

Abstract
A group of DAG-lactones with altered functionality (C=O → CH2 or C=O → C=S) at the sn-1 and sn-2 carbonyl pharmacophores was synthesized and used as probes to dissect the individual role of each carbonyl in binding to protein kinase C (PKC). The results suggest that the hydrated sn-1 carbonyl is engaged in very strong hydrogen bonding interactions with the charged lipid headgroups and organized water molecules at the lipid interface. Conversely, the sn-2 carbonyl has a more modest contribution to the binding process as a result of its involvement with the receptor (C1 domain) via conventional hydrogen bonding to the protein. The parent DAG-lactones, E-6 and Z-7, were designed to bind exclusively in the sn-2 binding mode to insure the correct orientation and disposition of pharmacophores at the binding site.
Introduction
The lipophilic second messenger, sn-1,2-diacylglycerol (DAG) plays a prominent role in cellular signal transduction.1-3 Generated through both G-protein coupled and tyrosine kinase activated isoforms of phospholipase C, as well as indirectly by phospholipase D, DAG binds to the C1 domains (C1a or C1b) of protein kinase C (PKC) isozymes and other non-kinase protein targets activating their downstream pathways.4,5 The importance of these pathways in cellular responses, including proliferation, differentiation, gene expression, and tumor promotion, has been well documented in the literature in studies with the phorbol esters, which function as potent and metabolically stable DAG surrogates.6
Both conventional (α, β1 and β2, and γ) and novel (δ, ε, η, and θ) PKC isozymes are thought to be activated as a result of association of the cytosolic enzyme with membranes containing acid phospholipids.7,8 This association is strongly facilitated by the liberation of DAG which causes the transient translocation of PKC to the inner leaflet of the cellular membrane.9-11
In order to accelerate our understanding of the structure-activity analysis of ligand and C1 domain interactions, we have developed a chemically accessible template in the form of a rigid lactone that contains a conformationally constrained glycerol backbone.12 The resulting DAG-lactones seem to overcome part of the entropic penalty associated with the binding of DAG, and nanomolar binding affinities in the range normally observed for the phorbol esters have been achieved in vitro.12
Ever since the X-ray structure of the binary complex of phorbol-13-O-acetate bound to the C1b domain of PK-Cδwas solved, the role of the C-9 OH pharmacophore in phorbol has remained elusive.13 This critical pharmacophore does not appear to be engaged at all with the receptor, but instead it forms an intramolecular hydrogen bond with the C-13 carbonyl ester of phorbol itself. Although it is possible that such an intramolecular hydrogen bond could be biologically relevant, the more likely explanation is that its formation is improperly facilitated by the absence of a lipid bilayer in the crystal structure. Indeed, recent molecular modeling studies performed by Miskovsky et al.14 on a binary complex of phorbol myristate (PMA) and a phosphatidyl choline (DPPC) bilayer (PMA-DPPC), or on the more relevant ternary C1b-PMA-DPPC complex, illustrate convincingly the important role of the furtive C-9 OH by uncovering strong hydrogen bonding interactions of this OH directly to the phosphate group, or with the water molecules surrounding the headgroups of DPPC.
In a similar manner, our modeling studies on binary complexes involving the C1 domain and DAG-lactones have also shown that for either one of the two binding modes identified (sn-1 or sn-2)15 there is an orphan carbonyl pharmacophore whose role we propose is equivalent to that of the C-9 OH of phorbol. These two apparently comparable binding modes for the DAG-lactones are able to form identical networks of hydrogen bonds with amino acids Thr242, Leu251, and Gly253, as was observed with phorbol-13-O-acetate.13 The sn-1 binding mode is defined as that in which the sn-1 carbonyl is hydrogen bonded to the C1 domain, and for the alternative sn-2 binding mode, it is the sn-2 carbonyl that appears directly engaged in hydrogen bonding to the protein.
In a preliminary study designed to determine the importance of these non-equivalent carbonyl moieties, we synthesized compounds 3, 4, and 5 with the intent to dissect the importance of each individual carbonyl relative to the parent DAG-lactones 1 (E-isomer) and 2 (Z-isomer).16 Compounds 4 and 5 were individually assayed as E- and Z-isomers, respectively, whereas compound 3 was evaluated as a mixture of the two geometric isomers. Because all the compounds showed an indistinct ca. 100-fold decrease in binding affinity relative to the parent compounds, it was impossible to assess the independent role of each carbonyl (sn-1 or sn-2) towards binding, and the only conclusion that could be drawn was that the presence of both groups was essential. However, in ensuing studies we were able to design DAG-lactones, such as 6 (E-isomer) and 7 (Z-isomer), that showed an unequivocal preference for the sn-2 binding mode due to the large branched alkyl chain being positioned adjacent to the lactone carbonyl (Figure 1).15,17 We proposed that utilizing this new and more potent DAG-lactone template could improve our chances of diagnosing the different roles played by each carbonyl in the binding process. Indeed, the difference in binding affinity between DAG-lactone 1 versus 6, and 2 versus 7, was respectively17- to12-fold higher, suggesting that compounds 6 and 7 were better candidates for the study.
Figure 1.
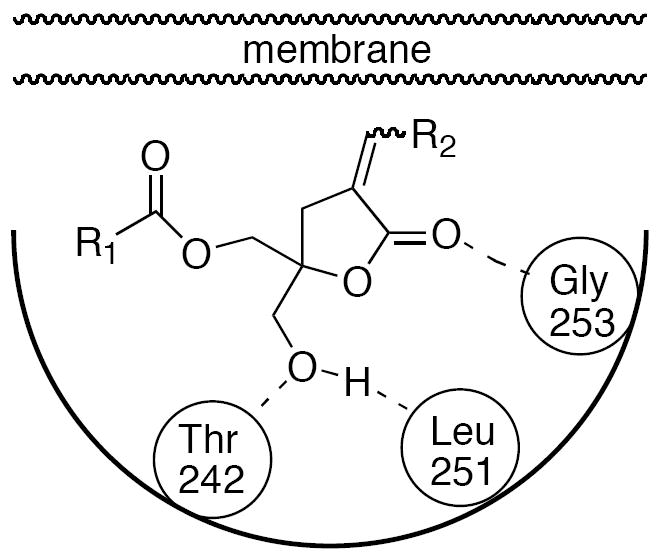
Schematic representation of the sn-2 binding mode of DAG-lactones.
Having the role of the lactone (sn-2) carbonyl defined as bound to the C1 domain (sn-2 binding mode, Figure 1), the hypothesis was that in the “real life” ternary complex the apparently orphan sn-1 carbonyl pharmacophore —as it appears in the binary complex— would be directed to the membrane interface where it would bind to either organized water molecules or the lipid headgroups. If our assumption were correct, elimination of either of these carbonyls, represented by compounds 8 (E,Z-isomers), 9 (E-isomer) and 10 (Z-isomer), or replacement by a thiocarbonyl moiety, as in compounds 11 (E-isomer) and 12 (E-isomer), would affect the binding affinities of the ligands as a function of the binding environment of each carbonyl.
The changes in binding affinities that were measured confirmed the above hypothesis and suggest that the two carbonyls indeed reside in different environments with the sn-1 carbonyl engaged in strong polar interactions at the interface and capable of playing a similar role to that proposed for the C-9 OH of phorbol.14
Chemistry
The two critical branched chain components for this project were the previously used aldehyde 1317-19 and 5-methyl-3-(2-methylpropyl)hexyl]triphenylphosphonium iodide (16) (Scheme 1). The latter compound was obtained from 13 via lithium aluminum hydride reduction to the alcohol (14), iodination with Ph3P/imidazole/I2 to give 4-(2-iodoethyl)-2,6-dimethylheptane (15), and final treatment with triphenylphosphine to afford the desired phosphonium salt (16) as a white solid.
Scheme 1.
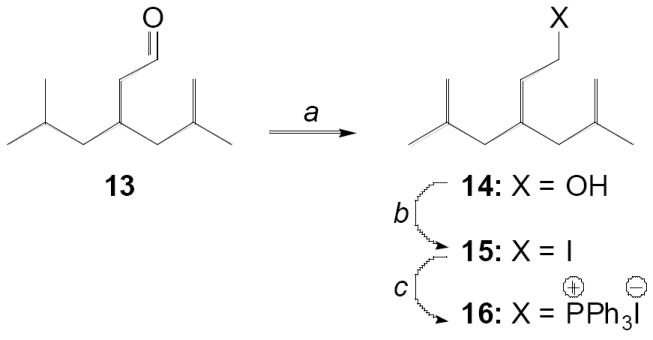
Reagents and conditions: (a) LiAlH4/THF, 0 °C; (b) PPh3, imidazole, I2, THF, rt; (c) PPh3, PhCH3, reflux.
The DAG-lactones missing the sn-2 carbonyl were synthesized via Wittig reaction with the known 5-[(4-methoxyphenoxy)methyl]-5-[(phenylmethoxy)methyl]-2,4,5-trihydrofuran-3-one (17)20 and the corresponding ylid generated from 16 with n-butyllithium (Scheme 2). Compound 18 was obtained as an inseparable mixture of geometric isomers. Removal of the p-methoxyphenyl (PMP) group with ammonium cerium(IV) nitrate afforded monoalcohol 19 and acylation with pivaloyl chloride followed by deprotection of the benzyl ether with BCl3 at −78 °C provided the target compound 8 as an inseparable mixture of geometric isomers. Judging from the integration of the pivaloyl methyl [C(O)C(CH3)3] signal, the ratio of isomers was estimated to be 14:1, but the exact geometry of the double bond of the predominant isomer could not be determined.
Scheme 2.

Reagents and conditions: (a) 16, n-BuLi/THF, 0 °C → reflux; (b) (NH4)2Ce(NO3)6/CH3CN-H2O, 0 °C; (c) (CH3)3C(O)Cl, Et3N/CH2Cl2 (0 °C); (d) BCl3, CH2Cl2, -78 °C.
The DAG-lactone targets devoid of the sn-1 carbonyl (E-9 and Z-10) were synthesized, respectively, from the isomers E-(21) and Z-(22),19 which were prepared according to our published method (Scheme 3). Removal of the benzyl ether with BCl3 at −78 °C gave the corresponding monoalcohols E-23 and Z-24, which were subsequently converted to the corresponding methylsulfonate esters E-25 and Z-26. Deprotection of the p-methoxyphenyl ether with ammonium cerium(IV) nitrate provided monoalcohols E-27 and Z-28; and displacement of the mesylate ester with neopentyl alcohol gave the desired targets E-9 and Z-10.
Scheme 3.

Reagents and conditions: (a) BCl3, CH2Cl2, -78 °C; (b) CH3SO2Cl, Et3N, CH2Cl2, 0 °C; (c) (NH4)2Ce(NO3)6/CH3CN-H2O, 0 °C; (d) (CH3)3CH2OH, Et3N, DMF, 0 °C.
The strategy for the synthesis of the thiolactone target E-11 started with the known lactone, 5-[(4-methoxyphenoxy)methyl]-5-[(phenylmethoxy)methyl]-3,4,5-trihydrofuran-2-one (29),19 which was converted to 31 in two easy steps (Scheme 4). Condensation of 31 with aldehyde 13, followed by in situ conversion of the intermediate aldol adduct to the olefin by the presence of triethylamine, DBU and methanesulfonyl chloride, afforded the individual geometric isomers E-32 and Z-33, which were individually separated by column chromatography. Consistent with previously synthesized DAG-lactones, the vinyl proton of the Z-isomer displayed a characteristic multiplet at δ6.12-6.18 in its 1H NMR spectrum, while the corresponding signal of the E-isomer appeared more downfield at δ= 6.72-6.77. In the following step, regardless of the geometry of the isomer selected as the starting material, the reaction with Lawesson’s reagent at 110 °C in toluene generated exclusively the thiolactone E-isomer (E-34). This assignment is based on the fact that the E-isomers are thermodynamically more stable than the Z-isomers, and also because of the appearance of the vinyl proton signal at δ7.05-7.10 appears is even further downfield compared to the lactone E-isomer (E-32). The diol generated from E-34 after treatment with BCl3 at −78 °C was immediately acylated with one equivalent of pivaloyl chloride to afford the target compound E-11 as a yellowish oil.
Scheme 4.

Reagents and conditions: (a) (NH4)2Ce(NO3)6, CH3CN-H2O (0 °C); (b) PhCH2Br, NaH, DMF, rt; (c) i. 13, [(CH3)3Si]2NLi/THF, -78 °C; ii. CH3SO2Cl, Et3N, DBU, CH2Cl2, rt; (d) Lawesson’s reagent, PhCH3, reflux; (e) i. BCl3, CH2Cl2, -78 °C; ii. (CH3)3COCl, Et3N, CH2Cl2, 0 °C.
DAG-lactone E-12, with the thiocarbonyl group at the sn-1 position, was accessible from diol E-35, which was easily obtained from E-32 (Scheme 5). According to the method of Salaby and Rapoport,21 a solution of E-35 was treated with 2,2-dimethyl-1-(6-nitrobenzotriazolyl)propane-1-thione (36) in the presence of DBU to give the desired target E-12 as a yellowish oil.
Scheme 5.

A few remarks about the chemistry are in order. In these DAG-lactones there is a single asymmetric carbon. However, because DAG-lactones E-6 and Z-7 possess a large branch chain at the sn-2 position they are extremely potent PK-C ligands with Ki values in the nanomolar range, and the difference between a pure enantiomer and its racemate is very small (ca. 2 nM versus 4 nM).22 Therefore, for this investigation only racemic mixtures were synthesized. The thermodynamically more stable isomer is usually the E-isomer, which is normally obtained in a higher ratio. Although some small differences in affinity have been detected between geometric isomers, these also tend to be small (≤ 2-fold).22 Because the thiolactone target E-11 could only be obtained as the E-isomer, we concentrated our synthetic effort in obtaining the complete E-isomer series for all the target compounds in order to perform a comparative SAR study (vide infra). The synthetic schemes that are described here are quite adaptable to the preference of the individual chemist for a particular protecting group or method of deprotection; thus, there are several alternatives to reach the target compounds besides the ones shown in the schemes.
A final point of interest is the stability of the thiolactone ring in compound E-11. When this compound was initially synthesized, the mass spectrum showed the corresponding MH+ ion peak at 399 for the thiolactone and a weak peak at 383 for the protonated lactone. Two months later, while standing at room temperature, the intensity of the peaks was reversed showing a ratio of products overwhelmingly in favor of the lactone. Conversion of the thiolactone ring to lactone could be catalyzed by trace amounts of acid and moisture. Under the same conditions, however, compound E-12 remained stable. Based on these observations, the biological assay of these samples was performed with freshly synthesized materials.
Biological results and discussion
The PK-C binding affinity for all the ligands is expressed as Ki, which reflects the ability of the compounds to displace [20-3H]-phorbol-12,13-dibutyrate (PDBU) from the enzyme, or isolated C1 domain, in a competition assay.23
As shown in Table 1, for the set of compounds where the carbonyl at either the sn-1 or sn-2 position was eliminated (C=O → CH2), removal of the sn-1 carbonyl from the parent DAG-lactones E-6 and Z-7 precipitated a more severe drop in binding affinity than the removal of the sn-2 carbonyl. This effect was most pronounced in case of compound E-9 (Table 1). Unfortunately, the compound devoid of the sn-2 carbonyl (8) could not be separated into its geometric isomers. The removal of these carbonyl groups follows a similar trend for either the intact isozyme αor the isolated C1bδdomain. For the set of compounds where the carbonyl function is replaced with a thiocarbonyl (C=O → C=S), the drop in binding affinity follows a similar trend as for the removal of the entire function, but the effects are less dramatic. The thiocarbonyl compounds were obtained only as E-isomers (vide supra).
Table 1.
Inhibition of [3H]PDBU binding to PK-Cαand to C1bδby DAG-lactones with modified sn-2 or sn-1 carbonyl functions.
| Compound | Structural change | Ki (PK-C_) (nM) | Ki (C1b_) (nM) |
|---|---|---|---|
| E-6 | — | 3.25 ± 0.15 | 0.90 ± 0.07 |
| Z-7 | — | 2.90 ± 0.35 | 1.16 ± 0.07 |
| E,Z-8 | sn-2 (CH2) | 622 ± 47 | 219 ± 23 |
| E-9 | sn-1 (CH2) | 14,290 ± 840 | 1,890 ± 190 |
| Z-10 | sn-1 (CH2) | 5,874 ± 20 | 2,140 ± 130 |
| E-11 | sn-2 (C=S) | 23.7 ± 2.2 | 3.13 ± 0.14 |
| E-12 | sn-1 (C=S) | 113 ± 12 | 20.6 ± 1.1 |
To study the effects of the lipid environment, we also compared the changes in binding affinity in the presence and absence of phosphatidyl serine (PS).24 In order to make a better comparison between the effects of removing the carbonyls or replacing them with the thiocarbonyl function in either the presence or absence of PS, we decided to analyze these effects on the smaller C1bδdomain using only the E-isomers: compounds E-6, E,Z-8, and E-9 (C=O → CH2) and compounds E-6, E-11 and E-12 (C=O → C=S) (Table 2). The isolated C1bδdomain has been shown to translocate to cellular membranes in response to DAG or phorbol signaling, suggesting that its ligand binding and membrane interactions are similar in isolation and in the full-length protein. Testing the effects of removing PS on the isolated C1bδdomain eliminates the confounding factor of the C2 domain, which also interacts with charged lipid membranes.
Table 2.
Inhibition of [3H]PDBU binding to C1bδby DAG-lactones with modified sn-2 or sn-1 carbonyl functions in the presence or absence of phosphatidyl serine (PS).
| Compound | Structural change | Ki (C1b_) (nM) +PS | Ki (C1b_) (nM) –PS |
|---|---|---|---|
| E-6 | — | 0.90 ± 0.07 | 103 ± 16 |
| E,Z-8 | sn-2 (CH2) | 219 ± 23 | 6,620 ± 640 |
| E-9 | sn-1 (CH2) | 1,890 ± 190 | 101,000 ± 10,000 |
| E-11 | sn-2 (C=S) | 3.13 ± 0.14 | 173.4 ± 4.8 |
| E-12 | sn-1 (C=S) | 20.6 ± 1.1 | 606 ± 50 |
In order to understand the observed changes in Table 2 in thermodynamic terms, we considered the Ki value to be equivalent to the dissociation constant (Kd) for the enzyme-ligand complex. In that case, the free energy for the binding of the parent DAG-lactone (E-6) can be expressed as:
This value corresponds to a reference state A, which could be compared to other states (B) representing structural or environmental changes in the following manner:
Using the value of R as 0.00198 kcal/mol•K and assuming room temperature conditions (300 K) we would have:
The Δ(ΔG°) values in a +PS environment will reflect the changes caused by the structural modifications in that medium, whereas Δ(ΔG°) values in a –PS environment will represent the changes caused by the removal of the phosopholipid for each of the molecular alterations as shown in Tables 3 and 4 for the sn-1 and sn-2 carbonyls. All the Ki values for these calculations were taken from Table 2.
Table 3.
Δ(ΔG°) variations caused by structural changes of the sn-1 carbonyl in DAG-lactones in the presence or absence of phosphatidyl serine (PS).
| +PS | −PS | ||||
|---|---|---|---|---|---|
| Kd(B)/Kd(A) | Δ(ΔG°) kcal/mol | Kd(B)/Kd(A) | Δ(ΔG°) kcal/mol | ||
| ↓ | Kd(CO)/KdCO) | 0 | → | Kd(CO)/Kd(CO) | 2.82 |
| ↓ | Kd(CS)/KdCO) | 1.86 | → | Kd(CS)/Kd(CS) | 2.01 |
| ↓ | Kd(CH2)/KdCO) | 4.54 | → | Kd(CH2)/Kd(CH2) | 2.36 |
Table 4.
Δ(ΔG°) variations caused by structural changes of the sn-2 carbonyl in DAG-lactones in the presence or absence of phosphatidyl serine (PS).
| +PS | −PS | ||||
|---|---|---|---|---|---|
| Kd(B)/Kd(A) | Δ(ΔG°) kcal/mol | Kd(B)/Kd(A) | Δ(ΔG°) kcal/mol | ||
| ↓ | Kd(CO)/KdCO) | 0 | → | Kd(CO)/Kd(CO) | 2.82 |
| ↓ | Kd(CS)/KdCO) | 0.74 | → | Kd(CS)/Kd(CS) | 2.38 |
| ↓ | Kd(CH2)/KdCO) | 3.26 | → | Kd(CH2)/Kd(CH2) | 2.03 |
When these results are plotted in a graph, the analysis of the data is enormously simplified (Figure 2). As can be seen along the X-axis in both Figures 2A and 2B, complete removal of either carbonyl results in poorer binding; however, the effect is much more pronounced when the sn-1 carbonyl is eliminated. Similarly, replacing the carbonyl with a thiocarbonyl also has a more dramatic affect at the sn-1 position than at the sn-2 position (Figure 2A). The effect of removing PS is approximately the same (within 1 kcal/mol) for all the structurally modified DAG-lactones, and slightly higher for the parent compound (E-6).
Figure 2.
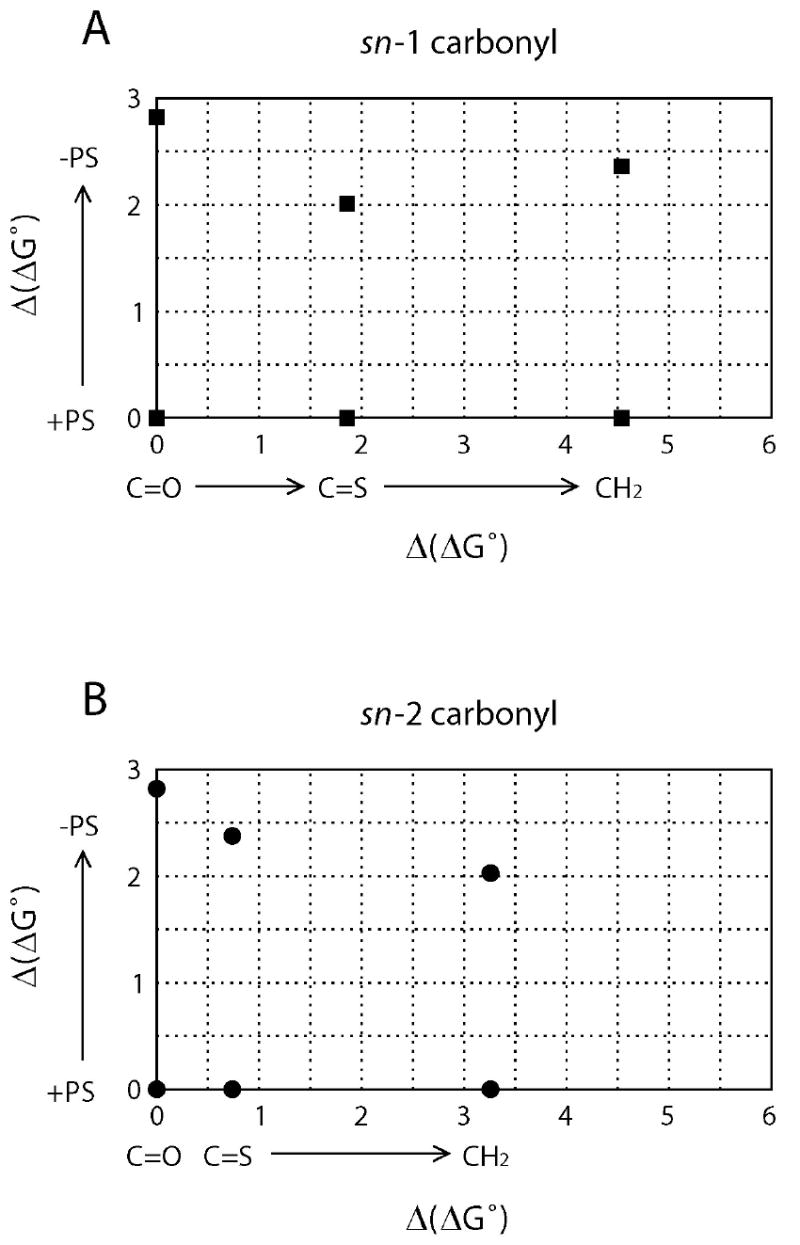
Δ(ΔG°) plots for normal and altered sn-1 (A) and sn-2 (B) carbonyls in the presence or absence of phosphatidyl serine (PS). Δ(ΔG°) changes along the x-axis correspond to structural changes, while Δ(ΔG°) changes along the y-axis reflect the effect of PS in the binding process.
The parent DAG-lactones, E-6 and Z-7, were designed to bind exclusively in the sn-2 binding mode to insure the correct orientation and disposition of pharmacophores during binding. In this sn-2 binding mode, the sn-2 carbonyl is engaged in a hydrogen bonding interaction with the C1 domain, while the sn-1 carbonyl is directed outward toward the solvent environment of the complex. Removing or altering the sn-2 carbonyl, therefore, will affect the interactions of the DAG-lactone with the C1 domain, whereas removing or altering the sn-1 carbonyl will affect the interactions of the DAG-lactone with surrounding solvent, presumably the interfacial region of the PS bilayer. These results show that removal of the sn-2 carbonyl, as in compound E,Z-8 appears to be less costly than removing the sn-1 carbonyl, as in compounds E-9 and Z-10. This suggests that weakening the interaction between the DAG-lactone and its receptor by removing the hydrogen bond formed by the sn-2 carbonyl is less important to the overall binding affinity of the complex than altering the interaction of the bound DAG-lactone with the bilayer interface. Using a similar argument, replacement of the C=O by the less polarized C=S,25,26 as in compound E-11, weakens proportionally the strength of the hydrogen bond of the thio-sn-2 carbonyl to the C1 domain. However, in the case of the sn-1 position, as in compound E-12, the less hydrated C=S bond interacts less effectively with water or the polar headgroups of the phospholipids. Since very strong hydrogen bonds are formed when one of the partners bears an electrostatic charge, the effect of this change is stronger at the bilayer interface where the sn-1 carbonyl resides. The negative effect of removing PS for the parent DAG-lactone (E-6), as well as the structurally modified compounds, also seems to reflect the importance of the interactions between the DAG-lactone:C1 domain complex and the bilayer interface for productive binding. The interaction of the sn-1 carbonyl on the DAG-lactone with the water and lipid headgroups in the bilayer interface environment may be important for the correct orientation of the molecule at the active site allowing the primary alcohol to engage in hydrogen bonding with Thr242 and Leu251. In the absence of the sn-1 carbonyl, the single polar sn-2 carbonyl might seek to position itself in the more polar environment of the interface, causing the molecule to flip out of the sn-2 binding mode and leading to a very unproductive binding mode with loss of the critical hydrogen bonds to Thr242 and Leu251 which will translate in much higher Ki values.
Additionally, or alternatively, the sn-1 carbonyl may be important in mediating the penetration of the C1 domain into the membrane as it binds to the DAG-lactones. Before PKC has a chance to access the inner hydrophobic core of the cell membrane, it must first encounter the polar head group layer of its constituent phospholipids. Although there have been many experimental and theoretical studies on the energetics of inserting small helical peptides into the bilayer interfacial region, very little work has been done on possible mechanisms for partial ß-sheet insertion, as must occur with the C1 domain.27 Measurements of partitioning of unfolded amino acids between water and POPC bilayers have shown that inserting the backbone amide bond into the lower-dielectric bilayer interface is thermodynamically unfavorable, with a cost of approximately 1.2 kcal/mol for each residue.28 Yet there are several non-hydrogen-bonded solvent-exposed backbone amide bonds and other polar groups in the C1 domain, even when the ligand is bound (Figure 3A). Several lines of evidence suggest that a certain degree of order is experienced by a few molecules of water that tend to penetrate the membrane’s surface. Some experiments suggest that between 5 and 20 water molecules are organized around each molecule of phospholipid.29 The sn-1 carbonyl retains some positional flexibility in the bound complex, and it can orient itself in such a way as to form a water-bridged hydrogen bond to several different backbone carbonyl groups in the C1 domain (Figure 3B, C). It is therefore possible that partial hydration of the sn-1 carbonyl, by even one or two water molecules, will provide an energetic advantage for the insertion of the C1 domain, by providing pre-positioned structural water as hydrogen-binding partners for the backbone amides.
Figure 3.
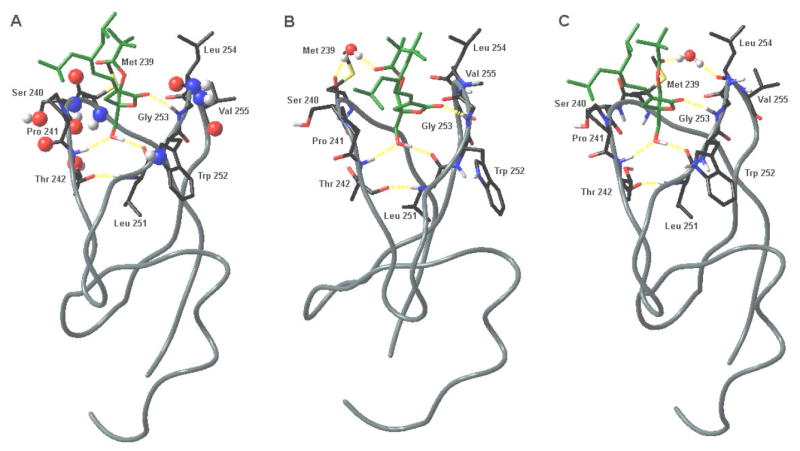
The DAG-lactone E-6 (shown in green) docked in the binding site of the C1bδdomain.13 Hydrogen bonds are drawn with dashed yellow lines. A. Solvent-exposed non-hydrogen-bonded polar atoms are indicated in larger ball-and-stick representation. B. Water-bridged hydrogen bonding between the sn-1 carbonyl of the DAG-lactone and the backbone carbonyl of residue Met 239. C. Water-bridged hydrogen bonding between the sn-1 carbonyl of the DAG-lactone and the backbone carbonyl of residue Leu 254. Models were built and energy minimized using the OPLS-2003 forcefield30 in MacroModel (Schrödinger, Inc.)
We conclude that the experiments presented here point to the existence of a third unknown binding site, which resides at the lipid interface. Although it is not possible to characterize this binding site in precise structural terms, it appears to be the source of very strong hydrogen bonding interactions between a hydrated sn-1 carbonyl in DAG-lactones – and most likely the C-9 OH in the case of phorbol esters – with the charged lipid headgroups and organized water at the lipid interface.
General Experimental Section
All chemical reagents were commercially available. Melting points were determined on MelTemp II apparatus, Laboratory Devices, USA, and are uncorrected. Column chromatography was performed on silica gel 60, 230-400 mesh (Bodman Ind.), and analytical TLC was performed on Analtech Uniplates silica gel GF. 1H and 13C NMR spectra were recorded on a Varian Unity Inova instrument at 400 and 100 MHz, respectively. Spectra are referenced to the solvent in which they were run (7.24 ppm for CDCl3). Infrared spectra were recorded on a Jasco model 615 FT-IR instrument. Positive-ion fast atom bombardment mass spectra (FABMS) were obtained on a VG 7070E-HF double-focusing mass spectrometer operated at an accelerating voltage of 6 kV under the control of a MASPEC-II data system for Windows (Mass Spectrometry Services, Ltd.). Either glycerol or 3-nitrobenzyl alcohol was used as the sample matrix and ionization was effected by a beam of xenon atoms generated in a saddle-field ion gun at 8.0 ± 0.5 kV. Nominal mass spectra were obtained at a resolution of 1200, and matrix-derived ions were background subtracted during data system processing. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA.
5-Methyl-3-(2-methylpropyl)hexan-1-one (13)
According to a general procedure previously reported from this lab,18 a stirred solution of commercially available 2,6-dimethylheptan-4-one (36 g, 0.25 mol) in THF (200 mL) was cooled to −78 °C and treated dropwise with vinylmagnesium bromide(1 M in THF, 500 mL). The reaction mixture was allowed to reach room temperature, stirred for 1 h, and quenched by the slow addition of a saturated aqueous solution of ammonium chloride (200mL). The resulting mixture was extracted with ethyl ether (300 mL), and the combined organic extract was washed with water and brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with ethyl ether:hexanes (1:10) as eluant to give intermediate 5-methyl-3-(2-methylpropyl)hex-1-en-3-ol (39 g, 92 %) as an oil which was oxidized directly in the next step. A solution of PCC (148 g, 0.69 mol) and 4 Å molecular sieves (148 g) in CH2Cl2 (1 L) was treated dropwise with a solution of 5-methyl-3-(2-methylpropyl)hex-1-en-3-ol (39 g 0.23 mol) in CH2Cl2 (100 mL). After stirring for 24 h at room temperature, the reaction mixture was diluted with ethyl ether (500 mL), filtered through a pad of silica gel, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with ethyl ether:hexanes (1:10) as eluant to give 5-methyl-3-(2-methylpropyl)hex-2-en-1-one as an oil (38.5 g, 95 %), which was then dissolved in CH2Cl2 (200 mL) and immediately reduced under a hydrogen-filled balloon in the presence of 10% Pd/C (4 g). After stirring for 3 h at room temperature, the reaction mixture was filtered through Celite® and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with ethyl ether:hexanes (1:10) as eluant to give 5-methyl-3-(2-methylpropyl)hexan-1-one (13) as an oil (23 g, 64 %) which was used directly without further purification.
5-Methyl-3-(2-methylpropyl)hexan-1-ol (14)
A solution of 13 (12 g, 0.07 mol) in THF (50 mL) was added dropwise over 10 min to a suspension of lithium aluminum hydride (5.3 g, 0.14 mol) in THF (300 mL) that was maintained at 0 °C. After the addition was complete, the reaction was allowed to reach room temperature. After stirring at room temperature for 2 h, the reaction was quenched by the careful addition of ice-water (12 mL), followed by 15% aqueous NaOH (12 mL), and finally a second addition of water (36 mL). The resulting mixture was filtered through Celite® and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with ethyl ether:hexanes (1:5) as eluant to give 5-methyl-3-(2-methylpropyl)hexan-1-ol (14) as an oil (10 g, 83 %) which was used directly without further purification. 1H NMR (CDCl3) δ3.55-3.59 (irr t, 2 H, CH2OH), 1.95 (br s, 1 H, OH), 1.60 (septuplet, 2 H, 2 ×CHMe2), 1.45 (m, 3 H, CH2CH2OH, CH(i-Bu)2), 1.05 (irr t, 4 H, 2 ×CH2CHMe2), 0.70-0.85 (singlets, 12 H, 4 ×CH3); 13C-NMR (CDCl3) δ 61.0, 44.6, 37.6, 30.0, 25.4, 23.2, 22.9.
4-(2-Iodoethyl)-2,6-dimethylheptane (15)
5-Methyl-3-(2-methylpropyl)hexan-1-ol (14) (3 g, 0.017 mol) was added to a solution of triphenylphosphine (4.9 g, 0.018 mol) and imidazole (2.5 g, 0.037 mol) in THF (15 mL) and cooled to 0 °C. Dropwise addition of iodine (4.5 g, 0.018 mol) to the resulting suspension at 0 °C was followed by stirring at room temperature for 2 h. The solvent was evaporated in vacuo and the residue was purified by flash column chromatography on silica gel with hexanes as eluant to give 4-(2-iodoethyl)-2,6-dimethylheptane (15) as an oil (3.75 g, 78 %); 1H NMR (CDCl3) δ3.21 (t, 2 H, J=7.6 Hz, CH2I), 1.75-1.81 (m, 2 H, CH2CH2I), 1.63 (septuplet, 2 H, 2 ×CHMe2), 1.51 (quintuplet, 1 H, CH(i-Bu)2), 1.06 (irr t, 2 × CH2CHMe2), 0.84-0.90 (s, 12 H, 4 × CH3); 13C-NMR (CDCl3) δ 43.7, 38.9, 34.3, 25.3, 23.2, 23.0; GC/MS(EI) m/z 282 (M+), 155 (M+-I). Anal. (C11H23I) C, H, I.
[5-Methyl-3-(2-methylpropyl)hexyl]triphenylphosphonium iodide (16)
A solution of 15 (3.75 g, 0.013 mol) in toluene (10 mL) was treated with triphenylphosphine (5.11 g, 0.019 mol) and heated to reflux for 24 h. After the reaction was completed by TLC analysis, the reaction mixture was concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with EtOAc as eluant to give 16 as a white solid (6.2 g, 84 %); mp 55-57°C; 1H NMR (CDCl3) δ7.70-7.86 (m, 15 H, Ph), 3.42-3.51 (m, 2 H, CH2(PPh3)3), 1.68 (m, 1 H, CH(i-Bu)2), 1.58 (m, 2 H, CH2CH2(PPh3)3), 1.46 (septuplet, 2 H, 2 ×CHMe2), 1.14 (m, 4 H, 2 × CH2CHMe2), 0.87, 0.84, 0.83 and 0.81 (s, 12 H, 4 ×CH3); FABMS m/z (relative intensity) 417 (C29H38P+, 100.0).
(E/Z)-4-Methoxy-1-({4-[5-methyl-3-(2-methylpropyl)hexylidene]-2-[(phenylmethoxy)methyl](2-2,3,5-trihydrofuryl)}methoxy)benzene (18)
n-Butyllithium (5.2 mL, 2 M in THF, 10.4 mmol) was added to a stirred solution of 16, (4.014 g, 7.4 mmol) in THF (20 mL). After stirring for 30 min. at room temperature, the mixture was cooled to 0 °C and a solution of 1720 (1.3 g, 3.7 mmol) in THF (10 mL) was added in one portion. The resulting solution was heated to reflux for 2 h, cooled to room temperature and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with hexanes:EtOAc (5:1) as eluant to give 18 as an oil (711 mg, 40 %) consisting of an inseparable mixture of geometric isomers. Only the signals for the dominant isomer are reported: 1H NMR (CDCl3) δ7.24-7.34 (m, 5 H, Ph), 6.79-6.86 (m, 4 H, PhOCH3), 5.30 (m, 1 H, >C=CH), 4.56 (AB q, 2 H, J = 12.3 Hz, OCH2Ph), 4.43 (br s, 2 H, H-5), 3.95 (AB q, 2 H, J = 9.2 Hz, CH2OAr), 3.76 (s, 3 H, OCH3), 3.58 (AB q, 2 H, J = 9.8 Hz, CH2OBn), 2.48-2.60 (m, 2 H, H-3), 1.96 (irr t, 2 H, >CH=CHCH2), 1.62 (septuplet, 2 H, 2 ×CHMe2), 1.53 (quintuplet, 1 H, CH(i-Bu)2), 1.02-1.08 (m, 4 H, 2 ×CH2CHMe2), 0.79-0.87 (m, 12 H, 4 ×CH3); 13C NMR (CDCl3) δ154.0, 153.4, 138.5, 138.3, 134.0, 133.8, 128.9, 128.7, 128.6, 127.7, 127.6, 119.1, 115.8, 114.7, 84.4, 83.7, 73.6, 71.8, 71.7, 71.5, 70.2, 55.9, 44.1, 34.3, 34.2, 33.3, 25.4, 23.2, 22.9; FABMS m/z (relative intensity) 481 (MH+, 19.9); 480 (M•+, 37). Anal. (C31H44O4 •0.6H2O) C, H.
(E/Z)-{4-[5-Methyl-3-(2-methylpropyl)hexylidene]-2-[(phenylmethoxy)methyl]-2-2,3,5-trihydrofuryl}methan-1-ol (19)
A solution of 18 (333 mg, 0.69 mmol) in CH3CN-H2O (4:1, 10 mL) was cooled to 0 °C and treated with ammonium cerium(IV) nitrate (1.1 g, 2.1 mmol). After stirring for 30 min at 0 °C, the reaction mixture was diluted with CH2Cl2. The organic layer was washed with water and brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with EtOAc:hexanes (1:2) as eluant to give 19 as an oil (159 mg, 60 %) consisting of an inseparable mixture of geometric isomers. Only the signals for the dominant isomer are reported: 1H NMR (CDCl3) δ7.25-7.36 (m, 5 H, Ph), 5.25-5.29 (m, 1 H, >C=CH), 4.55 (AB q, 2 H, J = 12.3 Hz, OCH2Ph), 4.37 (br s, 2 H, H-5), 3.65 (dd, 1 H, J = 11.32, 6.64, CHHOH), 3.58 (dd, 1 H, J = 11.32, 6.15, CHHOH), 3.48 (AB q, 2 H, J = 9.4 Hz, CH2OBn), 2.44 (m, 2 H, H-3), 2.12 (t, 1 H, J = 6.3, OH), 1.94 (irr t, 2 H, >CH=CHCH2), 1.64 (septuplet, 2 H, 2 ×CHMe2), 1.54 (quintuplet, 1 H, CH(i-Bu)2), 1.05 (m, 4 H, 2 ×CH2CHMe2), 0.80-0.87 (m, 12 H, 4 ×CH3); 13C NMR (CDCl3) δ138.3,138.2, 128.5, 127.8, 127.7, 119.2, 113.1, 84.9, 73.8, 72.7, 71.4, 65.5, 44.1, 34.3, 33.7, 33.3, 25.4, 23.2, 22.9; FABMS m/z (relative intensity) 375 (MH+, 12.1). Anal. (C24H38O3 •0.1H2O) C, H.
(E/Z)-{4-[5-methyl-3-(2-methylpropyl)hexylidene]-2-[(phenylmethoxy)methyl]-2-2,3,5-trihydrofuryl}methyl 2,2-dimethylpropanoate (20)
A stirred solution of 19 (159 mg, 0.42 mmol) in CH2Cl2 (4 mL) at 0 °C was treated with triethylamine (0.18 mL, 1.3 mmol) and pivaloyl chloride (78 μL, 0.63 mmol) for 10 min at the same temperature. Concentration in vacuo followed by flash column chromatography on silica gel with EtOAc:hexanes (1:10) as eluant gave 20 as an oil (152 mg, 82 %) consisting of an inseparable mixture of geometric isomers which was used directly in the next step without further purification. Only the signals for the dominant isomer are reported: 1H NMR (CDCl3) δ7.25-7.35 (m, 5 H, Ph), 5.25-5.30 (m, 1 H, >C=CH), 4.55 (AB q, 2 H, J = 12.3 Hz, OCH2Ph), 4.40 (m, 2 H, H-5), 4.13 (AB q, 2 H, J = 11.3 Hz, CH2OCO), 3.45 (AB q, 2 H, J = 9.3 Hz, CH2OBn), 2.44 (m, 2 H, H-3), 1.94 (irr t, 2 H, >CH=CHCH2), 1.62 (septuplet, 2 H, 2 ×CHMe2), 1.52 (quintuplet, 1 H, CH(i-Bu)2), 1.18 (br s, 9 H, CO(CH3)3), 1.01-1.09 (m, 4 H, 2 ×CH2CHMe2), 0.70-0.99 (m, 12 H, 4 ×CH3).
(E/Z)-{2-(Hydroxymethyl)-4-[5-methyl-3-(2-methylpropyl)hexylidene]-2-2,3,5-trihydrofuryl}methyl 2,2-Dimethylpropanoate (8)
A stirred solution of 20 (141 mg, 0.31 mmol) in CH2Cl2 (4 mL) was cooled to −78 °C and treated dropwise with BCl3 (1 M in CH2Cl2, 93 μL). After 30 min at −78 °C, the reaction was quenched with a saturated aqueous NaHCO3 solution (10 mL) and immediately partitioned between CH2Cl2 (10 mL) and the aqueous NaHCO3 solution. The organic layer was washed with water and brine, dried (MgSO4) and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with EtOAc:hexanes (1:2) as eluant to give 8 as an oil (66 mg, 60 %) consisting of an inseparable ca. 14:1 mixture of geometric isomers. Only the signals for the dominant isomer are reported: 1H NMR (CDCl3) δ5.75-5.81 (m, 1H, >C=CH), 4.27 (br s, 2 H, H-5), 4.22 (d, 1 H, J = 11.4 Hz, CHHOCO), 3.87 (d, 1 H, J = 11.4 Hz, CHHOCO, 3.38 (broad AB q, 2 H, CH2OH), 2.63 (br s, 1 H, CH2OH), 2.41 (AB q, 2 H, J = 14.4, H-3), 2.00 (irr t, 2 H, >CH=CHCH2), 1.62 (septuplet, 2 H, 2 ×CHMe2), 1.57 (quintuplet, 1 H, CH(i-Bu)2), 1.24 (s, 9 H, CO(CH3)3), 1.06 (overlapping triplets, 4 H, 2 ×CH2CHMe2), 0.79-0.90 (singlets, 12 H, 4 ×CH3); 13C NMR (CDCl3) δ179.6, 134.9, 132.3, 74.8, 65.8, 65.3, 52.2, 44.2, 39.1, 33.3, 31.5, 27.3, 25.4, 23.2, 23.1, 22.9; FABMS m/z (relative intensity) 369 (MH+, 11.7). Anal. (C 22H40O4• 0.5H2O) C, H.
(E)-5-[(4-Methoxyphenoxy)methyl]-3-[5-methyl-3-(2-methylpropyl)hexilidene]-5-[(phenylmethoxy)methyl]-4,5-dihydrofuran-2-one (21) and (Z)-5-[(4-methoxyphenoxy)methyl]-3-[5-methyl-3-(2-methylpropyl)hexilidene]-5-[(phenylmethoxy)methyl]-4,5-dihydrofuran-2-one (22)
These compounds were synthesized according to previously published methods.19
(Z)-5-(Hydroxymethyl)-5-[(4-methoxyphenoxy)methyl]-3-[5-methyl-3-(2-methylpropyl)hexylidene]-4,5-dihydrofuran-2-one (24)
A stirred solution of 22 (769 mg, 1.5 mmol) in CH2Cl2 (8 mL) was cooled to −78 °C and treated dropwise with BCl3 (1 M in CH2Cl2, 4.5 mL). After 20 min at −78 °C, the reaction was quenched with a saturated aqueous NaHCO3 solution (20 mL) and immediately partitioned between CH2Cl2 (15 mL) and the aqueous NaHCO3 solution. The organic layer was washed with water and brine, dried (MgSO4) and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with EtOAc:hexanes (1:4) as eluant to give 24 as an oil (530 mg, 86 %); 1H NMR (CDCl3) δ6.84 (s, 4 H, PhOCH3), 6.23-6.28 (tt, 1H, J = 7.6, 2.2 Hz, >C=CH), 4.00 (AB q, 2 H, J = 9.5, Hz, OCH2PhOCH3), 3.80 (br AB q, 2 H, J = 12.1, Hz, CH2OH), 3.76 (s, 3 H, OCH3), 2.98 (d of irr quartets, 1 H, J = 16.4 Hz, H-4a), 2.89 (d of irr quartets, 1 H, J = 16.4 Hz, H-4b), 2.62-2.74 (m, 2 H, >CH=CHCH2), 2.16 (br s, 1 H, CH2OH), 1.57-1.71 (overlapping septet and quintet, 3 H, 2 ×CHMe2, CH(i-Bu)2), 1.10 (irr t, 4 H, 2 ×CH2CHMe2), 0.83-0.88 (m, 12 H, 4 ×CH3); 13C NMR (CDCl3) δ169.1, 154.5, 152.6, 144.7, 125.0, 115.8, 114.8, 83.0, 70.3, 65.5, 55.9, 44.1, 33.4, 32.2, 25.3, 23.2, 22.9, 22.8; FABMS m/z (relative intensity) 405 (MH+, 57.3), 404 (M+•, 100). Anal. (C24H36O5) C, H.
(Z)-{2-[(4-Methoxyphenoxy)methyl]-4-[5-methyl-3-(2-methylpropyl)hexylidene]-5-oxo-2-2,3-dihydrofuryl}methyl Methylsulfonate (26)
A solution of 24 (510 mg, 1.3 mmol) in CH2Cl2 (5 mL) was cooled to 0 °C, treated with triethylamine (0.54 mL, 3.9 mmol) and methanesulfonyl chloride (0.15 mL, 1.95 mmol), and stirred for 10 min while slowly warming to room temperature. The reaction mixture was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel with EtOAc:hexanes (1:10) as eluant to give 26 as an oil (640 mg, 100 %); 1H NMR (CDCl3) δ6.82 (s, 4 H, PhOCH3), 6.29-6.34 (tt, 1 H, J = 7.6, 2.3 Hz, >C=CH), 4.43 (s, 2 H, CH2SO2CH3), 4.01 (AB q, 2 H, J = 9.7, Hz, OCH2PhOCH3), 3.76 (s, 3 H, OCH3), 3.04 (s, 3 H, SO2CH3), 3.02 (d of irr quartets, 1 H, J = 16.6 Hz, H-3a), 2.96 (d of irr quartets, 1 H, J = 16.6 Hz, H-3b), 2.62-2.75 (m, 2 H, >CH=CHCH2), 1.58-1.71 (m, 3 H, 2 ×CHMe2, CH(i-Bu)2), 1.04-1.16 (m, 4 H, 2 ×CH2CHMe2), 0.80-0.89 (m, 12 H, 4 ×CH3); 13C NMR (CDCl3) δ168.1, 154.8, 152.1, 145.9, 123.5, 115.8, 114.9, 80.3, 70.3, 70.0, 55.8, 44.1, 37.8, 33.7, 33.4, 32.3, 25.3, 23.2, 22.9, 22.8; FABMS m/z (relative intensity) 483 (MH+, 71.5), 482 (M•+, 100). Anal. (C25H38O7S) C, H.
(Z)-{2-(Hydroxymethyl)-4-[5-methyl-3-(2-methylpropyl)hexylidene-5-oxo-2-2,3-dihydrofuryl}methyl Methylsulfonate (28)
A solution of 26 (618 mg, 1.28 mmol) in CH3CN-H2O (4:1, 8 mL) was cooled to 0 °C and treated with ammonium cerium(IV) nitrate (2.1 g, 3.84 mmol). After 30 min stirring at the same temperature, the reaction mixture was diluted with EtOAc (10 mL). The organic layer was washed with water and brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with EtOAc:hexanes (1:4) as eluant to give 28 as an oil (453 mg, 93 %); 1H NMR (CDCl3) δ6.28-6.33 (tt, 1 H, J = 7.6, 2.3 Hz, >C=CH), 4.36 (AB q, 2 H, J = 11.1 Hz, CH2SO2CH3), 3.69-3.76 (m, 2 H, CH2OH), 3.07 (s, 3 H, SO2CH3), 2.85 (br AB q, 2 H, J = 17.1 Hz, H-3), 2.65-2.71 (m, 2 H, >CH=CHCH2), 2.18 (br s, 1 H, CH2OH), 1.60-1.71 (m, 3 H, 2 _CHMe2, CH(i-Bu)2), 1.02-1.15 (m, 4 H, 2 ×CH2CHMe2), 0.85-0.87 (m, 12 H, 4 ×CH3); 13C NMR (CDCl3) δ168.5, 146.0, 123.7, 81.8, 69.8, 64.6, 44.1, 37.7, 33.4, 33.0, 32.3, 25.3, 23.2, 23.1, 22.8; FABMS m/z (relative intensity) 377 (MH+, 100.0). Anal. (C18H32O6S) C, H.
(Z)-5-[(2,2-Dimethylpropoxy)methyl)-5-(hydroxymethyl)-3-[5-methyl-3-(2-methylpropyl)hexylidene]-4,5-dihydrofuran-2-one (10)
A cooled solution of neopentyl alcohol (190 mg, 2.2 mmol) in DMF (5mL) at 0 °C was treated with triethylamine (0.23 mL, 1.62 mmol). The cooling bath was removed and after 30 min stirring at room temperature, a solution of 28 (204 mg, 0.54 mmol) in DMF (5 mL) was added. The resulting solution was stirred for 1 h and the reaction was quenched by the slow addition of water (5 mL). Following several extractions with EtOAc (40 mL), the combined organic layers were washed with water and brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with ethyl ether:hexanes (1:10) as eluant to give 10 (59 mg, 30 %) as an oil; 1H NMR (CDCl3) δ6.01 (t, 1 H, J = 7.2 Hz, >C=CH), 3.86 (AB q, 2 H, J = 10.7, Hz, CH2OCH2C(CH3)3), 3.71 (dd, 1 H, J = 12.3, 4.9 Hz, CHHOH), 3.64 (dd, 1 H, J = 12.3, 7.8 Hz CHHOH), 2.83 (d, 1 H, J = 4.7 Hz, H-4a), 2.68 (s, 2 H, OCH2C(CH3)3), 2.66 (d, 1 H, J = 4.7 Hz, H-4b), 2.46 (irr t, 2 H, J ≈ 6.6 Hz, >CH=CHCH2), 1.98-2.01 (br dd, J ≈ 8.0, 5.2 Hz, 1 H, CH2OH), 1.54-1.66 (m, 3 H, 2 ×CHMe2, CH(i-Bu)2), 1.06-1.10 (m, 4 H, 2 ×CH2CHMe2), 0.98 (s, 9 H, OCH2C(CH3)3), 0.87 (br s, 6 H, 2 ×CH3), 0.85 (br s, 6 H, 2 ×CH3); 13C NMR (CDCl3) δ171.4, 168.5, 145.6, 127.0, 74.4, 63.5, 59.4, 49.7, 44.2, 36.4, 34.5, 33.4, 31.5, 26.8, 25.3, 23.2, 23.2, 22.9; FABMS m/z (relative intensity) 369 (MH+, 100.0). Anal. calcd for C22H40O4• 0.8H2O: C, 69.00; H, 10.95. Found: C, 68.82; H, 10.28.
(E)-5-(Hydroxymethyl)-5-[(4-methoxyphenoxy)methyl]-3-[5-methyl-3-(2-methylpropyl)hexylidene]-4,5-dihydrofuran-2-one (23)
Starting from 21 (592 mg, 1.2 mmol) and following the same procedure as for 24, compound 23 was obtained as an oil (360 mg, 74 %); 1H NMR (CDCl3) δ6.82 (s, 4 H, PhOCH3), 6.78-6.84 (m, 1H, >C=CH), 4.04 (AB q, 2 H, J = 9.6 Hz, OCH2Ph), 3.87 (dd, 1 H, J = 12.3, 7.2 Hz, CHHOH), 3.78 (dd, 1 H, J = 12.3, 6.4 Hz, CHHOH), 3.77 (s, 3 H, OCH3), 2.92 (dm, 1 H, J = 16.7 Hz, H-4a), 2.78 (dm, 1 H, J = 16.7 Hz, H-4b), 2.13-2.16 (irr t, 2 H, >CH=CHCH2), 1.98 (br t, 1 H, J ≈ 7 Hz, CH2OH), 1.72 (irr quintet, 1 H, CH(i-Bu)2), 1.64 (irr septet of doublets, 2 H, 2 ×CHMe2), 1.05-1.16 (m, 4 H, 2 ×CH2CHMe2), 0.79-0.89 (m, 12 H, 4 × CH3); 13C NMR (CDCl3) δ170.1, 154.6, 152.6, 141.1, 127.1, 115.8, 114.8, 83.7, 70.4, 65.6, 55.9, 44.0, 34.9, 33.0, 30.3, 25.4, 23.1, 22.8; FABMS m/z (relative intensity) 405 (MH+, 53.8), 404 (M•+, 100). Anal. (C24H36O5• 0.2H2O) C, H.
(E)-{2-[(4-Methoxyphenoxy)methyl]-4-[5-methyl-3-(2-methylpropyl)hexylidene]-5-oxo-2-2,3-dihydrofuryl}methyl Methylsulfonate (25)
Starting from 23 (347 mg, 0.86 mmol) and following the same procedure as for 26, compound 25 was obtained as an oil (415 mg, 100 %); 1H NMR (CDCl3) δ6.79 (s, 4 H, PhOCH3), 6.76-6.84 (m, 1H, >C=CH), 4.41 (s, 2 H, CH2SO2CH3), 3.98 (AB q, 2 H, J = 9.7 Hz, OCH2Ph), 3.72 (s, 3 H, OCH3), 3.01 (s, 3 H, SO2CH3), 2.90 (dm, 1H, J = 16 Hz, H-3a), 2.82 (dm, 1 H, J = 16 Hz, H-3b), 2.09-2.14 (m, 2 H, >CH=CHCH2), 1.68 (quintuplet, 1 H, CH(i-Bu)2), 1.59 (m, 2 H, 2 ×CHMe2), 1.00-1.13 (m, 4 H, 2 ×CHCH2CHMe2), 0.76-0.88 (m, 12 H, 4 ×CH3); 13C NMR (CDCl3) δ169.1, 154.8, 152.1, 142.3, 125.6, 115.8, 114.9, 80.9, 70.4, 70.2, 55.8, 44.0, 37.9, 35.1, 33.0, 30.7, 25.4, 23.1, 22.8, 22.7; FABMS m/z (relative intensity) 483 (MH+, 62.0), 482 (M•+, 100). Anal. (C25H38O7S) C, H.
(E)-{2-(Hydroxymethyl)-4-[5-methyl-3-(2-methylpropyl)hexylidene-5-oxo-2-2,3-dihydrofuryl}methyl methylsulfonate (27)
Starting from 25 (415 mg, 0.86 mmol) and following the same procedure as for 28, compound 27 was obtained as an oil (271 mg, 84 %); 1H NMR (CDCl3) δ6.72-6.77 (m, 1 H, >C=CH), 4.30 (AB q, 2 H, J = 11.1 Hz, CH2SO2CH3), 3.68 (br s, 2 H, CH2OH), 3.03 (s, 3 H, SO2CH3), 2.94 (br s, 1 H, CH2OH), 2.71 (m, 2 H, H-3), 2.07 (m, 2 H, >CH=CHCH2), 1.66 (irr quintet, 1 H, CH(i-Bu)2), 1.57 (irr septet, 2 H, 2 ×CHMe2), 0.97-1.10 (m, 4 H, 2 ×CH2CHMe2), 0.75-0.85 (m, 12 H, 4 ×CH3); 13C NMR (CDCl3) δ169.7, 142.3, 126.0, 82.7, 70.0, 64.7, 44.0, 37.7, 35.1, 32.9, 29.9, 25.4, 23.1, 22.8, 22.7; FABMS m/z (relative intensity) 377 (MH+, 100.0). Anal. (C18H32O6S) C, H.
(E)-5-[(2,2-Dimethylpropoxy)methyl)-5-(hydroxymethyl)-3-[5-methyl-3-(2-methylpropyl)hexylidene]-4,5-dihydrofuran-2-one (9)
Starting from 27 (147 mg, 0.39 mmol) and following the same procedure as for 10, compound 9 (100 mg, 70 %) was obtained as an oil; 1H NMR (DMSO) δ6.78 (t, 1 H, J = 7.2 Hz, >C=CH), 4.81 (t, 1 H, J = 6.0 Hz, CH2OH), 3.71 (AB q, 2 H, J = 10.5, Hz, CH2OCH2C(CH3)3), 3.43 (dd, 1 H, J = 11.9, 5.8, CHHOH), 3.32 (dd, 1 H, J = 11.9, 6.0 Hz, CHHOH), 2.67 (AB q, 2 H, J = 14.2, Hz, OCH2C(CH3)3), 2.51 (d, 1 H, J = 5.1 Hz, H-4a), 2.28 (d, 1 H, J = 5.1 Hz, H-4b), 2.09 (irr t, 2 H, J ≈ 6.7 Hz, >CH=CHCH2), 1.48-1.61 (m, 3 H, 2 ×CHMe2, CH(i-Bu)2), 0.93-1.05 (irr t, J ≈ 7.0 Hz, 4 H, 2 ×CH2CHMe2), 0.86 (s, 9 H, OCH2C(CH3)3), 0.77-0.79 (singlets, 12 H, 4 ×CH3); 13C NMR (CDCl3) δ168.4, 146.5, 127.2, 74.3, 64.0, 59.5, 50.4, 44.5, 44.4, 34.0, 33.2, 31.7, 28.6, 26.6, 25.4, 23.1, 22.8; FABMS m/z (relative intensity) 369 (MH+, 42.9). Anal. (C22H40O4) C, H.
5-(Hydroxymethyl)-5-[(phenylmethoxy)methyl]-3,4,5-trihydrofuran-2-one (30)
A stirred solution of 2919 (3.3g, 9.6 mmol) in CH3CN-H2O (4:1, 20 mL) was cooled to 0 °C and treated with ammonium cerium(IV) nitrate (15.0 g, 28.8 mmol). After further stirring for 30 min at the same temperature, the reaction mixture was diluted with CH2Cl2, the organic layer was washed with water and brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with EtOAc:hexanes (1:2) as eluant to give 30 as an oil (1.76 g, 78 %); 1H NMR (CDCl3) _7.25-7.36 (m, 5 H, Ph), 4.54 (s, 2 H, OCH2Ph), 3.75 (dd, 1 H, J = 12.1, 6.4 Hz, CHHOH), 3.62 (dd, 1 H, J = 12.1, 6.4 Hz, CHHOH), 3.55 (AB q, 2 H, J = 10.3 Hz, CH2OBn), 2.96 (t, 1 H, J = 6.4 Hz, CH2OH), 2.53-2.69 (m, 2 H, H-3), 2.09-2.20 (m, 2 H, H-4) ; 13C NMR (CDCl3) δ177.8,137.7, 128.7, 128.0, 127.8, 88.0, 73.8, 72.6, 65.5, 29.4, 25.8. FABMS m/z (relative intensity) 237 (MH+, 20.9). Anal. (C13H16O4) C, H.
5,5-bis[(phenylmethoxy)methyl]-3,4,5-trihydrofuran-2-one (31)
A solution of 30 (1.76 g, 7.5 mmol) in DMF (10 mL) at 0 °C was treated with NaH (60% dispersion in mineral oil, 600 mg, 15.0 mmol). After removing the cooling bath, stirring continued for 10 min at room temperature and then benzyl bromide (1.1 mL, 9.0 mmol) was added. The resulting solution was stirred for 1 h, then quenched by the slow addition of water (70 mL) and extracted with EtOAc (50 mL) several times. The combined organic layers were washed with water and brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with ethyl ether:hexanes (1:20) as eluant to give 31 (1.2 g, 50 %) as an oil; 1H NMR (CDCl3) δ7.26-7.38 (m, 5 H, Ph), 4.55 (s, 4 H, OCH2Ph), 3.58 (AB q, 4 H, J = 10.3 Hz, CH2OBn), 2.60 (irr t, 2 H, J ≈ 8.6 Hz, H-3), 2.15 (irr t, 2 H, J ≈ 8.6 Hz, H-4). This compound was used in the next step without further purification.
(E)-5,5-bis[Phenylmethoxy)methyl]-3-[5-methyl-3-(2-methylpropyl)hexylidene]-4,5-dihydrofuran-2-one (32) and (Z)-5,5-bis[Phenylmethoxy)methyl]-3-[5-methyl-3-(2-methylpropyl)hexylidene]-4,5-dihydrofuran-2-one (33)
A stirred solution of 31 (1.2 g, 3.8 mmol) in THF (10 mL) was cooled to −78 °C and treated dropwise with lithium bis(trimethylsilyl)amide (1 M in THF, 7.6 mL). After further stirring for 30 min at −78 °C, the mixture was treated with a solution of 13 (647 mg, 3.8 mmol) in THF (10 mL) and stirred for 1 h at the same temperature. The reaction was quenched by the slow addition of saturated aqueous NH4Cl (20 mL) and extracted with ether (20 mL) several times. The combined organic layers were washed with water and brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with EtOAc:hexanes (1:10) as eluant to give the intermediate ß-hydroxylactone as an oil (1.47 g, 2.9 mmol), which was then immediately dissolved in CH2Cl2 (10 mL), cooled to 0 °C and treated with triethylamine (1.2 mL, 8.7 mmol) and methanesulfonyl chloride (0.34 mL, 4.4 mmol). After warming to room temperature and further stirring at room temperature for 10 min, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 1.3 mL, 8.7 mmol) was added and the resulting solution was stirred for a total of 15 min. The reaction mixture was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel with EtOAc:hexanes (1:10) as eluant to give 32 (425 mg, 31 %) and 33 (599 mg, 45 %) as oils.
32: 1H NMR (CDCl3) δ7.25-7.36 (m, 5 H, Ph), 6.72-6.77 (br tt, J = 7.6 Hz, 1 H, >C=CH), 4.56 (AB s, 4 H, 2 ×OCH2Ph), 3.58 (AB q, 4 H, J = 10.1 Hz, 2 ×CH2OBn), 2.74 (br s, 2 H, H-4), 2.10 (t, 2 H, >CH=CHCH2), 1.56-1.72 (m, 3 H, 2 ×CHMe2, CH(i-Bu)2), 1.08 (t, 4 H, J = 7.0 Hz, 2 ×CH2CHMe2), 0.80-0.89 (m, 12 H, 4 ×CH3); FABMS m/z (relative intensity) 479 (MH+, 1.7). Anal. (C31H42O4•0.4H2O) C, H.
33: 1H NMR (CDCl3) δ7.25-7.35 (m, 5 H, Ph), 6.12-6.18 (tt, 1 H, J = 7.5, 2.3 Hz, >C=CH), 4.56 (AB s, 4 H, 2 ×OCH2Ph), 3.57 (AB q, 4 H, J = 10.1 Hz, 2 ×CH2OBn), 2.82 (AB q, J = 2.2 Hz, 2 H, H-4), 2.64-2.70 (m, 2 H, >CH=CHCH2), 1.56-1.72 (m, 3 H, 2 ×CHMe2, CH(i-Bu)2), 1.08 (irr t, J ≈ 7.1 Hz, 4 H, 2 ×CH2CHMe2), 0.78-0.88 (m, 12 H, 4 ×CH3); FABMS m/z (relative intensity) 479 (MH+, 1.1). Anal. (C31H42O4•0.2H2O) C, H.
(E)-5,5-bis[(Phenylmethoxy)methyl]-3-[5-methyl-3-(2-methylpropyl)hexylidene]-4,5-dihydrofuran-2-thione (34)
Lawesson’s reagent (2,4-bis(4-methoxyphenyl)-1,3-dithia-2,4-diphosphetane-2,4-disulfide,1.0 g, 2.6 mmol) and 32 (or 33) (599 mg, 1.3 mmol) were dissolved in toluene (6 mL) and stirred for 18 h at 110 °C. After cooling to room temperature, the solution was concentrated under reduced pressure until an oily material appeared, and then hexane was added to induce precipitation of excess Lawesson’s reagent. The precipitate was removed by filtration, and the filtrate was evaporated to dryness. Hexane was added again to the residue and the above filtration-evaporation process was repeated twice. The resulting residue was then subjected to column chromatography to give 34 (470 mg, 73%) as an oil; 1H NMR (CDCl3) δ7.25-7.36 (m, 5 H, Ph), 7.05-7.10 (tt, 1H, J = 7.8, 2.7 Hz, >C=CH), 4.57 (s, 4 H, 2 ×OCH2Ph), 3.64 (AB q, 4 H, J = 10.3 Hz, 2 ×CH2OBn), 2.86 (br s, 2 H, H-4), 2.09-2.12 (irr dd, 2 H, >CH=CHCH2), 1.75 (irr quintet, 1 H, CH(i-Bu)2), 1.64 (irr septet, 2 H, 2 × CHMe2), 1.09-1.17 (m, 4 H, 2 ×CH2CHMe2), 0.80-0.95 (m, 12 H, 4 ×CH3) ; 13C NMR (CDCl3) δ211.4, 142.9, 139.1, 137.6, 128.4, 127.7, 127.6, 92.4, 73.7, 71.5, 44.1, 35.8, 33.1, 31.4, 25.3, 22.9, 22.6; FABMS m/z (relative intensity) 495 (MH+, 2.7). Anal. (C31H42O3S•1.1H2O) C, H.
(E)-{2-(Hydroxymethyl)-4-[5-methyl-3-(2-methylpropyl)hexylidene]-5-thioxo-2-2,3-dihydrofuryl}methyl 2,2-Dimethylpropanoate (11)
A stirred solution of 34 (470 mg, 0.95 mmol) in CH2Cl2 (10 mL) was cooled to −78 °C and treated dropwise with BCl3 (1 M soln in CH2Cl2, 5.7 mL). After further stirring for 30 min at the same temperature, the reaction was quenched by the addition of an aqueous saturated NaHCO3 solution (10 mL), warmed to room temperature, and immediately partitioned between ether and the aqueous NaHCO3 solution. The organic layer was washed with water and brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with EtOAc:hexanes (1:2) as eluant to give a diol intermediate as a clear oil (242 mg, 81 %) that was used immediately in the following step.
A stirred solution of the diol (71 mg, 0.54 mmol) in CH2Cl2 (2 mL) was cooled to 0 °C and treated with triethylamine (48 μL, 0.35 mmol) and pivaloyl chloride (31 μL, 0.25 mmol) for 10 min. The reaction mixture was then concentrated in vacuo and the residue was purified by flash column chromatography on silica gel with EtOAc:hexanes (1:10) as eluant to give 11 as a yellowish oil (59 mg, 65 %); 1H NMR (CDCl3) δ7.06-7.11 (tt, 1H, J = 7.7, 2.8 Hz, >C=CH), 4.37 (d, 1 H, J = 12.1, Hz, CHHOCO), 4.20 (d, 1 H, J = 12.1, Hz, CHHOCO), 3.76 (AB q, 2 H, J = 12.3, Hz, CH2OH), 2.91 (dm, 1 H, J ≈ 16.5 Hz, H-3a), 2.76 (dm, 1 H, J ≈ 16.5 Hz, H-3b), 2.30-2.40 (br s, 1 H, CH2OH), 2.09-2.13 (m, 2 H, >CH=CHCH2), 1.75 (irr quintet, 1 H, CH(i-Bu)2), 1.63 (irr septet, 2 H, 2 ×CHMe2), 1.18 (s, 9 H, COCH(CH3)3), 1.07-1.18 (m, 4 H, 2 ×CH2CHMe2), 0.86-0.90 (m, 12 H, 4 _CH3); 13C NMR (CDCl3) δ210, 178.1, 143.9, 138.4, 91.8, 65.1, 64.4, 44.1, 35.9, 33.0, 30.9, 27.0, 25.3, 25.2, 22.9, 22.6 ; FABMS m/z (relative intensity) 399 (MH+, 20.1). Anal. (C22H38O4S• 0.33H2O) C, H.
(E)-5-[(2,2-Dimethyl-1-thioxopropoxy)methyl]-5-(hydroxymethyl)-3-[5-methyl-3-(2-methylpropyl)hexylidene]-4,5-dihydrofuran-2-one (12)
BCl3 (1M in CH2Cl2, 5.3 mL) was added slowly to a −78 °C solution of 32 (425 mg, 0.89 mmol) in CH2Cl2 (10 mL) and stirred for 1 h. Aqueous saturated NaHCO3 (10 mL) was added slowly and the mixture was then diluted with CH2Cl2 (4 mL). The layers were separated and the aqueous layer was further extracted with CH2Cl2. The combined organic extract was dried (MgSO4), concentrated in vacuo, and the residue was purified by silica gel column chromatography to give diol 35 (200 mg, 75%) as a clear oil that was used directly for the following step. [FABMS m/z (relative intensity) 299 (MH+, 100)]
A solution of 35 (59 mg, 0.2 mmol) in THF (10 mL) at room temperature was treated with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 56 μL, 0.40 mmol) and 2,2-dimethyl-1-(6-nitrobenzotriazolyl)propane-1-thione (36)21 (53 mg, 0.25 mmol) and stirred for 10 min. The reaction mixture was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel with EtOAc:hexanes (1:40) as eluant to give 12 as a yellowish oil (50 mg, 60 %); 1H NMR (CDCl3) δ6.80-6.85 (tt, 1 H, J = 7.6, 2.8 Hz, 1 H, >C=CH), 4.55 (AB q, 2 H, J = 11.9 Hz, CH2OCS), 3.84 (dd, 1 H, J = 12.1, 6.4 Hz, CHHOH), 3.71 (dd, 1 H, J = 12.1, 6.4 Hz, CHHOH), 2.87 (dm, 1 H, J ≈ 12.5 Hz, H-4a), 2.73 (dm, 1 H, J ≈ 12.5 Hz, H-4b), 2.19 (t, 1 H, J = 6.4 Hz, CH2OH), 2.13 (m, 2 H, >CH=CHCH2), 1.72 (irr quintet, 1 H, CH(i-Bu)2), 1.64 (irr septet, 2 H, 2 × CHMe2), 1.27 (s, 9 H, CS(CH3)3), 1.05-1.15 (m, 4 H, 2 ×CH2CHMe2), 0.80-0.90 (m, 12 H, 4 ×CH3); FABMS m/z (relative intensity) 399 (MH+, 28.2), 101 ((CH3)3C≡S+, 66). Anal. (C22H38O4S) C, H.
[3H]PDBu Binding Assay
[3H]PDBu was obtained from PerkinElmer (no longer available as a catalog item). PDBu was purchased from LC Laboratories (Woburn, MA). The recombinant full-length PKCα was purchased from Invitrogen (Carlsbad, CA). The recombinant plasmid of GST-δC1b was constructed as described previously.31 The δC1b protein was expressed and purified from BL-21 (DE3) E. coli (Stratagene, La Jolla, LA) using a B-PER GST spin purification kit (Pierce Biotechnology, Rockford, IL), according to the manufacturer’s instruction. The purity of the protein was verified by SDS-PAGE and staining with Coomassie Blue.
Binding of [3H]PDBu to PKCα and the isolated δC1b domain was measured using the polyethylene glycol precipitation assay.23 Briefly, an assay mixture (250 μL) containing 50 mM Tris-HCl (pH 7.4), 100 μg/ml phosphatidylserine (PS), 4 mg/ml bovine IgG, [3H]PDBu, 0.1 mM CaCl2 (for PKCα, or 1 mM EGTA for δC1b), various concentrations of competing ligand and the receptor was incubated for 5 min at 37°C (for PKCα) or 10 min at 18°C (for δC1b). The samples were then chilled on ice for 7 min, and 200 μL of 35% polyethylene glycol in 50 mM Tris-HCl (pH 7.4) was added. The tubes were incubated on ice for an additional 10 min and then centrifuged at 4 °C. A 100 μL aliquot of the supernatant was removed for the determination of the free [3H]PDBu concentration, and the pellet was carefully dried. The tip of the tube was cut off and the pellet was counted in a scintillation counter to determine the total bound [3H]PDBu. Specific binding was calculated as the difference between the total and the nonspecific binding, which was determined in the presence of 64 _M non-radioactive PDBu. For measuring the competitive binding of the compound with [3H]PDBu in the absence of PS, a similar protocol was used except that the PS was omitted.
Supplementary Material
Combustion analysis for compounds 8, 9, 10, 11, 12, 15, 18, 19, 23, 24, 25, 26, 27, 28, 30, 32, 33, 34. This material is available free of charge via the Internet at http://pubs.acs.org.

Structures of compounds 1-12
Acknowledgments
This publication has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. NO1-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- 1.Nishizuka Y. Intracellular Signaling by Hydrolysis of Phospholipids and Activation of Protein-Kinase-C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 2.Hodgkin MN, Pettitt TR, Martin A, Michell RH, Pemberton AJ, Wakelam MJO. Diacylglycerols and phosphatidates: which molecular species are intracellular messengers? Trends Biochem Sci. 1998;23:200–204. doi: 10.1016/s0968-0004(98)01200-6. [DOI] [PubMed] [Google Scholar]
- 3.Newton AC. Diacylglycerol’s affair with protein kinase C turns 25. Trends Pharmacol Sci. 2004;25:175–177. doi: 10.1016/j.tips.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Yang CF, Kazanietz MG. Divergence and complexities in DAG signaling: looking beyond PKC. Trends Pharmacol Sci. 2003;24:602–608. doi: 10.1016/j.tips.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Shirai Y, Saito N. Activation mechanisms of protein kinase C: maturation, catalytic activation, and targeting. J Biochem (Tokyo) 2002;132:663–668. doi: 10.1093/oxfordjournals.jbchem.a003271. [DOI] [PubMed] [Google Scholar]
- 6.Brose N, Rosenmund C. Move over protein kinase C, you’ve got company: Alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci. 2002;115:4399–4411. doi: 10.1242/jcs.00122. [DOI] [PubMed] [Google Scholar]
- 7.Newton AC, Keranen LM. Phosphatidyl-L-Serine Is Necessary for Protein-Kinase Cs High-Affinity Interaction with Diacylglycerol-Containing Membranes. Biochemistry. 1994;33:6651–6658. doi: 10.1021/bi00187a035. [DOI] [PubMed] [Google Scholar]
- 8.Newton AC. Regulation of protein kinase C. Curr Opin Cell Biol. 1997;9:161–167. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- 9.Oancea E, Teruel MN, Quest AFG, Meyer T. Green fluorescent protein (GFP)-tagged cysteine-rich domains from protein kinase C as fluorescent indicators for diacylglycerol signaling in living cells. J Cell Biol. 1998;140:485–498. doi: 10.1083/jcb.140.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakai N, Sasaki K, Ikegaki N, Shirai Y, Ono Y, Saito N. Direct visualization of the translocation of the gamma- subspecies of protein kinase C in living cells using fusion proteins with green fluorescent protein. J Cell Biol. 1997;139:1465–1476. doi: 10.1083/jcb.139.6.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oancea E, Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- 12.Marquez VE, Blumberg PM. Synthetic diacylglycerols (DAG) and DAG-lactones as activators of protein kinase C (PK-C) Acc Chem Res. 2003;36:434–443. doi: 10.1021/ar020124b. [DOI] [PubMed] [Google Scholar]
- 13.Zhang GG, Kazanietz MG, Blumberg PM, Hurley JH. Crystal-Structure of the Cys2 Activator-Binding Domain of Protein-Kinase C-Delta in Complex with Phorbol Ester. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 14.Hritz J, Ulicny J, Laaksonen A, Jancura D, Miskovsky P. Molecular interaction model for the C1B domain of protein kinase C-gamma in the complex with its activator phorbol-12-myristate-13-acetate in water solution and lipid bilayer. J Med Chem. 2004;47:6547–6555. doi: 10.1021/jm049786s. [DOI] [PubMed] [Google Scholar]
- 15.Sigano DM, Peach ML, Nacro K, Choi Y, Lewin NE, Nicklaus MC, Blumberg PM, Marquez VE. Differential binding modes of diacylglycerol (DAG) and DAG lactones to protein kinase C (PK-C) J Med Chem. 2003;46:1571–1579. doi: 10.1021/jm020476o. [DOI] [PubMed] [Google Scholar]
- 16.Benzaria S, Bienfait B, Nacro K, Wang S, Lewin NE, Beheshti M, Blumberg PM, Marquez VE. Conformationally constrained analogues of diacylglycerol (DAG). 15. The indispensable role of the sn-1 and sn-2 carbonyls in the binding of DAG-lactones to protein kinase C (PK-C) Bioorg Med Chem Lett. 1998;8:3403–3408. doi: 10.1016/s0960-894x(98)00614-3. [DOI] [PubMed] [Google Scholar]
- 17.Lee J, Han KC, Kang JH, Pearce LL, Lewin NE, Yan S, Benzaria S, Nicklaus MC, Blumberg PM, Marquez VE. Conformationally constrained analogues of diacylglycerol. 18. The incorporation of a hydroxamate moiety into diacylglycerol-lactones reduces lipophilicity and helps discriminate between sn-1 and sn-2 binding modes to protein kinase C (PK-C). Implications for isozyme specificity. J Med Chem. 2001;44:4309–4312. doi: 10.1021/jm0103965. Erratum published in J. Med. Chem. 2003, 46 (13): 2794. [DOI] [PubMed] [Google Scholar]
- 18.Nacro K, Bienfait B, Lee J, Han KC, Kang JH, Benzaria S, Lewin NE, Bhattacharyya DK, Blumberg PM, Marquez VE. Conformationally constrained analogues of diacylglycerol (DAG). 16. How much structural complexity is necessary for recognition and high binding affinity to protein kinase C? J Med Chem. 2000;43:921–944. doi: 10.1021/jm9904607. [DOI] [PubMed] [Google Scholar]
- 19.Choi Y, Kang JH, Lewin NE, Blumberg PM, Lee J, Marquez VE. Conformationally constrained analogues of diacylglycerol. 19. Synthesis and protein kinase C binding affinity of diacylglycerol lactones bearing an N-hydroxylamide side chain. J Med Chem. 2003;46:2790–2793. doi: 10.1021/jm030082c. [DOI] [PubMed] [Google Scholar]
- 20.Lee J, Kang JH, Lee SY, Han KC, Torres CM, Bhattacharyya DK, Blumberg PM, Marquez VE. Protein kinase C ligands based on tetrahydrofuran templates containing a new set of phorbol ester pharmacophores. J Med Chem. 1999;42:4129–4139. doi: 10.1021/jm980713g. [DOI] [PubMed] [Google Scholar]
- 21.Shalaby MA, Rapoport H. A general and efficient route to thionoesters via thionoacyl nitrobenzotriazoles. J Org Chem. 1999;64:1065–1070. doi: 10.1021/jo981985u. [DOI] [PubMed] [Google Scholar]
- 22.Kang JH, Siddiqui MA, Sigano DM, Krajewski K, Lewin NE, Pu YM, Blumberg PM, Lee J, Marquez VE. Conformationally constrained analogues of diacylglycerol. 24. Asymmetric synthesis of a chiral (R)-DAG-Lactone template as a versatile precursor for highly functionalized DAG-lactones. Org Lett. 2004;6:2413–2416. doi: 10.1021/ol0492041. [DOI] [PubMed] [Google Scholar]
- 23.Lewin NE, Blumberg PM. [3H]-Phorbol 12,13-Dibutyrate Binding Assay for Protein kinace C and Related Proteins. Methods Mol Biol. 2003;233:129–156. doi: 10.1385/1-59259-397-6:129. [DOI] [PubMed] [Google Scholar]
- 24.Wang QMJ, Fang TW, Nacro K, Marquez VE, Wang SM, Blumberg PM. Role of hydrophobic residues in the C1b domain of protein kinase C delta on ligand and phospholipid interactions. J Biol Chem. 2001;276:19580–19587. doi: 10.1074/jbc.M010089200. [DOI] [PubMed] [Google Scholar]
- 25.Abboud JLM, Roussel C, Gentric E, Sraidi K, Lauransan J, Guiheneuf G, Kamlet MJ, Taft RW. Studies on Amphiprotic Compounds .3. Hydrogen-Bonding Basicity of Oxygen and Sulfur-Compounds. J Org Chem. 1988;53:1545–1550. [Google Scholar]
- 26.Hadad CM, Rablen PR, Wiberg KB. C-O and C-S bonds: Stability, bond dissociation energies, and resonance stabilization. J Org Chem. 1998;63:8668–8681. [Google Scholar]
- 27.Yin M, Ochs RS. A Mechanism for the Partial Insertion of Protein Kinase C into Membranes. Biochem Biophys Res Commun. 2001;281:1277–1282. doi: 10.1006/bbrc.2001.4500. [DOI] [PubMed] [Google Scholar]
- 28.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 29.Jain MK. Introduction to Biological Membranes. 2. John Wiley & Sons; New York: 1988. pp. 51–85. [Google Scholar]
- 30.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 31.Kazanietz MG, Barchi JJ, Jr, Omichinski JG, Blumberg PM. Low affinity binding of phorbol esters to protein kinase C and its recombinant cysteine-rich region in the absence of phospholipids. J Biol Chem. 1995;270:14679–14684. doi: 10.1074/jbc.270.24.14679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Combustion analysis for compounds 8, 9, 10, 11, 12, 15, 18, 19, 23, 24, 25, 26, 27, 28, 30, 32, 33, 34. This material is available free of charge via the Internet at http://pubs.acs.org.