

Summary
During acute stress in the endoplasmic reticulum (ER), mammalian prion protein (PrP) is temporarily prevented from translocation into the ER and instead routed directly for cytosolic degradation. This ‘pre-emptive’ quality control (pQC) system benefits cells by minimizing PrP aggregation in the secretory pathway during ER stress. However, the potential toxicity of cytosolic PrP raised the possibility that persistent pQC of PrP contributes to neurodegeneration in prion diseases. Here, we find evidence of ER stress and decreased translocation of nascent PrP during prion infection. Transgenic mice expressing a PrP variant with reduced translocation at levels expected during ER stress was sufficient to cause several mild age-dependent clinical and histological manifestations of PrP-mediated neurodegeneration. Thus, an ordinarily adaptive quality control pathway can be contextually detrimental over long time periods. We propose that one mechanism of prion-mediated neurodegeneration involves an indirect ER stress-dependent effect on nascent PrP biosynthesis and metabolism.
Introduction
Several neurodegenerative diseases are caused by aberrant metabolism of the widely expressed cell surface glycoprotein PrP (reviewed in Prusiner, 1998; Collinge and Clarke, 2007; Aguzzi et al., 2008). These diseases can be inherited through PrP mutations or acquired via a transmissible agent composed largely of a misfolded isoform of PrP termed PrPSc. Exogenous PrPSc is capable of converting the normal cellular isoform (PrPC) into additional PrPSc molecules, leading to its accumulation and generating additional transmissible agent. In the familial diseases, PrP mutations appear to cause accumulation of misfolded PrP through poorly understood mechanisms that in some cases also generate PrPSc. Thus, altered PrP folding, metabolism, and accumulation are the proximal causes of both familial and transmissible prion diseases. However, the downstream events that culminate in selective neuronal death in any of these diseases are unknown.
Although it was originally assumed that the accumulation of misfolded PrP aggregates (e.g., PrPSc) would be intrinsically damaging to neurons, this view proved overly simplistic. Early tissue grafting experiments demonstrated that brain regions knocked out for the Prnp gene were immune to degeneration caused by PrPSc deposition (Brandner et al., 1996). More recently, selective post-natal knockout of Prnp in neurons halted and even reversed the damage caused by either pre-existing PrPSc or newly generated PrPSc made by adjacent non-neuronal cells (Mallucci et al., 2003; Mallucci et al., 2007). These observations have led to the conclusion that ongoing PrP expression is obligate for neuronal damage caused by PrPSc, suggesting that neurotoxic molecule(s) are actively generated from newly synthesized cellular PrP.
Two non-mutually exclusive models can explain this requirement for PrP expression. The most widely considered possibility is that conversion of PrPC to PrPSc, or perhaps the clearance of newly synthesized PrPSc, generates an intermediate species or byproduct that is neurotoxic (e.g., as proposed by Collinge and Clarke, 2007). Thus, toxicity is a cell-autonomous consequence of ongoing PrPSc replication and clearance, a process absolutely dependent on PrPC expression. At present however, PrPSc production or clearance in a cellular context is poorly understood and direct evidence for a hypothetical neurotoxic intermediate or byproduct is lacking. A less obvious model is one in which PrPSc accumulation alters cellular metabolism in a manner that cause nascent PrPC to be made in a neurotoxic form (e.g., as proposed in Hegde et al., 1999). One example of such altered metabolism may be ER stress, a commonly observed feature of various neurodegenerative diseases (Lindholm et al., 2006) including those caused by PrP (reviewed in Hetz and Soto, 2006). Furthermore, simply applying PrPSc to cultured cells causes dysregulation of ER Ca+2 homeostasis and leads to ER stress (Hetz et al., 2003). While these observations highlight at least one adverse consequence of PrPSc on cellular function, it has been unclear how such a general effect could cause neurodegeneration that is both cell type specific and dependent on active PrP expression.
A plausible way to link PrPSc-mediated ER stress to changes in PrPC metabolism recently emerged through the discovery of pre-emptive quality control (pQC). The pQC pathway selectively aborts the ER translocation of certain secretory and membrane proteins during acute ER stress to allow their direct proteasome-mediated degradation in the cytosol (Kang et al., 2006). This pathway protects cells from excessive nascent protein entry into and misfolding within the ER lumen during conditions of compromised ER function. Because PrP is an excellent pQC substrate, its translocation into the ER is partially attenuated by ER stress caused by a variety of independent mechanisms (Kang et al., 2006; Orsi et al., 2006). Thus, at least one potential effect of PrPSc accumulation during prion disease progression is to cause ER stress-induced routing of at least some nascent PrP through the pQC pathway. Whether such chronic PrP degradation via pQC actually occurs during prion disease, and if this re-routing might contribute to the pleiotropic neurodegenerative phenotypes in prion diseases is unclear.
Intriguingly, forced cytosolic expression of a PrP construct lacking ER targeting and GPI-anchoring signals (ΔSS-PrP) can cause severe neurodegeneration (Ma et al., 2002). However, the significance of this observation to neurodegeneration observed in prion diseases has been unclear (Fioriti et al., 2005; Roucou et al., 2003). Not only is expression of ΔSS-PrP non-physiological, but the phenotype and neuropathology are highly atypical for either transmissible or genetic prion diseases in animals or humans. Furthermore, evidence for cytosolic PrP generation by retrotranslocation from the ER lumen is minimal and disputed (Drisaldi et al., 2003; Rane et al., 2004; Kang et al., 2006). Even if retrotranslocation of PrP did occur, ΔSS-PrP expression is not an especially close mimic since this substrate would be handled by cytosolic quality control pathways that involve different machinery and are mechanistically distinct from ER-associated degradation. Thus, while the atypical neurodegeneration caused by enforced cytosolic PrP expression is provocative, its physiologic or pathologic relevance has been uncertain and leaves open the issue of what pathway(s) contribute to the neuronal dysfunction and phenotypes in prion diseases.
Nonetheless, the toxicity of ΔSS-PrP expression in mice (Ma et al., 2002), together with the possibility of PrPSc-mediated ER stress (Hetz et al., 2003; Hetz and Soto, 2006) and stress-induced translocational attenuation of PrPC (Kang et al., 2006; Orsi et al., 2006), raised a testable hypothesis for a neurotoxic mechanism during prion disease pathogenesis: persistent elevated routing of PrP through the pQC pathway during chronic ER stress induced by PrPSc accumulation leads to damage of selected subsets of neurons and corresponding clinical symptoms. Such a model make two key predictions. First, PrPSc accumulation should lead to ER stress and reduced translocation of newly synthesized PrP into the ER lumen. And second, at least a subset of the neurodegenerative sequelae of prion infection should be directly inducible, even in the absence of PrPSc or misfolded PrP aggregates, solely by changing the metabolism of PrPC to that occurring during chronic ER stress. In this study, we have performed experiments to test these predictions. Our results provide a means to link PrPSc-mediated ER stress to a specific change in PrP metabolism that contributes to neurodegenerative disease.
Results
PrPSc induces ER stress and influences PrP translocation
Brain tissue from scrapie-inoculated hamsters (Sc-Ha) at the end stages of disease were compared to normal hamsters (Ha) for expression of various proteins, including markers of ER stress. When equal amounts of brain homogenate were analyzed (see total protein stain, Fig. 1A), total PrP levels were increased in the scrapie-infected tissue. The increased PrP was due to the accumulation of PrPSc, as confirmed by its proteinase K (PK) resistance (Fig. 1B). Elevated GFAP levels, corresponding to the increased astrogliosis seen in hamster prion disease, further verified that the Sc-Ha samples represented late stages of disease. When analyzed for various ER markers, the Sc-Ha sample contained elevated levels (by ∼2 to 3 fold) of BiP, a major lumenal chaperone whose upregulation is tightly correlated with ER stress (Ron and Walter, 2007). In addition, another ER chaperone, the oxidoreductase GRP58/ERp57, was also elevated. By contrast, other ER proteins, such as Calnexin (a transmembrane chaperone) and TRAPα (a component of the protein translocon) were not detectably affected. These proteins serve as internal controls for non-specific ER expansion. Thus, late stages of hamster scrapie containing high levels of PrPSc display markers of ER stress.
Fig. 1. PrPSc accumulation induces ER stress and reduces PrP translocation into the ER.
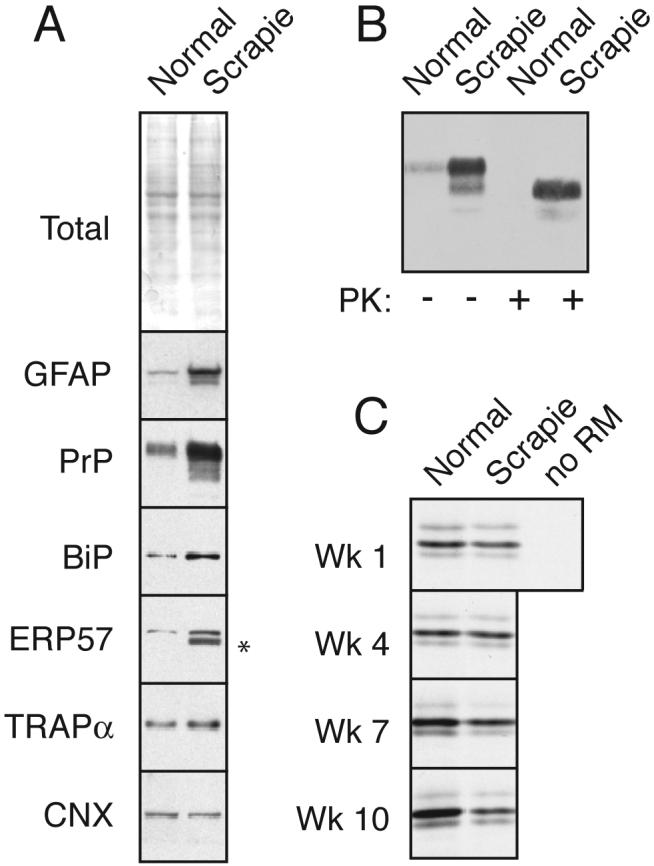
(A) Total brain homogenate from normal and PrPSc-infected hamsters (‘normal’ and ‘scrapie’) were analyzed by staining for total proteins or immunoblotted for the indicated antigens. Asterisk indicates trace IgG heavy chain that occassionally contaminates tissue homogenates from residual blood.
(B) Analysis of PrP from the samples in panel A for resistance to digestion by proteinase K (PK). No protease-resistant PrP was detected in normal tissue even upon gross overexposure of the blot (not shown).
(C) ER microsomes from normal and PrPSc-infected hamsters were used for in vitro translocation assays for PrP. After synthesis with 35S-methionine, the samples were treated with PK to digest non-translocated products, and the protease-protected PrP (indicative of its successful translocation into the ER microsomes) was recovered by immunoprecipitation. Shown are autoradiographs of the translocated PrP. Note that in the absence of membranes (‘-’), full length PrP is not protected. In samples from animals 1 and 4 weeks post-inoculation (before PrPSc accumulation), no difference is observed in normal and scrapie microsomes. By contrast, at a time when PrPSc accumulation is high (7 and 10 weeks; see Sup. Fig. 1), translocation is significantly lower in infected microsomes relative to the uninfected control. Note that each pair of microsomes at every time point were isolated and analyzed in parallel; however, comparisons may not be valid between time points, so the apparent increase in translocation in normal microsomes over time may not be meaningful.
To determine whether this ER stress seen in scrapie-infected hamsters impacts the translocation of newly synthesized PrP into the ER, we isolated microsomes from normal and infected brain tissue and tested their functional activity for PrP import. In this experiment, hamsters were inoculated with either saline or PrPSc. At weekly intervals following the inoculations, one each of the control and PrPSc-inoculated animals was sacrificed and brain tissue used for rough microsome (RM) isolation. At the end of ten weeks (the approximate incubation time to death), all of the RMs were analyzed in parallel. Note that each pair of control versus infected RM at every time point represents matched and directly comparable samples. Furthermore, since the first four pairs of microsomes are from animals before any phenotype is observed and before any PrPSc is detectable biochemically (Sup. Fig. 1A), they represent four independent comparisons comprising a ‘pre-disease’ set. By contrast, the last four pairs of microsomes have readily detectable PrPSc accumulation in samples from the inoculated animals (Sup. Fig. 1A) and constitute a ‘disease’ set.
Each of the RM samples was incubated with an in vitro translation extract supplemented with PrP transcript and 35S-Methionine to allow both endogenous mRNAs and exogenously added PrP transcript to be translated into radiolabeled proteins. Analysis of the total products by autoradiography showed very similar profiles and amounts of each band (Sup. Fig. 1B). This confirmed the uniformity of RM recovery, RNA integrity, and translation efficiency in all of the samples. To assess PrP translocation, samples were then subjected to a protease protection assay in which only the translocated products are protected from digestion. The protease-protected PrP that had been translocated into the RMs was then recovered by immunoprecipitation and visualized by autoradiography. As seen in Fig. 1C, the amount of newly synthesized (i.e., radiolabeled) and translocated PrP was the same in RMs from control and PrPSc-inoculated brains at the 1 and 4 week pre-disease time points. By contrast, the amount of PrP translocated into RMs from PrPSc-inoculated brains at the 7 and 10 week disease time points was noticeably less (roughly half) than that seen in the matched control-inoculated RMs. Averaging the four comparisons in the ‘pre-disease’ set showed no difference in PrP translocation into PrPSc-inoculated RMs (103% of control; p=0.9). By contrast, the ‘disease’ set showed a statistically lower level of translocation capacity into PrPSc-inoculated RMs (72% of control; p < 0.05).
These results indicate that at a gross level (in which the entire brain is averaged and treated as a single entity), PrPSc accumulation induces at least some limbs of the ER stress response (as judged by BiP and GRP58 upregulation), consistent with recent observations in both mice and cultured cells (Hetz et al., 2005; Hetz and Soto, 2006). More importantly, the stressed ER isolated from these diseased brains were reduced slightly (to ∼70% of control) in their translocation capacity relative to ER from non-diseased brain. This is consistent with experiments in cultured cells showing that several types of ER stressors all lead to decreased translocation of PrP (Kang et al., 2006; Orsi et al., 2006), a phenomenon that can also be recapitulated in vitro upon perturbation of ER lumenal proteins (Kang et al., 2006). Clearly, new in situ methodologies for measuring translocation will be needed to assess this effect of PrPSc on a finer scale in vivo. That notwithstanding, the above data supports the hypothesis that one effect of PrPSc is to cause reduced translocation of newly synthesized PrP into the ER. This effect may be a secondary consequence of ER stress, during which PrP is routed at least partially into the pQC pathway to generate (at least transiently) cytosolic PrP.
Design of a PrP variant constitutively degraded selectively by pQC
To investigate whether increased routing of PrP into the pQC pathway might be a contributing factor in neurodegeneration, we needed a means to cause this re-routing for PrP without PrPSc accumulation or ER stress (which presumably has many other pleiotropic effects). The molecular steps comprising pQC include SRP-dependent targeting of the nascent polypeptide to the ER membrane, transfer to the translocon, rejection from translocation, and release into the cytosol for proteasomal degradation (Fig. 2A). This series of events is spatially and mechanistically distinct from other pathways of proteasomal degradation (Meusser et al., 2005; Bukau et al., 2006) including failed targeting (in which nascent PrP would interact with chaperones in the cytosol rather than SRP) or retrotranslocation (in which processed PrP would be extracted from the ER lumen by the cytosolic VCP/p97 complex for degradation). Importantly, among these three potential routes for cytosolic PrP degradation, only the pQC pathway is clearly and unambiguously utilized upon ER stress (Kang et al., 2006; Orsi et al., 2006). No evidence exists for PrP ever being directly synthesized in the cytosol (except upon artificial deletion of its signal sequence), and pulse-chase studies of PrP during both normal and stressed conditions failed to detect its retrotranslocation from the ER lumen to cytosol (Drisaldi et al., 2003; Rane et al., 2004; Kang et al., 2006). Thus, pQC appears to be the principal regulatory point of early PrP biosynthesis during ER stress. Hence, to mimic the consequences of ER stress on PrP metabolism, it was important that PrP be routed selectively into the pQC pathway and not other routes of proteasomal degradation.
Fig. 2. Design and mechanistic analysis of a constitutive pQC variant of PrP.

(A) Schematic diagram of cytosolic quality control (cQC), pre-emptive quality control (pQC), and ER-associated degradation (ERAD). Of these, only the pQC pathway has been demonstrated to be utilized by PrP during ER stress (Kang et al., 2006; Orsi et al., 2006).
(B) The signal sequences and cleavage site (arrowhead) for constructs used in this study. Lysine residues used for crosslinking analyses are in bold.
(C) Crosslinking to cytosolic proteins of ribosome-associated nascent chains (RNCs) synthesized up to PrP residue 150. PrP (P), Ifn-PrP (I), Opn-PrP (O), and ΔSS-PrP (D) are analyzed. The arrowhead indicates crosslinks to SRP54, confirmed by immunoprecipitation (right panel). Asterisk indicates the position of uncrosslinked nascent chains.
(D) Crosslinking to ER proteins of RNCs synthesized to PrP residue 150. After crosslinking, the products were fractionated into membrane-associated and lumenal proteins, shown in the left and middle panels, respectively. Open arrow indicates crosslinks to the translocon component Sec61α (verified by immunoprecipitation; not shown), and closed arrow indicates crosslinks to the lumenal chaperone PDI, identified by immunoprecipitation in the right panel.
(E) PrP was synthesized in the absence or presence of ER-derived rough microsomes (RM) in a lysate supplemented with His-tagged ubiquitin. Ubiquitin-conjugated products were captured on immoblized Co+2. The positions of PrP species representing precursor (pre), signal-cleaved (s.c.), glycosylated (glyc) and ubiquitinated (Ub) products are indicated. Also shown are the Ubiquitin-conjugated products for ΔSS-PrP, illustrating its relatively poor ubiquitination.
(F) Ubiquitination analysis (as in panel E) of PrP, Ifn-PrP, and Opn-PrP in the absence and presence of RMs. The lower panel shows the total products and the upper panel the Ubiquitin-conjugated species captured via the His-tagged ubiquitin.
To accomplish this, we replaced the PrP signal sequence with the signal sequence from Interferon-γ (Ifn; see Fig. 2B) shown previously to be competent for ER targeting, but inefficient in translocon gating (Kim et al., 2002). As a matched control, we also used another signal sequence [(from Osteopontin (Opn)] that is comparable in length but whose targeting and gating efficiency are much higher (Kim et al., 2002). Analysis of 150-mer nascent PrP chains in vitro by crosslinking revealed that PrP, Ifn-PrP, and Opn-PrP, but not ΔSS-PrP, all interact with SRP in the cytosol (Fig. 2C). Upon targeting to ER-derived microsomes (which did not occur for ΔSS-PrP; data not shown), PrP, Ifn-PrP, and Opn-PrP each released from SRP and was transferred to the Sec61 translocon as judged by crosslinking (Fig. 2D). However, Ifn-PrP did not make contacts with lumenal chaperones (such as protein disulfide isomerase) as efficiently as either PrP or Opn-PrP (Fig. 2D). Thus, Ifn-PrP is targeted to the ER translocon via the SRP pathway, but is inefficient in its access to the ER lumen.
To determine whether this poor access to the ER lumen leads to its subsequent release into the cytosol for degradation, we analyzed PrP ubiquitination. When PrP is synthesized in vitro in the absence of ER-derived rough microsomes (RM), PrP precursors become poly-ubiquitinated (Fig. 2E). By contrast, inclusion of RM in the translation reaction results in PrP translocation (as evidenced by its glycosylation) rather than cytosolic ubiquitination (which was reduced by over 80%). In contrast to either PrP or Opn-PrP, Ifn-PrP showed a substantial amount (∼40%) of ubiquitinated products even when synthesized in the presence of RM (Fig. 2F). Curiously, ΔSS-PrP was poorly ubiquitinated in this same assay (Fig. 2E), indicating that its recognition and/or metabolism is different than cytosolic full length PrP (see Fig. 3C and 4C below). Thus, Ifn-PrP mimics the pQC pathway by displaying efficient SRP-dependent targeting to the translocon, poor access to the ER lumen, release into the cytosol, and ubiquitination.
Fig. 3. Ifn-PrP metabolism is distinct from CtmPrP and ΔSS-PrP.

(A) Wild type PrP, Ifn-PrP, and Ifn-PrP(A120L) were analyzed by in vitro translation and translocation assays. An inhibitor of glycosylation was included in all reactions to simplify the banding pattern. Half of each sample was analyzed directly, while the remainder was digested with PK. The positions of full length (FL) PrP, and the proteolytic fragments corresponding to CtmPrP and NtmPrP are indicated. Note that Ifn-PrP makes comparable amounts of CtmPrP as wild type, while Ifn-PrP(A120L) makes substantially more.
(B) Wild type PrP, Ifn-PrP, PrP(A117V) and PrP(AV3) were expressed in N2a cells, and microsomes isolated from these cells were subjected to analysis for CtmPrP by limited PK digestion. Shown are different relative amounts of undigested sample, as well as the products after digestion under ‘mild’ and ‘harsh’ conditions (see Hegde et al., 1998). The PK-digested samples were deglycosylated with PNGase before analysis. In this assay, PK digestion under mild conditions generates an ∼18 kD fragment corresponding to CtmPrP (indicated by asterisk). A smaller band corresponding to the C-terminal globular domain of PrP is indicated by the arrowheads. Note that Ifn-PrP levels are very low due to its constitutive degradation (see Fig. 4B), even though its rate of expression was verified to be comparable to wild type PrP by pulse-labeling experiments as in Fig. 4A (data not shown). A band at ∼14 kD seen in the Ifn-PrP samples appears to be a degradation intermediate that is sometimes observed.
(C) PrP and ΔSS-PrP were synthesized in vitro in the absence of ER membranes and analyzed by sucrose gradient sedimentation. An aliquot of the total translation products is also shown. Note that PrP is ubiquitinated significantly more efficiently than ΔSS-PrP, and that the two proteins have different sedimentation profiles indicative of associations with different complexes.
Fig. 4. Ifn-PrP mimics pQC in vivo in the absence of ER stress.

(A) ER translocation of the indicated PrP constructs in transiently transfected Hela cells subjected to acute ER stress (15 min) by Ca+2 depletion using thapsigargin (Tg). Translocation was quantified using relative glycosylation efficiency and is indicated below the respective lanes. The positions of unglycosylated (-CHO) and glycosylated (+CHO) species of PrP are indicated. Note that protein synthesis is reduced in stressed cells due to PERK-mediated phosphorylation of eIF2α.
(B) N2a cells transiently transfected with Ifn-PrP were treated with proteasome inhibitor (10 uM MG132) for 0, 2, or 4 h as indicated and analyzed by immunoblotting. Samples were separated into detergent-soluble (S) and insoluble (P) fractions before analysis. ‘4+20’ indicates samples from cells treated with inhibitor for 4 hours, and cultured in the absence of inhibitor for an additional 20 h. The last lane is a marker for mature PrP from cells expressing wild type PrP.
(C) N2a cells transiently transfected with ΔSS-PrP were treated with proteasome inhibitor (10 uM MG132) for 4 h as indicated, and either harvested immediately, or cultured for an additional 4 to 24 h in the absence of inhibitor. All samples were analyzed for ΔSS-PrP by immunoblotting with 3F4 antibody.
(D) N2a cells transiently transfected with Ifn-PrP or empty vector were separated into detergent-soluble (S) and insoluble (P) fractions before analysis by immunoblot using a PrP antibody that detects both endogenous PrP and Ifn-PrP. Note the lack of changes to endogenous PrP in cells expressing Ifn-PrP (most of which is found in the insoluble fraction as unglycosylated species).
Relationship between pQC and other pathways of altered PrP biosynthesis
Additional experiments illustrated that the Ifn-PrP paradigm is distinct from other models of cytosolic (Ma et al., 2002) or cytosolically exposed transmembrane PrP (Hegde et al., 1998). A transmembrane form (termed CtmPrP) is also generated at the ER during PrP biosynthesis and its over-representation can lead to neurodegeneration (Hegde et al., 1998; Hegde et al., 1999). However, previous mechanistic analyses suggest that increased generation of CtmPrP typically requires a mutation that raises the hydrophobicity of the central hydrophobic region that serves as the transmembrane domain (Kim et al., 2001; Kim and Hegde, 2002; Stewart and Harris, 2003). While decreasing the efficiency of the signal sequence can facilitate CtmPrP generation, this alone is not sufficient (see Sup. Fig. 2). Direct analysis of PrP and Ifn-PrP showed that both proteins generate comparable amounts of CtmPrP (Fig. 3A). However, there is less of the fully translocated isoform for Ifn-PrP, indicating that a higher proportion was cytosolic (consistent with the increased ubiquitination of Ifn-PrP seen in Fig. 2F). Only in the context of an additional mutation in the transmembrane domain does the Ifn signal lead to increased CtmPrP (Fig. 3A; Kim et al., 2002). Similar results were obtained in cultured cells (Fig. 3B), where we observed that Ifn-PrP does not generate increased CtmPrP (relative to wild type PrP). Instead, the majority of the protein is degraded in the cytosol (see Fig. 4), leading to its low steady state expression levels. This is in contrast to previously characterized transmembrane domain mutants (A117V and AV3; see Hegde et al., 1998), where increased CtmPrP could be seen by the same assay.
These results illustrate two important points. First, compared to wild type PrP, Ifn-PrP does not generate significantly increased levels of CtmPrP in vitro or in vivo; rather, a substantial proportion of Ifn-PrP is cytosolic, where it is ubiquitinated (Fig. 2F) and degraded by a proteasome-dependent pathway (see Fig. 4B). And second, the cytosolically released population of Ifn-PrP must have initially been at the translocon since it has the potential to be inserted into the membrane, had the transmembrane domain been sufficiently hydrophobic [e.g., Ifn-PrP(A120L) in Fig. 3A]. This is consistent with previous in vitro analyses that demonstrated that Ifn-PrP targets efficiently to the ER membrane, but does not mediate efficient translocation (Fons et al., 2003). Thus, we can conclude that Ifn-PrP mimics the pQC pathway to generate cytosolically localized PrP that, despite targeting to the ER membrane, remains unprocessed and is not membrane inserted in the CtmPrP configuration.
We also considered whether ΔSS-PrP, which results in cytosolically localized PrP, is comparable to or different from cytosolic PrP generated by the pQC pathway. We found that PrP containing a signal sequence interacts with different proteins (including SRP) than ΔSS-PrP interacts with during its synthesis on the ribosome (as judged by crosslinking; Fig. 2C). More importantly, PrP and ΔSS-PrP were found to be in different sized complexes in the cytosol when analyzed by sedimentation through sucrose density gradients (Fig. 3C). In this experiment, it is also readily apparent that cytosolic full length PrP is ubiquitinated significantly more efficiently than ΔSS-PrP, a conclusion confirmed by direct ubiquitination assays (see Fig. 2E). These results collectively indicate that non-translocated PrP made by the pQC pathway (in which N- and/or C-terminal signals are unprocessed) is distinct from cytosolic PrP artificially generated by deletion of its signal sequences. Importantly, the former species is clearly physiologically relevant since it is generated during different types of ER stress (Kang et al., 2006; Orsi et al., 2006), including potentially the stress induced by PrPSc accumulation (Fig. 1). Because Ifn-PrP faithfully mimics the pQC pathway taken by PrP during ER stress, this construct represents a valid model for one (of presumably many) consequences of PrPSc accumulation.
Validation of the Ifn-PrP model of pQC in cultured cells
Analysis of Ifn-PrP in pulse-labeled cultured cells confirmed that in vivo, its translocation into the ER lumen (as judged by its glycosylation) during non-stressed conditions is comparable to the level observed for PrP during acute stress induced by ER calcium depletion (Fig. 4A). Opn-PrP translocation was comparable to or slightly higher than PrP, and changed little during ER stress. That non-translocated Ifn-PrP was indeed being degraded in the cytosol was evidenced by accumulation of unglycosylated precursor molecules upon proteasome inhibition and its subsequent degradation upon removal of the inhibitor (Fig. 4B). Of note, ΔSS-PrP aggregates generated during proteasome inhibition appear to be relatively refractory to degradation even when inhibition is alleviated (Fig. 4C). This is consistent with its decreased efficiency of recognition by the ubiquitination machinery in vitro (Fig. 3D), further confirming that the metabolism of ΔSS-PrP is distinct from the pathway taken by Ifn-PrP. Importantly, the constitutive degradation of Ifn-PrP via pQC did not affect the expression, metabolism, or properties of endogenous PrP, whose levels, solubility, and glycosylation pattern remained unaltered (Fig. 4D). This further illustrates that Ifn-PrP routed into the pQC pathway remains topologically distinct from and does not interact, either physically or functionally with PrP routed into the secretory pathway. Thus, Ifn-PrP represents a version of PrP whose biosynthesis, trafficking, and metabolism under normal conditions closely mimics that seen for PrP during ER stress. We could therefore now ask whether elevated and chronic degradation of PrP via the pQC pathway, an event that occurs during ER stress such as that induced by PrPSc accumulation (Fig. 1), could be a contributing factor in neurodegeneration.
The consequences of constitutive pQC of PrP in transgenic mice
The Opn-PrP and Ifn-PrP coding regions in a well-characterized cosmid containing ∼35 kB of the PrP promoter (Scott et al., 1992) were used to generate transgenic mice. In contrast to Opn-PrP, the Ifn-PrP transgene apparently had adverse consequences during early development and produced fewer relatively small founder animals, several of which died within two months after birth (see Supplementary Note). These observations suggest that generation of cytosolic PrP during development via the pQC pathway might be detrimental for reasons that remain to be studied. This effect precluded the generation of transgenic mice expressing Ifn-PrP at high or even wild-type levels (see Sup. Fig. 3). Nonetheless, we obtained several transgenic founders, one of which allowed the generation of a stable breeding line that could be analyzed further for age-dependent neurologic phenotypes.
While transgenic Ifn-PrP mice were noticeably smaller than either non-transgenic littermates or Opn-PrP transgenics, they had a comparable lifespan (over 700 days) and did not show any increased rate of early death relative to Opn-PrP controls (Fig. 5A). Note that the death of some animals (in both transgenic lines) by ∼200 days is unrelated to the PrP transgenes or to neurodegenerative illnesses. Because the potential phenotypes of these animals could not necessarily be predicted, we have plotted all deaths including those resulting from animal fighting and intercurrent illnesses such as infections and tumors. Consistently however, Ifn-PrP mice showed a progressive age-dependent phenotype characterized by a rough hair coat, slight ataxia that worsened over time, hunched posture (kyphosis), and occasional movement disorder and seizure (Fig. 5B, C, and Sup. Movies 1-6). Young mice (2-3 months old) showed only subtle defects in coordination and hind limb strength. More obvious impairments including relative unresponsiveness to external stimuli and altered gait were only apparent after ∼18-24 months. Comparable results were obtained with an independent founder animal carrying the Ifn-PrP transgene, arguing for the specificity of the phenotype to the transgene rather than an unrelated effect. However, other stable breeding lines were not obtained despite repeated attempts at breeding encompassing over 15 litters (see Supplementary Note).
Fig. 5. Phenotype of Ifn-PrP transgenic mice with constitutive pQC of PrP.

(A) Lifespans of Ifn-PrP and Opn-PrP mice. All causes of death are included in the analysis except those mice that were sacrificed prematurely for analysis.
(B) Representative Opn-PrP and Ifn-PrP mice at ∼2 months. Note rough hair coat and smaller size of Ifn-PrP mouse.
(C) Representative Ifn-PrP mouse at ∼2 years. Note kyphosis (arrow), rough hair coat, and abnormal gait (Sup. Movie 2, 3, and 6).
At the level of gross pathology, the brains of Ifn-PrP mice were notably smaller than Opn-PrP mice, consistent with their overall smaller size (Fig. 6A). Surprisingly however, little or no pathologic changes were observed in adolescent or young adult mice (Fig. 6B-H). Only after ∼18 months were very modest age-dependent spongiform changes observed in the cerebellum, hippocampus, and midbrain (Fig. 6B-D). These neuropathologic effects were only marginally greater than that seen simply as a consequence of old age, despite the readily apparent clinical phenotype. Notably, no gross degeneration of any of the cerebellar layers was observed at any age, and the number and morphology of Purkinje cells remained normal throughout life (Fig. 6E). Analysis for reactive astrocytosis by GFAP staining showed very modest, age-dependent increases in the number and size of astrocytes in the hippocampal region of Ifn-PrP mice relative to Opn-PrP transgenic mice (Fig. 6F). Staining of 2 year old mice with Fluoro-Jade C, a commonly used marker for degenerating neurons, showed increased signal for Ifn-PrP relative to Opn-PrP in both the hippocampus and in parts of the cerebral cortex (Fig. 6G). No Fluoro-Jade C staining in any brain regions was observed in younger Ifn-PrP mice (less than 1 year old; data not shown). Thus, the phenotype of Ifn-PrP mice is distinct from the highly atypical early onset cerebellar degeneration in ΔSS-PrP-expressing mice (Ma et al., 2002), suggesting that the different pathways of trafficking and degradation of these two PrP variants (Fig. 2A) have different functional consequences. More importantly, the progressive age-dependent clinical phenotype of Ifn-PrP is more akin to the clinical picture of both genetic and transmissible prion diseases.
Fig. 6. Histologic analysis of Ifn-PrP mice reveals mild neurodegeneration.

(A-D) H&E stained saggital brain sections show no alterations to gross brain morphology or development in Ifn-PrP or Opn-PrP mice at either ∼2 months or ∼2 years of age. Green boxes indicate regions shown in greater detail in panels B-D.
(E) Immunostaining for calbindin to visualize Purkinje cells (brown). Note that neither the Purkinje cells, granular layer (left) or molecular layer (right) are affected grossly in Ifn-PrP mice.
(F) GFAP staining of Ifn-PrP or Opn-PrP mice at either ∼2 months or ∼2 years of age. Shown is a region of hippocampus where age-dependent increase in reactive astrocytes is observed in Ifn-PrP mice beyond that seen in old Opn-PrP mice.
(G) Fluoro-Jade C staining of Ifn-PrP or Opn-PrP mice at ∼2 years of age. Shown are regions where increased staining is observed in Ifn-PrP mice. Note that no staining was observed in young mice of either genotype (data not shown).
Quantitation of pQC in Ifn-PrP transgenic mice
Biochemical analyses on brain tissue and cells from transgenic Ifn-PrP mice showed that Ifn-PrP expression had no effect on the levels or modification of endogenous PrP (data not shown; see also Fig. 7B), consistent with the results from cell culture (Fig. 4D). Furthermore, steady state Ifn-PrP expression levels in whole brain, and even neurons cultured from transgenic newborns, was extremely low (Sup. Fig. 4 and Fig. 7A). In addition, focal accumulations were not detected by PrP immunohistochemistry of either cultured neurons or brain sections (data not shown). This is consistent with the expectation from in vitro studies that a substantial fraction of Ifn-PrP should be constitutively degraded by the very rapid and efficient pQC pathway. Indeed, proteasome inhibition of neuronal cultures caused a progressive accumulation of non-glycosylated Ifn-PrP, but no change in the mature cell surface population (Fig. 7A).
Fig. 7. Quantitation of pQC in Ifn-PrP transgenic mice.
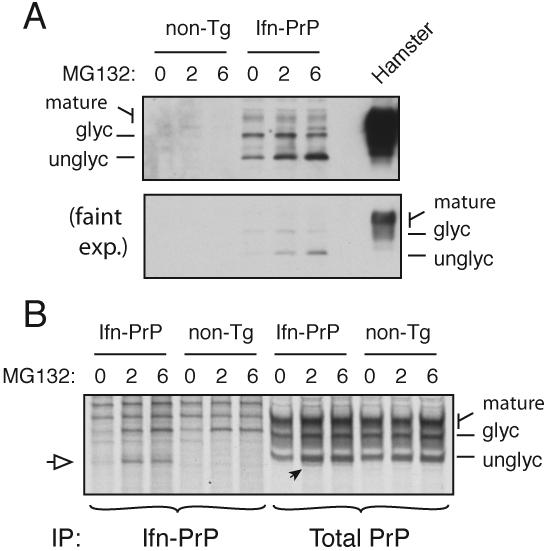
(A) Expression of Ifn-PrP in mixed cortical cell cultures prepared from newborn transgenic and non-transgenic mice after treatment with proteasome inhibitor (10 μM MG132) for the indicated times. For comparison, PrP expression in normal hamster brain is shown. Detection was with the 3F4 monoclonal antibody selective to hamster (and not mouse) PrP. Two exposures of the blot are shown to illustrate the very low level steady state expression of Ifn-PrP, and the selective increase in the unglycosylated cytosolic form of PrP upon proteasome inhibition.
(B) Cortical cultures as in panel A were pre-treated with MG132 as indicated, pulse-labeled for 1 h with 35S-methionine in the absence or presence of MG132, and immunoprecipitated with either 3F4 (to selectively recover the transgenically expressed Ifn-PrP) or a pan-PrP antibody to recover both endogenous and transgenic PrPs. The white arrow indicates the position of unglycosylated (and non-translocated) Ifn-PrP, seen selectively when the proteasome is inhibited. This is also seen in the total PrP immunoprecipitates, where it represents ∼10% of total PrP synthesized.
To quantify the rate of pQC-mediated degradation in Ifn-PrP mice, we analyzed primary neurons cultured from transgenic newborns. Pulse-labeling and immunoprecipitation revealed Ifn-PrP expression predominantly in a non-glycosylated form that was stabilized by proteasome inhibition (Fig. 7B). Using an antibody that recognizes both the transgene-expressed Ifn-PrP and endogenous mouse PrP, we could deduce that ∼5-10% of total PrP was represented by Ifn-PrP. This is almost certainly an underestimate because not only is Ifn-PrP rapidly degraded, but even in the presence of proteasome inhibitors, Ifn-PrP would migrate heterogeneously due to its poly-ubiquitination.
As a second approach to quantify the level of Ifn-PrP expression and provide an upper limit for the extent of pQC, we determined the relative abundances of Ifn-PrP, Opn-PrP, and endogenous PrP mRNAs in brain tissue (Sup. Fig. 3). Total mRNA was isolated from whole brain tissue, quantified, and used in RT-PCR reactions containing serial dilutions of template. Synthetic mRNAs generated by in vitro transcription served as standards to determine absolute mRNA abundances. From these analyses, we determined that Opn-PrP and Ifn-PrP mRNAs are expressed at ∼ 4 × 107 and 3.3 × 106 copies per mg total brain tissue, respectively. Based on semi-quantitative immunoblotting (Sup. Fig. 4), we know that Opn-PrP protein levels are ∼2-fold higher than endogenous PrP. Given that their 5′ and 3′ UTRs are comparable and they are expressed in the same sets of cells by the transgenic promoter used in our study, we can reasonably infer that the rates of synthesis of the respective proteins mirror the mRNA levels. We therefore estimate endogenous PrP mRNA levels to be ∼ 2 × 107 copies per mg brain tissue (roughly 6-fold higher than Ifn-PrP mRNA levels). These values places an upper limit on the absolute amount of PrP routed into pQC in the Ifn-PrP mice at ∼15-20% (if all of the Ifn-PrP was being degraded by this pathway). Analysis of an independent Ifn-PrP founder animal that developed a very similar phenotype showed comparable (within a two-fold) mRNA levels by this same analysis. Thus, in both cases, Ifn-PrP is not being over-expressed relative to endogenous PrP. Using the above biochemically determined value of ∼10% as a minimum (Fig. 7B), we can estimate that the overall level of pQC in our Ifn-PrP mouse line must be in the range of 10-20% of endogenous PrP levels.
These results indicate that Ifn-PrP mice mimic the situation expected under relatively mild to moderate ER stress conditions: the majority of synthesized PrP continues to be translocated into the ER, while a small proportion is degraded via pQC in the cytosol. This routing of PrP through the pQC degradation pathway at a modest rate for prolonged times in cells that normally express PrP causes phenotypic and histologic changes that partially overlap with the wide spectrum of neuropathologic sequelae in prion diseases. This is consistent with our analysis of scrapie-infected hamsters where ER stress was evident (Fig. 1A) and corresponded to a small but significant decrease in PrP translocation capacity (Fig. 1C). Interestingly, the phenotype of Ifn-PrP mice despite the absence of PrP accumulation illustrates that PrP-mediated neuronal dysfunction can be uncoupled from either PrPSc or aggregate deposition.
Discussion
The most important implication of this study is a potential means to link PrPSc, via ER stress and pQC, to a PrP-dependent pathway of neurotoxicity. What has been vexing thus far is how rather generic consequences of PrPSc accumulation such as reduced proteasome activity (Kristiansen et al., 2007) or ER stress (Hetz et al., 2003) could cause selective pathology that is dependent on active PrP expression. Our results provide one answer to this problem by demonstrating that the consequence of ER stress for nascent PrP, increased routing through pQC, is in itself sufficient to cause cell type-selective neuronal damage. However, it should be emphasized that an effect of PrPSc on nascent PrP translocation is not likely to be the only pathway that contributes to neurodegeneration. Indeed, Ifn-PrP mice show a relatively mild neurodegenerative phenotype that recapitulates only a subset of pathology seen with a bona fide prion disease.
One can therefore conclude that an effect on PrP translocation is only one of several consequences of prion infection and PrPSc accumulation. Importantly however, other putative mechanisms of neurotoxicity must necessarily be dependent on active synthesis of new PrP (Brandner et al., 1996; Mallucci et al., 2003; Mallucci et al., 2007). Such contributing factors could include a hypothetical neurotoxic intermediate generated by the PrPC to PrPSc conversion process (Collinge and Clarke, 2007), increased CtmPrP (Hegde et al., 1999), or decreased proteasome activity (Kristiansen et al., 2007). The latter might further exacerbate the consequences of pQC by reducing degradation of non-translocated PrP, leading to its increased accumulation and toxicity. Each of these and other contributing factors is likely to affect different subsets of cell types to differing extents. This could explain why the phenotypes of prion diseases are not only diverse and complex, but also more severe than models that recapitulate only one downstream consequence like reduced PrP translocation (this study) or increased CtmPrP production (Hegde et al., 1998). Thus, the pleiotropic cell biological consequences of PrPSc accumulation are likely to influence nascent PrP biosynthesis, trafficking, and metabolism in multiple ways, each of which could contribute to the overall phenotype.
Our results underscore that even a modest deviation from normal PrP biosynthesis at the ER can have tangible neurologic consequences in certain cell types over the life of an organism. This is analogous to the finding that a very slight increase in the production of CtmPrP at the ER can cause region-selective neurodegeneration in both mouse models and inherited human disease (Hegde et al., 1998; Hegde et al., 1999). In the Ifn-PrP model, elevated CtmPrP could not be detected in either cell culture (Fig. 3A) or brain tissue (Sup. Fig. 4C), indicating that the primary cause of the phenotype in these mice is non-translocated PrP. In both cases, neurodegeneration is not simply a consequence of aggregation or PrP accumulation, but instead seems highly selective to minor and/or transiently generated forms of PrP that would easily elude detection. Hence, our results are not inconsistent with previous difficulty in detecting cytosolic PrP in prion infected tissue using an anti-signal sequence antibody (Stewart and Harris, 2003). Because non-translocated PrP does not accumulate due to its rapid ubiquitination and degradation (Kang et al., 2006), it is not surprising that its presence was not detected by an antibody of limited sensitivity. Nonetheless, cytosolic PrP generated via the pQC pathway can cause neurodegeneration at levels that are essentially undetectable at steady state.
At present it is unclear why routing of PrP through pQC leads to the observed neurodegenerative phenotype. One intriguing possibility is that transient exposure to the cytosol permits inappropriate interactions between PrP and proteins that are ordinarily not accessible. Such putative interacting proteins may be selectively expressed or functionally more critical in some cell types than others, thereby explaining the region-specific phenotype. In support of such a model, it is worth noting that CtmPrP is also partially exposed to the cytosol and causes region selective degeneration despite its widespread overexpression (Hegde et al., 1998). It will therefore be interesting to determine whether the N-terminus of PrP, which is exposed to the cytosol in both CtmPrP and cytosolic PrP, makes inappropriate interactions that lead to cellular dysfunction.
It is noteworthy that a slight inefficiency in the signal sequence of PrP underlies both the routing of PrP through pQC during stress (Kang et al., 2006) and generation of CtmPrP caused by mutations in the potential transmembrane domain (Kim and Hegde, 2002). Because the signal-translocon interaction appears to be modulatable in trans by either specific factors (Voigt et al., 1996; Fons et al., 2003) or changes in cellular conditions (Levine et al., 2005; Kang et al., 2006), this specific step in biosynthesis of PrP may be especially susceptible to perturbation. Conversely, generation of both CtmPrP and cytosolic PrP (even during pQC) can be bypassed by improving signal sequence efficiency (Kim and Hegde, 2002; Rane et al., 2004; Kang et al., 2006). It may therefore be possible to alleviate at least some of the neurotoxic consequences of PrPSc or certain mutations by enforced translocation into the ER to avoid cytosolic exposure. While this may be beneficial under some circumstances, constitutive translocation is clearly detrimental under conditions of ER stress when the maturation capacity of the ER lumen is compromised (Kang et al., 2006). The ability of PrP to be routed into the pQC pathway appears to have evolved for avoiding its misfolding in the ER during stress. Thus, at least some features of the pathogenesis of prion diseases may be an adverse consequence of a normally beneficial quality control pathway dependent on translocational regulation.
More generally, ER stress induced by other diverse causes could over time contribute to cellular dysfunction in part by its effect on protein translocation into the ER. Intriguingly, woozy mutant mice that are deficient in the BiP cochaperone Sil1 also result in neurodegeneration, albeit with different features than the Ifn-PrP mice (Zhao et al., 2005). This mutation seems both to cause ER stress, and in a yeast model system, reduced protein translocation efficiency (Tyson and Stirling, 2000). It is attractive to speculate that a subtle deficiency in ER function by any of several mechanisms may lead to a modest increase in pQC, reduced translocation of PrP (and other proteins whose cytosolic localization could be detrimental), and regional neurodegeneration over time. Consistent with such a model, ER stress is a commonly observed feature of various neurodegenerative diseases (Lindholm et al., 2006; Hetz and Soto, 2006). Thus, mislocalization of proteins at very low levels for extended time periods might be a more general mechanism of cellular dysfunction in slowly progressing neurodegenerative diseases.
Experimental Procedures
Constructs and antibodies
PrP, Ifn-PrP, Opn-PrP, and ΔSS-PrP constructs all contained the mature domain of Hamster PrP. The Ifn and Opn signal sequences were of porcine and rat origin and have been characterized previously (Kim et al., 2002). ΔSS-PrP encodes an initiating methionine and residues 23-230 of Hamster PrP (Ma et al., 2001). Ifn-PrP(A120L) is identical to Ifn-PrP except a Leu for Ala change at residue 120 (Kim et al., 2002). PrP(A117V) and PrP(AV3) have been characterized previously (Hegde et al., 1998). Antibodies were from the following sources: 3F4 mouse monoclonal against Hamster PrP (Signet); GFAP (Novus); BiP, GRP58/ERP57, Calnexin, and PDI (Stressgen); Calbindin (Sigma); TRAPα (Fons et al., 2003); PrP-A (raised by us against a synthetic peptide encoding KKRPKPGGWNTGGSRYC conjugated to KLH).
In vitro and cell culture analysis
Translation of PrP (and related constructs) in a cell-free system derived from reticulocyte lysate, translocation into canine pancreatic RMs, analysis of nascent chain interactions with crosslinking, and analysis of topology were performed using previously published methods (Kim et al., 2002; Kim and Hegde, 2002; see Supplementary Methods for more details). Analysis of ubiquitination was aided by inclusion in the translation system of excess His-Ubiquitin (5 uM, from Boston Biomed). Deubiquitination enzymes were inhibited with Ubiquitin-aldehyde (0.5 uM) and proteasome activity inhibited by the Hemin present in the translation extract. After the translation reaction, samples were denatured in 1% SDS, and the ubiquitinated products captured using Talon resin (Invitrogen). Sucrose gradient analysis of in vitro translation products was through 10-50% (w/v) gradients (55,000 rpm, 1 h, 4°C, in Beckman TLS-55 rotor) containing 100 mM KAc, 2 mM MgAc2, 50 mM Hepes. Cell culture, transfections, induction of ER stress with Thapsigargin, and analysis of PrP translocation under non-stressed and stressed conditions were as described (Kang et al., 2006). Analysis of PrP products for accumulation upon proteasome inhibition and fractionation by their solubility in non-denaturing detergents was as before (Rane et al., 2004). Microsomes were isolated from cultured cells or brain tissue by standard subcellular fractionation (Hegde et al., 1998). Analysis for CtmPrP by limited protease digestion was as before (Hegde et al., 1998).
Transgenic mice
The Ifn-PrP and Opn-PrP constructs were subcloned into a well-characterized cosmid in which ∼35 kb of the PrP promoter is used to drive transgene expression in physiologically relevant tissues and cells (Scott et al., 1992). Transgenic mice were made in the FVB background by standard methods, founders identified by southern blotting (using the transgene as a probe), and positive progeny in subsequent breedings identified by PCR genotyping using transgene-specific primers. Histologic analysis was as described (Hegde et al., 1998). Fluoro Jade C was obtained from Chemicon International and staining was as recommeded by the manufacturer. Purkinje cells were detected in formalin-fixed sections using anti-Calbindin D28k (Sigma) and HRP-conjugated secondary antibodies. Mixed population neuronal cultures for analysis of expression were prepared using minor modifications of standard methods (Lu et al., 1998), and analyzed after ∼7 to 10 days. Labeling was with 0.1 mCi per ml of 35S-Methionine for 1 h in Methionine-free media on cells pre-treated with 10 uM MG132 as indicated in the figure legends. Harvesting of cells and analysis by immunoprecipitation and immunoblots was as before (Rane et al., 2004).
Miscellaneous
Inoculation of Hamsters with Sc237 prions and analysis for PrPSc by PK digestion was as before (Hegde et al., 1998; Hegde et al., 1999). PrPSc digestions were for 1 h at 37°C with 0.1 mg/ml PK. Isolation of microsomes from freshly harvested brain tissue was as before (Hegde et al., 1998). The final microsome pellet from one half of the hamster brain was resuspended in 50 ul of physiologic salt buffer (PSB: 100 mM KAc, 2 mM MgAc2, 50 mM Hepes, pH 7.4, 1 mM DTT, 250 mM Sucrose), frozen in liquid N2, and stored at -80°. Translocation assays used 1 ul brain microsomes and 9 ul of a standard translation mix (Fons et al., 2003) containing 35S-Methionine, reticulocyte lysate, an acceptor peptide to inhibit glycosylation, and in vitro synthesized PrP transcript. Reactions were allowed to proceed for 40 min at 26°C before placing on ice. An aliquot (1 ul) was removed and analyzed directly to assess endogenous mRNA translation products (see Sup. Fig. 1B). The remainder was digested with 0.5 mg/ml PK on ice for 1 h to remove any non-translocated products. The samples were then inactivated with PMSF, boiled in 10 volumes of 1% SDS, and the translocated PrP products were recovered by immunoprecipitation with 3F4 antibody.
Supplementary Material
Acknowledgements
We are grateful to Christine Winters for advice on preparation of primary neuronal cultures, Yoseph Abebe for animal care, Heather Eshleman for optimizing the incorporation of His-Ubiquitin in vitro, and Heather Cameron for suggesting the use of Fluoro Jade C. We are especially indebted to Vishu Lingappa, in whose lab the experiments with PrPSc were performed. This work was supported by the Intramural Research Programs of the NICHD and NCI at the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- Drisaldi B, Stewart RS, Adles C, Stewart LR, Quaglio E, Biasini E, Fioriti L, Chiesa R, Harris DA. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem. 2003;278:21732–21743. doi: 10.1074/jbc.M213247200. [DOI] [PubMed] [Google Scholar]
- Fioriti L, Dossena S, Stewart LR, Stewart RS, Harris DA, Forloni G, Chiesa R. Cytosolic prion protein (PrP) is not toxic in N2a cells and primary neurons expressing pathogenic PrP mutations. J Biol Chem. 2005;280:11320–11328. doi: 10.1074/jbc.M412441200. [DOI] [PubMed] [Google Scholar]
- Fons RD, Bogert BA, Hegde RS. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J Cell Biol. 2003;160:529–539. doi: 10.1083/jcb.200210095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Tremblay P, Groth D, DeArmond SJ, Prusiner SB, Lingappa VR. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature. 1999;402:822–826. doi: 10.1038/45574. [DOI] [PubMed] [Google Scholar]
- Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003;22:5435–5445. doi: 10.1093/emboj/cdg537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Russelakis-Carneiro M, Walchli S, Carboni S, Vial-Knecht E, Maundrell K, Castilla J, Soto C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J Neurosci. 2005;25:2793–2802. doi: 10.1523/JNEUROSCI.4090-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz CA, Soto C. Stressing out the ER: a role of the unfolded protein response in prion-related disorders. Curr Mol Med. 2006;6:37–43. doi: 10.2174/156652406775574578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J, Hegde RS. Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell. 2006;127:999–1013. doi: 10.1016/j.cell.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Hegde RS. Cotranslational partitioning of nascent prion protein into multiple populations at the translocation channel. Mol Biol Cell. 2002;13:3775–3786. doi: 10.1091/mbc.E02-05-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Mitra D, Salerno JR, Hegde RS. Signal sequences control gating of the protein translocation channel in a substrate-specific manner. Dev Cell. 2002;2:207–217. doi: 10.1016/s1534-5807(01)00120-4. [DOI] [PubMed] [Google Scholar]
- Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, Naumann H, Clarke AR, van Leeuwen FW, Menendez-Benito V, Dantuma NP, et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell. 2007;26:175–188. doi: 10.1016/j.molcel.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Levine CG, Mitra D, Sharma A, Smith CL, Hegde RS. The efficiency of protein compartmentalization into the secretory pathway. Mol Biol Cell. 2005;16:279–291. doi: 10.1091/mbc.E04-06-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- Lu Z, McLaren RS, Winters CA, Ralston E. Ribosome association contributes to restricting mRNAs to the cell body of hippocampal neurons. Mol Cell Neurosci. 1998;12:363–375. doi: 10.1006/mcne.1998.0723. [DOI] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- Mallucci GR, White MD, Farmer M, Dickinson A, Khatun H, Powell AD, Brandner S, Jefferys JG, Collinge J. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron. 2007;53:325–335. doi: 10.1016/j.neuron.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- Orsi A, Fioriti L, Chiesa R, Sitia R. Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. J Biol Chem. 2006;281:30431–30438. doi: 10.1074/jbc.M605320200. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane NS, Yonkovich JL, Hegde RS. Protection from cytosolic prion protein toxicity by modulation of protein translocation. EMBO J. 2004;23:4550–4559. doi: 10.1038/sj.emboj.7600462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem. 2003;278:40877–40881. doi: 10.1074/jbc.M306177200. [DOI] [PubMed] [Google Scholar]
- Scott MR, Kohler R, Foster D, Prusiner SB. Chimeric prion protein expression in cultured cells and transgenic mice. Protein Sci. 1992;1:986–997. doi: 10.1002/pro.5560010804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RS, Harris DA. Mutational analysis of topological determinants in prion protein (PrP) and measurement of transmembrane and cytosolic PrP during prion infection. J Biol Chem. 2003;278:45960–45968. doi: 10.1074/jbc.M307833200. [DOI] [PubMed] [Google Scholar]
- Tyson JR, Stirling CJ. LHS1 and SIL1 provide a lumenal function that is essential for protein translocation into the endoplasmic reticulum. EMBO J. 2000;19:6440–6452. doi: 10.1093/emboj/19.23.6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt S, Jungnickel B, Hartmann E, Rapoport TA. Signal sequence-dependent function of the TRAM protein during early phases of protein transport across the endoplasmic reticulum membrane. J Cell Biol. 1996;134:25–35. doi: 10.1083/jcb.134.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Longo-Guess C, Harris BS, Lee JW, Ackerman SL. Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nat Genet. 2005;37:974–979. doi: 10.1038/ng1620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.