determined by conversion to the Mosher amide of 7 (crude) and analysis by 1H and 13C NMR.
The D-amino aldehyde D-5b was also olefinated to give D-6b (81%) and desilylated D-7b (65%, 99% ee).
The amines 7 were converted to the corresponding Mosher amides 8. These were rigorously analyzed by 13C and 1H NMR. In each case, the racemic amino acids (±)-1 were carried through the reaction sequence 1 → 8 for comparison and analysis purposes. No kinetic resolution was observed in the acylation of 7 to provide 8. The limits of detection for this method were determined to be <0.5%, thereby clearly revealing that no more than 2% racemization (i.e., 7 ≥98% ee) had occurred in any of the representative systems studied. This includes the particularly challenging examples, serinal 5g and phenylglycinal 5h. We also prepared the pure D-amino compounds for the b series (R = Pr) (see footnotes, Tables 1, 2). The Mosher amide 8a (R = Me) was purified to obtain a crystalline derivative whose single crystal X-ray structure is pictured in Figure 2.
Figure 2.

X-Ray structure of the Mosher amide 8a
In summary, a new borane-based route to N-TIPS-α-amino aldehydes 5 from amino acids 1 was demonstrated for eight (8) representative examples through a simple three-step process. First, N,O-bisTIPS-α-amino esters 2 were prepared (72–87%) through a new silylation protocol which avoids product racemization. Next, an unprecedented borane-mediated semi-reduction strategy was devised to convert 2 to stable, isolable oxazaborolidines 3 (100%) containing the mixed acetal functional group. The controlled hydrolysis of 3 gives 5 (49–60%) through a hemiacetal intermediate 4. These aldehydes possess high thermal stability, can be filtered through neutral alumina and resist both racemization and chemical degradation upon handling and storage! The Wittig olefination of 5 with Ph3PCHCOOEt provides a simple entry to N-TIPS-γ-amino-α, β-unsaturated amino esters 6 (73–90%) which are readily deprotected converted to provide the free amines 7 (62–71%). Analysis of the Mosher amide derivatives of these amines 7 reveals them to be ≥98% ee in every case, even with the traditionally difficult serine and phenylglycine examples. Thus, the TIPS group not only facilitates the preparation and isolation of 5 in pure form without racemization, but also permits these aldehydes to be used for further synthetic conversions without loss of optical purity. These meritorious features should dramatically increase the number of applications for these polyfunctional intermediates in chemical synthesis.
Supplementary Material
Full experimental procedures, characterization data, selected spectra for 2–3, 4–7 and derivatives and x-ray crystallographic data for 8a (PDF). This material is available free of charge via the Internet at http://pubs.acs.org
Scheme 2.
Acknowledgments
The support of the NSF (CHE0517194) and NIH SCORE (S06GM8102) Program is gratefully acknowledged. We thank Dr. Hong Zhao (University of Puerto Rico) for the X-ray structure of the Mosher amide 8a. The help of Dr. Iveliz Kock, Ms. Carmen Garcia, Mr. Jose Guzman and Ms. Raquel Nieves (University of Puerto Rico) with this study is also gratefully acknowledged.
Footnotes
Dedicated to the memory of a fine chemist, leader and good friend, the late Dr. Clinton F. Lane.
References
- 1.(a) Jurczak J, Golebiowski A. Chem Rev. 1989;89:149. and ref. cited therein. [Google Scholar]; (b) Reetz MT. Chem Rev. 1999;99:1121. doi: 10.1021/cr980417b. [DOI] [PubMed] [Google Scholar]; c) Liang X, Andersch Bols M. J Chem Soc, Perkin Trans 1. 2001:2136. [Google Scholar]
- 2.(a) Kwon S, Myers AG. J Am Chem Soc. 2005;127:16796. doi: 10.1021/ja056206n. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Hummel CW, Pitsinos EN, Nakada M, Smith AL, Shibayama K, Saimoto H. J Am Chem Soc. 1992;114:10082. [Google Scholar]; (c) Corey EJ, Reichard GA. J Am Chem Soc. 1992;114:10677. [Google Scholar]; (d) Vedejs E, Naidu BN, Klapars A, Warner DL, Li V, Na Y, Kohn H. J Am Chem Soc. 2003;125:15796. doi: 10.1021/ja030452m. [DOI] [PubMed] [Google Scholar]; (e) Myers AG, Kung DW. J Am Chem Soc. 1999;121:10828. [Google Scholar]; (f) Lin S, Yang Z, Kwok BHB, Koldobskiy M, Crews CM, Danishefsky SM. J Am Chem Soc. 2004;126:6347. doi: 10.1021/ja049821k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Boger DL, Colleti SL, Honda T, Menezes RF. J Am Chem Soc. 1994;116:5607. [Google Scholar]; (h) Hagihara M, Schreiber SL. J Am Chem Soc. 1992;114:6570. [Google Scholar]
- 3.(a) Andersson L, Isley TC, Wolfenden R. Biochemistry. 1982;21:4177. doi: 10.1021/bi00260a040. [DOI] [PubMed] [Google Scholar]; (b) Otto HH, Schirmeister T. Chem Rev. 1997;97:133. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]; (c) Leung D, Abbenante G, Fairlie DP. J Med Chem. 2000;43:305. doi: 10.1021/jm990412m. [DOI] [PubMed] [Google Scholar]; (d) Fu Y, Bieschke J, Kelly JW. J Am Chem Soc. 2005;127:15366. doi: 10.1021/ja0551382. [DOI] [PubMed] [Google Scholar]
- 4.(a) Garner P, Park JM. Org Synth. 1992;70:18. [Google Scholar]; (b) Roush WR, Hunt JA. J Org Chem. 1995;60:798. [Google Scholar]
- 5.(a) Myers AG, Zhong B, Movassaghi M, Kung DW, Lanman BA, Kwon S. Tetrahedron Lett. 2000;41:1359. doi: 10.1021/ol006427s. [DOI] [PubMed] [Google Scholar]; (b) Concellón JM, Mejica C. Eur J Org Chem. 2007:5250. doi: 10.1021/jo050655o. [DOI] [PubMed] [Google Scholar]
- 6.Hyun SI, Kim YG. Tetrahedron Lett. 1998;39:4299. [Google Scholar]
- 7.(a) Lubell WD, Rapoport H. J Am Chem Soc. 1987;109:236. [Google Scholar]; (b) Lubell WD, Rapoport H. J Org Chem. 1989;54:3824. [Google Scholar]; (c) Jamison TF, Rapoport H. Org Synth. 1993;71:226. [Google Scholar]
- 8.(a) Rücker C. Chem Rev. 1995;95:1009. and refs. cited therein. [Google Scholar]; (b) Soderquist JA, Vaquer J, Díaz MJ, Bordwell FG, Zhang S. Tetrahedron Lett. 1996;37:2561. [Google Scholar]; (c) Justo de Pomar JC, Soderquist JA. Tetrahedron Lett. 1998;39:4409. [Google Scholar]; (d) Justo de Pomar JC, Soderquist JA. Tetrahedron Lett. 2000;41:3285. [Google Scholar]; (e) Soderquist JA, Justo de Pomar JC. Tetrahedron Lett. 2000;41:3537. [Google Scholar]
- 9.(a) Gehrke CW, Leimer K. J Chromatogr. 1971;57:219. doi: 10.1016/0021-9673(71)80035-3. [DOI] [PubMed] [Google Scholar]; (b) Venkateswaran PS, Bardos TJ. J Org Chem. 1967;32:1256. doi: 10.1021/jo01279a101. [DOI] [PubMed] [Google Scholar]
- 10.For a similar system, see: Corey EJ, Bakshi RK, Shibata S. J Am Chem Soc. 1987;109:5551.
- 11.The major (1S, 2S)-4d diastereomer was identified through: (1) OH and NHTIPS 1H NMR signals at δ 3.91 and 0.20 which were exchangeable with D2O, (2) 13C NMR data showing diastereotopic isopropyl methyl groups for both the OTIPS (δ 17.78, 17.87) and NHTIPS (δ 18.43, 18.48) signals at δ 56.28 (C2) and 92.13 (C1), and (3) its clean conversion to 5d and TIPSOH (see Supporting Information).
- 12.For 5f and 5g, the aldehydes are generated through the distillation process from the crude hydrolysate. Once isolated, as for the other examples of 5, they are stable (> 1 week) when stored in the cold (−20 °C).
- 13.Garner’s aldehyde is isolated by distillation, but in neither chemically nor enantiomerically pure form. Therefore, its thermal stability in the distillation process has not been established.4a
- 14.For example, 1H NMR δ 0.5–0.7 for NH proton in 2, 5 and 6.
- 15.Generated with the Spartan 06 V112 program.
- 16.(a) Hagihara M, Anthony NJ, Stout TJ, Clardy J, Schreiber SL. J Am Chem Soc. 1992;114:6568. [Google Scholar]; (b) Baldauf C, Gunther R, Hofmann HJ. J Org Chem. 2005;70:5351. doi: 10.1021/jo0480489. [DOI] [PubMed] [Google Scholar]
- 17.Powers JC, Asgian JL, Ekici OD, James KE. Chem Rev. 2002;102:4639. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental procedures, characterization data, selected spectra for 2–3, 4–7 and derivatives and x-ray crystallographic data for 8a (PDF). This material is available free of charge via the Internet at http://pubs.acs.org