

Abstract
It has become clear in recent years that periodontitis is an inflammatory disease initiated by oral microbial biofilm. This distinction implies that it is the host response to the biofilm that destroys the periodontium in the pathogenesis of the disease. As our understanding of pathways of inflammation has matured, a better understanding of the molecular basis of resolution of inflammation has emerged. Resolution of inflammation is an active, agonist-mediated, well-orchestrated return of tissue homeostasis. There is an important distinction between anti-inflammation and resolution; anti-inflammation is pharmacologic intervention in inflammatory pathways, whereas resolution is biologic pathways restoring homeostasis. A growing body of research suggests that chronic inflammatory periodontal disease involves a failure of resolution pathways to restore homeostasis. This article reviews the resolution of inflammation in the context of periodontal disease and the potential for the modification of resolution pathways for the prevention and treatment of periodontal diseases. Proof-of-concept studies in the 1980s demonstrated that pharmacologic anti-inflammation prevented and slowed the progression of periodontal diseases in animals and man. However, the side-effect profile of such therapies precluded the use of non-steroidal anti-inflammatory drugs or other enzyme inhibitors or receptor antagonists in periodontal therapy. The isolation and characterization of resolving agonist molecules has opened a new area of research using endogenous lipid mediators of resolution as potential therapeutic agents for the management of inflammatory periodontitis. Work in animal models of periodontitis has revealed the potential of this therapeutic approach for its prevention and treatment and forced the reconsideration of our understanding of the pathogenesis of human periodontal diseases.
Keywords: Anti-inflammatory, lipoxins, Porphyromonas gingivalis, periodontal disease, resolvin E1
Infection by Gram-negative microorganisms is now well-recognized as the primary etiologic determinant of periodontal disease.1 However, the precise spectrum of microbial flora within the biofilm that is directly responsible for eliciting periodontitis has not been established.2 Although cross-sectional studies2 associated certain pathogens, such as Porphyromonas gingivalis, with periodontitis, the cause-and-effect relationship of specific microbes remains to be determined in longitudinal studies. However, it is increasingly clear that when the host-mediated response to the biofilm is left unchecked, tissue and bone of the periodontium are destroyed in the pathogenesis of the disease.1,3 Although an acute inflammatory response that is resolved in a timely manner prevents tissue injury, inadequate resolution and failure to return tissue to homeostasis results in neutrophil-mediated destruction and chronic inflammation.1
Attempts to intervene and inhibit an exuberant inflammatory response in tissues have traditionally involved the use of pharmacologic agents, such as non-steroidal anti-inflammatory drugs (NSAIDs), which antagonize proinflammatory pathways and/or signaling.4 However, a greater understanding of the pathways and processes underlying resolution of inflammation has led to increased interest in proresolving lipid mediators that contribute to the restoration of tissue homeostasis. It is now clear that resolution of inflammation is an active process, rather than a passive decay of proinflammatory signals,2,3 requiring the activation of proresolving molecules to neutralize and eliminate inflammatory leukocytes and, thereby, prevent pathology.1,3,4 Such proresolving molecules include lipoxins that are endogenously produced from the metabolism of arachidonic acid (AA) and are responsible, at least in part, for the restoration of homeostasis.2 Accordingly, these lipoxins, or their more stable aspirin-triggered form (ATL), act as agonists that control the resolution phase of acute inflammation and promote healing of the lesion.2,5,6 A number of other proresolving molecules have been elucidated that derive from omega-3 polyunsaturated fatty acids (PUFAs) rather than AA. Such molecules include resolvins and protectins, which share similar proresolving properties as lipoxins, acting as agonists stimulating the resolution of inflammation.5 These newly identified lipid mediators represent a potentially powerful intervention that stimulates resolution pathways leading to the restoration of homeostasis, possibly without the multiple side effects ascribed to traditional anti-inflammatory antagonist-based treatments.7
PHYSIOLOGIC PROCESSES INVOLVED IN THE RESTORATION OF TISSUE HOMEOSTASIS
It is increasingly apparent that an essential goal for intervention in inflammatory disease is the return of tissue to homeostasis. As such, the rapid and complete elimination of invading leukocytes from a lesion is the ideal outcome following an inflammatory event.2 Such a task becomes more daunting when an acute inflammatory response has progressed to chronic inflammation with associated destruction of the cellular matrix, scarring, fibrosis, and features characteristic of incomplete healing. As such, a timely and appropriate host response is necessary to prevent the progression of an acute inflammatory response to chronic disease and possibly, increase the likelihood of restoration of homeostasis. In periodontal disease, typical intervention procedures, such as root planing, boast some success in removing etiologic agents associated with inflammation, thereby helping to arrest periodontal disease. However, such procedures do not offer the necessary resolution of inflammation to restore tissue homeostasis.
Perhaps counterintuitively, restoration of tissue homeostasis through resolution pathways is initiated following an acute inflammatory response that generates lipid mediators of inflammation, including the classic eicosanoids, prostanoids (e.g., prostaglandins and thromboxanes) and prostacyclins, as well as leukotrienes (LTs). Such lipid mediators are produced upon agonist stimulation of G-protein receptors in the cell membrane, triggering the conversion of phosphatidylcholine to AA by the enzyme phospholipase A2.8 The AA released is metabolized by one of two pathways: a cyclooxygenase (COX)-1 and -2–dependent pathway that results in the generation of prostanoids or a 5-lipoxygenase (5-LO)-dependent pathway that results in LT synthesis (Fig. 1).8 The proinflammatory actions of prostanoids and LTs are implicated in a wide range of pathophysiologic processes associated with periodontal disease, including inflammatory cell recruitment, edema, pain, collagen destruction, and bone resorption.9
Figure 1.

Phospholipase A2 (PLA2) catalyzes phosphatidylcholine into AA in response to agonist stimulation of a G-protein–coupled cell surface receptor. AA acts as a substrate for COX activity to produce prostanoids (e.g., prostaglandins and thromboxanes) and prostacyclin, whereas leukotrienes are generated by 5-LO. The LO-LO pathway produces lipoxins, whereas aspirin triggers the production of 15-epi-lipoxins from AA through acetylation of COX-2. Through this pathway, 15-epi-H(p)ETEs are produced, which are then converted to lipoxins by transcellular biosynthesis using 5-LO from neutrophils. Nonsteroidal anti-inflammatory drugs (NSAIDs) act through antagonism of COX-1 and -2 enzymes. Reprinted with permission from the International and American Associations for Dental Research.8
Late in the inflammatory process when there is a high concentration of cells containing lipoxygenases and corresponding proinflammatory products, such as prostaglandin E2 (PGE2), a “class switch” may occur within neutrophils.2,10 This class switch gives rise to the synthesis of proresolving molecules through pathways that are spatially and temporally distinct from those involved in the generation of proinflammatory lipid mediators.2,10 Activation of these pathways that promote resolution occurs through cell–cell interactions in which 15-S-hyroxy-(p)-eicosatetraenoic acid (15-S-H(p)ETE) is produced from oxidation of AA by 15-LO through the process of transcellular biosynthesis. Accordingly, 15-S-H(p)ETE is further acted on by 5-LO to induce the synthesis of lipoxins, such as lipoxins A4 (LXA4) and B4 (LXB4).1,8 The lipoxins produced act as agonists to stimulate the resolution of inflammation and promote the restoration of tissue homeostasis through a number of mechanisms involving the regulation of leukocyte function. These include limiting polymorphonuclear neutrophil (PMN) migration into sites of inflammation, activating monocytes with a nonphlogistic phenotype (e.g., without the generation of a superoxide anion), and stimulating the uptake of apoptotic PMNs by macrophages.11-13
Aspirin (ASA) is unique from other NSAIDs: when the lipoxin pathway is activated in the presence of ASA, acetylation of the COX-2 enzyme occurs to inhibit further production of prostanoids from AA metabolism while inducing the synthesis of 15-R-H(p)ETE.1 With release of this 15-R-H(p)ETE, transformation to 5(6)-epoxytetraene occurs through transcellular biosynthesis in association with 5-LO activity. The 5(6)-epoxytetraene intermediate is then transformed to 15-epi-LXs or ATLs,1 which are a more bioactive form of native lipoxin and possess more powerful proresolving properties.1,14,15
The generation of lipoxins or ATLs triggered by “first-phase” proinflammatory lipid mediators may explain the potentially serious cardiovascular consequences of the chronic use of selective COX-2 antagonists. Inflammation is a multifactorial process, and a single “pan-inflammatory”4 agent does not exist that antagonizes all deleterious pathways involved while preserving the resolution pathways.3 As such, one of the unfortunate consequences of antagonism of a single enzymatic pathway, such as through NSAIDs specifically inhibiting COX-2 function (e.g., rofecoxib, valdecoxib, and celecoxib), is the prolongation of the resolution of inflammation. Although inhibition of the COX-2 pathway through use of these NSAIDs may attenuate acute inflammatory effects, the corresponding reduction in lipid mediators, such as PGE2, would fail to generate proresolving lipoxins that are required for restoring homeostasis in tissues.10 The resulting chronic low-grade inflammation may explain findings of increased myocardial risk in individuals receiving long-term treatment with selective COX-2 inhibitors.16
LIPOXIN ANALOGS PREVENT POLYMORPHONUCLEAR NEUTROPHIL–INDUCED INFLAMMATORY CHANGES IN A MOUSE MODEL OF ACUTE INFLAMMATION
With the generation of lipoxins suggested to stimulate resolution of inflammation in tissues and possibly restore homeostasis, the question arises as to whether “adding back” analogs of these molecules may offer some therapeutic advantage. Stable analogs of lipoxins prepared through total organic synthesis selectively interacted with lipoxin receptors in neutrophils in vitro, and the potential of these analogs as proresolving compounds was subsequently explored in an animal model of PMN-mediated tissue injury.17,18 Lipoxin analogs (e.g., LXA4, LXB4, and ATL) or vehicle control (acetone) were applied topically to ear skin in a mouse, followed by application of the proinflammatory mediator leukotriene B4 (LTB4) to the same site to determine the effects on induction of an inflammatory response.17 Although significant alterations in PMN-induced vascular permeability, as well as PMN infiltration, occurred at the site with application of vehicle control, such inflammatory changes were not apparent with lipoxin-analog treatment.17 As such, the topical application of LXA4, LXB4, and ATL to the ear skin profoundly inhibited the infiltration of PMN, as well as the vascular permeability changes associated with LTB4 application.
RESOLVINS AND PROTECTINS AS NOVEL PRORESOLVING LIPID MEDIATORS
New classes of proresolving lipid mediators have been identified in addition to endogenously derived lipoxins and ATLs.2,5 Such novel lipid molecules include resolvins and protectins and are derived from the omega-3 PUFAs, eicosapentanoic acid (EPA) and docosahexanoic acid (DHA), rather than AA.2 Although EPA is metabolized to the resolvin E series (RvEs), DHA forms docosatrienes (also known as the resolvin D series [RvDs]) as well as protectins (or neuroprotectins in neural tissues) (Fig. 2).2,5 Although resolvins and protectins stimulate anti-inflammatory and proresolving pathways similar to the lipoxins, their mechanisms of action differ,5 with binding occurring to distinct sites on inflammatory cells.2,5,19 As with the lipoxin generation of ATL, the coadministration of aspirin increased the stability and duration of the action of resolvins and protectins.2,20
Figure 2.
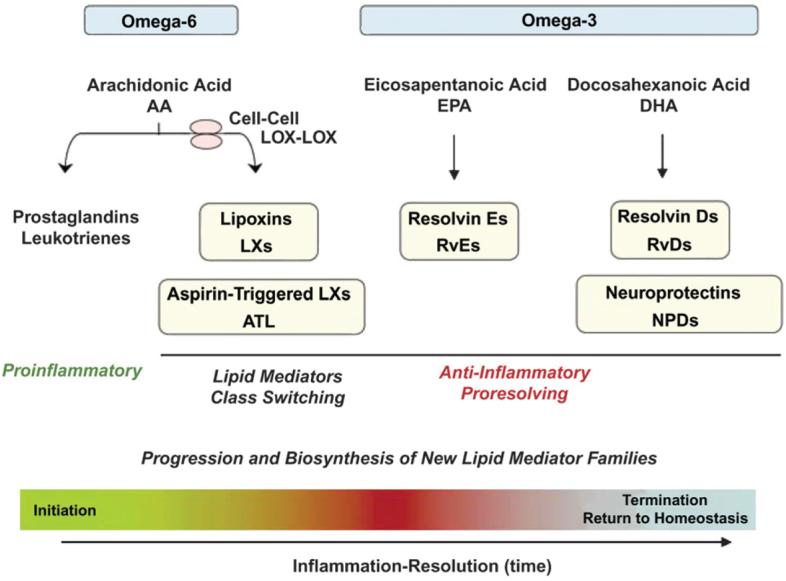
Chemical mediators involved in the initiation of acute inflammation, such as prostaglandins (PGs) and LTs, induce “class switching” toward proresolving lipid mediators. The proresolving mediators include ω-6 PUFAs, AA-derived LXs, ATLs, ω-3 PUFA EPA-derived RvEs, docosahexaenoic acid (DHA)-derived RvDs, and protectins (PDs) (or neuroprotectins in neural tissues). Reprinted with permission from Macmillan Publishers, copyright 2008.5
Resolvins stimulate the resolution of inflammation through multiple mechanisms, including preventing neutrophil penetration, phagocytosing apoptotic neutrophils to clear the lesion, and enhancing clearance of inflammation within the lesion to promote tissue regeneration.7,21-23 Such proresolving functions result in a shift in inflammatory response to a shorter resolution interval, which may help to prevent progression of an acute inflammatory response to chronic inflammation.7 The more rapid resolution of acute inflammation associated with resolvin and protectin function may underlie the beneficial effects of dietary consumption of the omega-3 PUFAs, EPA and DHA, in some inflammatory human diseases.24,25
P. GINGIVALIS–INDUCED EXPERIMENTAL MODEL OF PERIODONTITIS
With resolvins and protectins associated with proresolving properties that may serve to stimulate restoration of tissue homeostasis, the possibility that synthetic analogs of these molecules represent effective intervention was explored further.2,7,26 As such, an experimental model of periodontitis was established in which ligatures were placed bilaterally to the second premolars of both mandibular quadrants.26 To induce an inflammatory response, P. gingivalis was subsequently applied to the ligatured sites three times a week (alternate days) for 6 weeks. Initial characterization of the rabbit model involved intramuscular administration of the anti-infective agent systemic metronidazole (MTZ; 50 mg, every other day) to determine the infectious properties of the lesion.
An inflammatory response was apparent with application of P. gingivalis to the ligatures over a 6-week period, giving rise to profound inflammation and considerable tissue destruction.26 Histologic and radiologic findings supported frank inflammation and a profound loss of collagen and bone on infected ligatured teeth, as well as infrabony (vertical bone loss) defects. In control animals, ligature placement alone was not sufficient to induce disease, with bone and the supracrestal fiber network largely intact and only a mild inflammatory infiltrate over the 6-week period. Systemic MTZ administered at the time of ligature placement along with P. gingivalis induced bone loss to a similar extent as ligature alone (16.0% ± 2.2% and 10.0% ± 1.5% for MTZ and control groups, respectively), whereas significantly greater bone loss was observed in the group that received P. gingivalis applied to the ligature (43.0% ± 4.6%; P <0.05) over the 6-week period.
RESOLVIN E1 (RvE1) PREVENTS INFLAMMATION AND BONE LOSS IN EXPERIMENTAL PERIODONTITIS
Following characterization of the P. gingivalis–induced rabbit model of periodontal disease, the possibility that topical treatment with RvE1 prevents inflammation-induced tissue and bone loss was explored.2,26 RvE1, prepared by total organic synthesis,27,28 was topically applied to P. gingivalis–ligatured teeth (4 μg/tooth) three times a week in the rabbit model over a 6-week study period. Control groups received the same frequency of topical application of vehicle (ethanol) together with P. gingivalis application to ligatured teeth or placement of ligature alone.26 Macroscopic evaluation at the end of the 6-week period demonstrated frank inflammation, as well as tissue and bone loss, in the buccal and lingual mandibular region of rabbits receiving non-RvE1 vehicle control, suggesting significant progression of periodontal disease. In addition, a profound increase in bone-resorbing osteoclasts was observed histologically, with osteoclast proliferation increasing with closer proximity to the ligature, as well as prominent leukocyte infiltrates and collagen loss. This was in contrast to findings with RvE1 application, in which the development and progression of periodontal disease seemed to be prevented, with essentially no neutrophils or tissue damage observed. There was a remarkable absence of inflammatory changes and osteoclast proliferation, and bone remained largely intact. Correspondingly, mean alveolar bone loss was significantly reduced with RvE1 treatment or ligature alone compared to vehicle control.2 Radiographic evidence also demonstrated a significantly reduced percentage of bone loss associated with RvE1 treatment (<5%) over the 6-week period compared to vehicle controls (∼35%) or ligature alone (∼12%) (P <0.05).26
TREATMENT WITH RvE1 CONTROLS INFLAMMATION AND RESTORES BONE LOSS IN EXPERIMENTAL PERIODONTITIS
Having established that RvE1 has the potential to prevent the onset and progression of periodontitis, the question of whether RvE1 had positive actions in the treatment of established periodontitis was addressed.2,7 As before, experimental periodontitis was induced for 6 weeks through placement of ligatures tied bilaterally to mandibular quadrants, with topical application of P. gingivalis on alternate days, three times a week.7 A first group of animals was then sacrificed, representing the non-treatment periodontal disease group (baseline disease). The remaining groups received RvE1 treatment or vehicle control three times a week for an additional 6 weeks, without further application of P. gingivalis, prior to sacrifice at week 12.
Rabbits sacrificed after the first 6 weeks of P. gingivalis application to the ligature demonstrated significant periodontal inflammation, soft tissue and bone loss, and infrabony pocketing.7 Further progression of periodontal disease was observed with vehicle control over the subsequent 6-week period, with inflammation and bone loss comparable to baseline disease but with more severe infrabony pocketing, as well as fibrosis and tissue loss adjacent to the ligatures. Profound staining of osteoclasts and osteoclast-like cells was also detected in periodontal tissues of the baseline disease and vehicle control groups. In contrast, treatment with RvE1 was associated with essentially complete resolution of inflammation, resulting in the restoration of soft and hard tissues. As such, there was a remarkable absence of inflammation, as well as restoration of all interproximal tissues and bone with RvE1 treatment and an absence of osteoclasts associated with bone. Such findings translated to a significant reduction in soft tissue probing depth (the distance between the crest of the gingival and soft tissue attachment on the tooth surface), hard tissue crestal distance measurements, and infrabony defect depth compared to the baseline disease and vehicle control groups (P <0.01). In addition, RvE1 treatment almost completely restored the ∼30% bone loss detected in baseline periodontal disease (Fig. 3), whereas a further 13% bone loss was incurred with application of vehicle control over the subsequent 6-week period. Such bone loss with vehicle control was comparable to that induced by lipid mediators of similar structure to RvE1 topically applied following the induction of baseline disease at 6 weeks (i.e., an additional ∼18% and ∼9% bone loss with PGE2 and LTB4, respectively). Accompanying bone growth with RvE1 treatment was the formation of new periodontal ligament, connective tissue, and cementum, which suggested regeneration of the natural architecture of the periodontal organ.
Figure 3.

Although baseline periodontal disease at 6 weeks resulted in ∼30% bone loss, RvE1 treatment over the subsequent 6 weeks restored ∼95% of the lost bone. Groups treated with vehicle alone, LTB4, and PGE2 showed an additional ∼13%, 9%, and 18% bone loss, respectively. *P <0.05. Copyright 2007 The American Association of Immunologists.7
EFFECTS OF RvE1 ON BIOFILM COMPOSITION IN A RABBIT MODEL OF EXPERIMENTAL PERIODONTITIS
With RvE1 treatment appearing to stimulate the resolution of inflammation and the restoration of bone loss in a rabbit model of experimental periodontitis, the possibility that a shift occurs in the balance of microflora in the biofilm was investigated.7 There is a precedent for the highly dynamic nature of the biofilm composition, with factors, such as those that modify pH, shown to induce significant changes in the balance of microbial species present.29 Nevertheless, a significant reduction in viable counts of oral microflora as a result of the direct actions of RvE1 was not anticipated based on previous studies9 that failed to demonstrate significant antibacterial action over vehicle (ethanol) treatment alone.
Experiments were performed using DNA-DNA checkerboard hybridization analysis using probes to organisms isolated from the human periodontium; hence, positive signals represent the same or similar species that colonize the rabbit. Results demonstrated that microbial species present in rabbit periodontium prior to ligature placement included anaerobic and aerobic bacteria and largely consisted of Actinomyces viscosus– and Parvimonas micra (previously Peptostreptococcus micros or Micromonas micros)–like organisms.7 Introduction of P. gingivalis to the ligatures over a 6-week period, followed by application of vehicle control for an additional 6 weeks, induced the expression of this microorganism in 90% of plaque samples tested and resulted in greater complexity of microflora in the biofilm (Table 1). A corresponding shift in composition to more pathogenic, anaerobic flora, as well as a significant increase in several microbial species (e.g., A. viscosus–, P. micra–, Prevotella intermedia–, Streptococcus intermedius–, and Eikenella corrodens–like organisms), the emergence of previously undetected species in addition to P. gingivalis (e.g., Aggregatibacter actinomycetemcomitans [previously Actinobacillus actinomycetemcomitans]– and Fusobacterium nucleatum–like organisms), a reduction in the concentrations of certain microbes (e.g., Capnocytophaga curva– and C. rectus–like organisms), and an overall increase in bacterial load occurred during this time. With RvE1 treatment following the induction of baseline disease, the complexity of the microflora was greatly simplified in association with the reduction in inflammation and periodontal tissue destruction observed in this animal model. A reversion in P. gingivalis concentrations was observed, back to the negligible levels originally detected in healthy tissues prior to ligature placement. In addition, the composition of the biofilm overall generally approximated that observed in healthy tissues, with only one exception (i.e., an increase in a P. intermedia–like organism).
Table 1.
Modification of Composition of Flora Within Biofilm With the Introduction of P. gingivalis to Healthy Tissues, Baseline Periodontitis, and Subsequent RvE1 Treatment in a Rabbit Model of Experimental Periodontitis
| Amount of Detected Bacteria* |
|||
|---|---|---|---|
| Microbial Species | Healthy | Baseline Periodontitis |
RvE1 Treatment |
| A. actinomycetemcomitans | 0 | 1.5 ± 0.7† | 0 |
| P. gingivalis | 0 | 4.7 ± 1.3† | 0 |
| A. odontolyticus | 0 | 0 | 0 |
| A. viscosus | 2.4 ± 0.7 | 3.9 ± 1.6† | 2.2 ± 1.1 |
| A. israelii | 0 | 0 | 0 |
| P. micra | 2.6 ± 0.9 | 4.1 ± 1.4† | 2.4 ± 0.9 |
| P. intermedia | 0.1 ± 0.4 | 1.7 ± 0.9† | 1.6 ± 0.7† |
| P. nigrescens | 0 | 0 | 0 |
| C. curva | 0.3 ± 0.8 | 0 | 0 |
| C. rectus | 0.7 ± 0.6 | 0 | 0 |
| S. oralis | 0 | 0 | 0 |
| S. intermedius | 0.5 ± 0.4 | 2.1 ± 0.8† | 1.1 ± 0.7 |
| T. forsythia‡ | 0 | 0 | 0 |
| T. denticola | 0 | 0 | 0 |
| E. corrodens | 1.5 ± 0.7 | 3.8 ± 1.2† | 1.4 ± 0.8 |
| F. nucleatum | 0 | 2.4 ± 1.1† | 1.0 ± 0.6† |
| E. coli | 0 | 0 | 0 |
| E. faecalis | 0 | 0 | 0 |
Copyright 2007. The American Association of Immunologists.7
Chemiluminescence units (scale of 0 to 5 with 0 indicating no signal): 1 = undetectable <104 bacteria; 2 = 105 bacteria; 3 = >105 but <106 bacteria; 4 = 106 bacteria; and 5 = >106 bacteria.
P <0.05 compared to healthy.
Previously T. forsythensis.
The reasons for a reduction in the complexity of biofilm composition with RvE1 treatment remain unclear, given the absence of direct antibacterial properties. One possibility is that resolvin molecules promote the release of antimicrobial peptides, such as defensins and bactericidal/permeability-increasing protein, resulting in the destruction of select microorganisms.30 A second possibility is that P. gingivalis, being an asaccharolytic organism that requires essential amino acids as its food source, survives on peptides formed by the degradation of collagen fragments, such as those produced during an inflammatory response.2 As such, an inflammatory state is a more hospitable environment for bacteria, with formation of deep pockets favoring the survival of certain species. Such an explanation would suggest that the magnitude of inflammation generated by the host determines the composition of flora within the biofilm, which is a corollary to the hypothesis that the microbialspecies present sets the threshold for the inflammatory response.
CONCLUSIONS
Although the primary etiologic basis for periodontal disease seems to be bacterial, the excessive host inflammatory response and/or inadequate resolution of inflammation may be critical to the pathogenesis of periodontitis. Emphasis has traditionally been placed on the deleterious actions of lipid mediators, such as prostanoids and LT, in propagating the inflammatory response and enhancing tissue destruction. However, a class shift is now apparent that results in the production of proresolving compounds, such as lipoxins, and their more stable and bioactive form, ATL. Such endogenous production of lipoxins and ATL, in addition to the more recently elucidated resolvins and protectins, seems to be important for stimulating resolution pathways and restoring tissues to homeostasis. The proresolving functions of these lipid mediators in stimulating the restoration of homeostasis through agonist actions on neutrophils differ from the traditional pharmacologic approach of antagonizing select proinflammatory pathways. In a P. gingivalis–induced rabbit model of experimental periodontitis, RvE1 was potent when topically applied, demonstrating remarkable efficacy in the prevention and treatment of periodontal disease. As such, targeted agents, such as RvE1, that stimulate the resolution of inflammation may offer some therapeutic advantage in the treatment of periodontitis compared to more traditional pharmacologic interventions.
ACKNOWLEDGMENTS
Boston University is assigned patents on resolvins that are licensed for clinical development and are subject to consulting agreements for Dr. Van Dyke. This work was supported by United States Public Health Service grant P50-DE16191. The initial draft of this manuscript was developed by a medical writer (Axon Medical Communications Group, Toronto, Ontario) based on content provided solely by the author. The final manuscript submitted was under the sole control of the author.
REFERENCES
- 1.Van Dyke TE, Serhan CN. Resolution of inflammation: A new paradigm for the pathogenesis of periodontal diseases. J Dent Res. 2003;82:82–90. doi: 10.1177/154405910308200202. [DOI] [PubMed] [Google Scholar]
- 2.Van Dyke TE. Control of inflammation and periodontitis. Periodontol 2000. 2007;45:158–166. doi: 10.1111/j.1600-0757.2007.00229.x. [DOI] [PubMed] [Google Scholar]
- 3.Serhan CN. Novel omega–3-derived local mediators in anti-inflammation and resolution. Pharmacol Ther. 2005;105:7–21. doi: 10.1016/j.pharmthera.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Serhan CN, Brain SD, Buckley CD, et al. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: A new pharmacologic genus. Br J Pharmacol. 2008;153(Suppl 1):S200–S215. doi: 10.1038/sj.bjp.0707489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serhan CN. Lipoxin biosynthesis and its impact in inflammatory and vascular events. Biochim Biophys Acta. 1994;1212:1–25. doi: 10.1016/0005-2760(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 7.Hasturk H, Kantarci A, Goguet-Surmenian E, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. 2007;179:7021–7029. doi: 10.4049/jimmunol.179.10.7021. [DOI] [PubMed] [Google Scholar]
- 8.Kantarci A, Van Dyke TE. Lipoxins in chronic inflammation. Crit Rev Oral Biol Med. 2003;14:4–12. doi: 10.1177/154411130301400102. [DOI] [PubMed] [Google Scholar]
- 9.Pouliot M, Clish CB, Petasis NA, Van Dyke TE, Serhan CN. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: A role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry. 2000;39:4761–4768. doi: 10.1021/bi992551b. [DOI] [PubMed] [Google Scholar]
- 10.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 11.Serhan CN, Fiore S, Brezinski DA, Lynch S. Lipoxin A4 metabolism by differentiated HL-60 cells and human monocytes: Conversion to novel 15-oxo and dihydro products. Biochemistry. 1993;32:6313–6319. doi: 10.1021/bi00076a002. [DOI] [PubMed] [Google Scholar]
- 12.Maddox JF, Serhan CN. Lipoxin A4 and B4 are potent stimuli for human monocyte migration and adhesion: Selective inactivation by dehydrogenation and reduction. J Exp Med. 1996;183:137–146. doi: 10.1084/jem.183.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maddox JF, Hachicha M, Takano T, Petasis NA, Fokin VV, Serhan CN. Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J Biol Chem. 1997;272:6972–6978. doi: 10.1074/jbc.272.11.6972. [DOI] [PubMed] [Google Scholar]
- 14.Serhan CN, Maddox JF, Petasis NA, et al. Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry. 1995;34:14609–14615. doi: 10.1021/bi00044a041. [DOI] [PubMed] [Google Scholar]
- 15.Claria J, Lee MH, Serhan CN. Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Mol Med. 1996;2:583–596. [PMC free article] [PubMed] [Google Scholar]
- 16.FitzGerald GA. COX-2 in play at the AHA and the FDA. Trends Pharmacol Sci. 2007;28:303–307. doi: 10.1016/j.tips.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Takano T, Clish CB, Gronert K, Petasis N, Serhan CN. Neutrophil-mediated changes in vascular permeability are inhibited by topical application of aspirin-triggered 15-epi-lipoxin A4 and novel lipoxin B4 stable analogues. J Clin Invest. 1998;101:819–826. doi: 10.1172/JCI1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takano T, Fiore S, Maddox JF, Brady HR, Petasis NA, Serhan CN. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: Evidence for anti-inflammatory receptors. J Exp Med. 1997;185:1693–1704. doi: 10.1084/jem.185.9.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serhan CN, Gotlinger K, Hong S, Arita M. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: An overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat. 2004;73:155–172. doi: 10.1016/j.prostaglandins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 20.Serhan CN, Hong S, Gronert K, et al. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bannenberg GL, Chiang N, Ariel A, et al. Molecular circuits of resolution: Formation and actions of resolvins and protectins. J Immunol. 2005;174:4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 22.Ariel A, Fredman G, Sun YP, et al. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol. 2006;7:1209–1216. doi: 10.1038/ni1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447:869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Caterina R, Endres S, Kristensen SD, Schmidt EB, editors. n-3 Fatty Acids and Vascular Disease. Springer-Verlag; London: 1993. pp. 1–10. [Google Scholar]
- 25.Albert CM, Campos H, Stampfer MJ, et al. Blood levels of long-chain n-3 fatty acids and the risk of sudden death. N Engl J Med. 2002;346:1113–1118. doi: 10.1056/NEJMoa012918. [DOI] [PubMed] [Google Scholar]
- 26.Hasturk H, Kantarci A, Ohira T, et al. RvE1 protects from local inflammation and osteoclast-mediated bone destruction in periodontitis. FASEB J. 2006;20:401–403. doi: 10.1096/fj.05-4724fje. [DOI] [PubMed] [Google Scholar]
- 27.Arita M, Yoshida M, Hong S, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitro-benzene sulfonic acid-induced colitis. Proc Natl Acad Sci USA. 2005;102:7671–7676. doi: 10.1073/pnas.0409271102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arita M, Bianchini F, Aliberti J, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. 2005;201:713–722. doi: 10.1084/jem.20042031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marsh PD, Bradshaw DJ. The effect of fluoride on the stability of oral bacterial communities in vitro. J Dent Res. 1990;69 doi: 10.1177/00220345900690S129. Spec No:668-671; discussion 682-683. [DOI] [PubMed] [Google Scholar]
- 30.Levy O, Canny G, Serhan CN, Colgan SP. Expression of BPI (bactericidal/permeability-increasing protein) in human mucosal epithelia. Biochem Soc Trans. 2003;31:795–800. doi: 10.1042/bst0310795. [DOI] [PubMed] [Google Scholar]