

Abstract
Parkinson's disease (PD), a progressive neurodegenerative disease characterized by bradykinesia, rigidity, and resting tremor, is the most common neurodegenerative movement disorder. Although the majority of PD cases are sporadic, some are inherited, including those caused by leucine-rich repeat kinase 2 (LRRK2) mutations. The substitution of serine for glycine at position 2019 (G2019S) in the kinase domain of LRRK2 represents the most prevalent genetic mutation in both familial and apparently sporadic cases of PD. Because mutations in LRRK2 are likely associated with a toxic gain of function, destabilization of LRRK2 may be a novel way to limit its detrimental effects. Here we show that LRRK2 forms a complex with heat shock protein 90 (Hsp90) in vivo and that inhibition of Hsp90 disrupts the association of Hsp90 with LRRK2 and leads to proteasomal degradation of LRRK2. Hsp90 inhibitors may therefore limit the mutant LRRK2-elicited toxicity to neurons. As a proof of principle, we show that Hsp90 inhibitors rescue the axon growth retardation caused by overexpression of the LRRK2 G2019S mutation in neurons. Therefore, inhibition of LRRK2 kinase activity can be achieved by blocking Hsp90-mediated chaperone activity and Hsp90 inhibitors may serve as potential anti-PD drugs.
Keywords: Hsp90, LRRK2, G2019S, Parkinson's disease, protein degradation, chaperone
Introduction
Parkinson's disease (PD) is a common neurodegenerative movement disorder characterized clinically by bradykinesia, rigidity, and resting tremor and pathologically by the loss of dopaminergic neurons in the substantia nigra and the formation of Lewy bodies (Moore et al., 2005; Hardy et al., 2006). Although the majority of cases are sporadic, dominantly inherited mutations in α-synuclein and leucine-rich repeat kinase 2 (LRRK2) and recessively inherited mutations in parkin, DJ-1, and PINK1 have been linked to rare familial forms of PD. Mutations in LRRK2 have been linked to both familial and apparently sporadic forms of PD (Paisan-Ruiz et al., 2004; Zimprich et al., 2004; Farrer et al., 2005; Skipper et al., 2005). The LRRK2 protein, also known as Dardarin, contains multiple functional domains, including a LRR domain, a GTPase domain, a kinase domain, and a WD40 domain (Mata et al., 2006), may likely function as both an active GTPase and kinase (Greggio et al., 2006; Smith et al., 2006; Guo et al., 2007; Li et al., 2007; West et al., 2007). The most common mutation in LRRK2 is the G2019S substitution at the conserved Mg2+-binding motif within the kinase domain (Goldwurm et al., 2005; Bonifati, 2006), which likely increases the kinase activity of LRRK2 (Greggio et al., 2006; Smith et al., 2006; Jaleel et al., 2007; Luzon-Toro et al., 2007). Furthermore, mutant forms of LRRK2 are toxic, and the toxicity depends on LRRK2 having kinase activity. Because specific kinase inhibitor, which would potentially block mutant LRRK2 toxicity, are not yet available (Greggio and Singleton, 2007), alternative strategies might be more immediately useful as a potential therapeutic strategy for PD.
Here, we develop a novel approach to regulate the stability of LRRK2 via inhibiting the chaperone activity of heat shock protein 90 (Hsp90). Hsp90 is known to regulate the stability and activity of various signaling proteins, including protein kinases (Pearl and Prodromou, 2006). Two recent in vitro experiments have showed that LRRK2 forms a complex with Hsp90 via its kinase domain (Gloeckner et al., 2006; Dachsel et al., 2007). As an extension of these previous observations, we found that Hsp90 coimmunoprecipitated with LRRK2 in vivo from brain tissues. Furthermore, we demonstrated that inhibition of Hsp90 chaperone activity dramatically destabilized and increased the proteasome-mediated degradation of both endogenous and G2019S mutant LRRK2 in neurons. Our findings suggest that Hsp90 may serve as a useful target to suppress the accumulation and pathogenic activity of LRRK2 mutations. As a proof of principle, we showed that the treatment of Hsp90 inhibitor rescued the axon growth retardation defect caused by overexpression of the LRRK2 G2019S mutation in neurons, suggesting that Hsp90 inhibitors are valid therapeutic candidates for treatment of LRRK2-related PD.
Materials and Methods
Generation of LRRK2 G2019S conditional transgenic mice.
To develop a conditional LRRK2 G2019S transgenic mouse model, a cDNA fragment encoding the C-terminal hemagglutinin (HA)-tagged G2019S mutant LRRK2 protein was inserted into the mouse prior protein (pPrP)–tetP gene expression vector (a gift from Dr. David Borchelt, University of Florida, Gainesville, FL), which is controlled by the tetracycline-responsive promoter (tetP) (Jankowsky et al., 2005). The LRRK2 expression construct was then purified and microinjected into fertilized oocytes derived from C57BL6/J mice. The founder mice were crossed with wild-type (WT) C57BL6/J mice to produce the F1 generation. The F1 LRRK2 G2019S mutant mice were mated with calcium/calmodulin-dependent kinase II (CaMKII)–tTA mice (Mayford et al., 1996) to achieve high expression of LRRK2 in forebrain regions, including the olfactory bulb, striatum, hippocampus, and cortex. With the tet-off system, the expression of human LRRK2 was almost completely (>90%) suppressed after feeding the mouse with doxycycline (Dox)-treated pellets for 4 weeks (data not shown).
Immunoprecipitation.
Mouse forebrains were dissected out and homogenized in sucrose buffer [0.32 m sucrose, 1 mm NaHCO3, 1 mm MgCl2, 0.5 mm CaCl2, and protease inhibitor mixture (Roche, Indianapolis, IN) and phosphatase inhibitor cocktail (Pierce, Rockford, IL)]. Protein extracts (1 mg) were used for immunoprecipitation (IP), which were diluted in 1000 μl of cold IP buffer (20 mm HEPES, pH7.5, 1% NP-40, 5% glycol, 150 mm NaCl, 1 mm DTT, 1 mm PMSF, protease inhibitor mixture, and phosphatase inhibitor cocktail) and then incubated with 20 μl of HA matrix (anti-HA antibody-conjugated agarose beads; Roche) for 3 h at 4°C. The beads were washed with cold IP buffer six times and then eluted with 20 μl of triethylamine buffer (100 mm, pH 11). The eluted samples were neutralized by 5 μl of Tris-HCl (1 m), pH 7.4, and then size fractioned by SDS-PAGE. For immunoprecipitation from human embryonic kidney-293 (HEK-293) cells, cells were homogenized in cold IP buffer, and 500 μg of protein was used for immunoprecipitation using HA matrix as described previously. The beads were boiled in the SDS-PAGE sample buffer for 5 min, and the supernatants were size fractioned by SDS-PAGE.
Silver staining and mass spectrometry.
The LRRK2-binding proteins isolated by HA affinity purification was visualized by silver staining using the Silver Quest Silver Staining kit (Invitrogen, Carlsbad, CA) according to the instructions of the manufacturer. Visible protein bands were excised from the silver-stained gel and cut into 1 mm pieces. Proteins were identified by mass spectrometry performed by the Taplin Biological Mass Spectrometry Facility at Harvard Medical School (Boston, MA). A total of 14 Hsp90α-related peptides and 5 Hsp90β-related peptides were identified.
Western blot.
Neurons and HEK-293 cells were lysed in 20 mm HEPES, pH 7.4, 0.5% Triton X-100, 2% SDS, protease inhibitor mixture, and phosphatase inhibitor cocktail. Protein was size fractioned by 4–12% NuPage BisTris-PAGE (Invitrogen) using 3-(N-morpholino)-propanesulfonic acid running buffer (Invitrogen) and transferred to polyvinylidene difluoride membranes. Antibodies specific for LRRK2 (1:1000, JH5514), Hsp90 (1:3000; BD Biosciences Transduction Laboratories, Lexington, KY), cell division cycle 37 (Cdc37) (1:1000; BD Biosciences Transduction Laboratories), Hsp70 (1:3000; BD Biosciences Transduction Laboratories), HA (1:1000; Roche), α-synuclein (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), and β-actin (1:3000; Sigma) were used in this study. The enhanced chemiluminescence method was used to visualize the bound antibodies.
Cell culture and treatment.
Procedures for primary cultured cortical and hippocampal neuron cultures from postnatal day 1 pups and for HEK-293 cells were as described previously (Cai et al., 2005; Lai et al., 2006; Wang et al., 2008). When indicated, proteasome, lysosome, and protein synthesis inhibitors were preincubated with cells before treatment of Hsp90 inhibitors and kept in the culture medium. Geldanamycin (GA) (Sigma) and MG132 (Sigma) were dissolved in DMSO. Chloroquine (Sigma) was dissolved in water. PU-H71 and PU-DZ8 were dissolved in PBS buffer.
LRRK2 expression constructs and transfection.
The C-terminal HA-tagged wild-type and G2019S mutant LRRK2 cDNA were cloned into the pPrP–tetP tetracycline-regulated gene expression vector as indicated above. The cytomegalovirus (pCMV)–reverse tetracycline transactivator (rtTA) vector is a gift from Dr. Hermann Bujard (University of Heidelberg, Heidelberg, Germany). FuGene6 reagent (Roche) was used for cell transfection as suggested by the manufacturer. Dox (Sigma) was added into culture medium at the final concentration of 250 ng/L to turn on the LRRK2 expression in pPrP–tetP–LRRK2 and pCMV–rtTA cotransfected cells. In the PU-H71 dose-dependent study, HEK-293 cells were transfected with constructs for 24 h and then treated with different concentration of PU-H71 for 24 h. To disrupt the LRRK2–Hsp90 complex, culture cells were exposed to 1 μm GA for 4 h and then homogenized for immunoprecipitation.
Pulse chase.
HaloTag Interchangeable Labeling technology (Promega, Madison, WI) was used to study the stability of LRRK2 protein. Wild-type and G2019S mutant LRRK2 cDNA were cloned into the HaloTag pHT2 vector. After transfection with HaloTag wild-type or G2019S mutant LRRK2 vector for 36 h, HEK-293 cells were pulse-labeled with 5 μm HaloTag TMR Biotin ligand (Promega) in DMEM plus 10% FBS for 3 h. After washing three times with PBS, cells were chased in DMEM plus 10% FBS in the presence or absence of 1 μm PU-H71 for the indicated time. Cell were lysed in 20 mm Tris-HCl, pH 7.5, 10 mm NaCl, 1mm Na2EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 mg/ml leupeptin, and protease inhibitor cocktail (Roche). The biotin-labeled LRRK2 was captured by streptavidin-coated particles and Magnesphere Paramagnetic particles (Promega) for 2 h at 4°C and then washed three times with PBS containing 150 mm NaCl and 1% Triton X-100 and once with PBS only. Biotinylated LRRK2 was eluted by heating in SDS-PAGE sample buffer for 5 min at 95°C. Proteins were resolved on SDS-PAGE gel and detected by immunoblot using Halo antibody (Promega).
RNA extraction and reverse transcriptase-PCR.
Total RNA was extracted using RNAeasy Mini kit according to the instructions of the manufacturer (Qiagen, Valencia, CA). Reverse transcriptase (RT)-PCR was performed using SuperScript III first-strand synthesis system for RT-PCR (Invitrogen). RNase-treated cDNA was used for real-time PCR using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) in an ABI Prism 7700 Sequence Detection system. The cDNA of β-actin was used as an internal control. The following primers were used for LRRK2 (5′-AGCAGGACAAAGCCAGCCTCA-3′ and 5′-GATGGCAGCATTGGGATACAG-3′) and β-actin (5′-CGTTGACATCCGTAAAGACC-3′ and 5′-GCTAGGAGCCAGAGCAGTAA-3′).
Quantification of axonal outgrowth.
Hippocampal neurons were fixed after 2 d in culture and probed with TuJ1, a monoclonal neuron-specific antibody against βIII-tubulin. The images were captured using a Zeiss (Thornwood, NY) confocal microscope (LSM 510 META), and the lengths of axons were measured by using the segmented line function of NIH ImageJ software. In each experimental group, the lengths of axons were represented as a mean of axons from 50–100 neurons sampled from two to three randomly selected microscopic fields of three to four independent cultures.
Statistical analysis.
Data on drug-dose dependency and LRRK2 half-life (t1/2) were analyzed by nonlinear regression analysis using GraphPad Prism 4.0 (GraphPad Software, San Diego, CA). The IC50 values were expressed as mean ± 95% confidence intervals (CIs). Statistical significances were determined by comparing datasets of different groups using F test of the best-fit values for three parameters (logIC50, top and bottom). Statistical analysis of axon length was performed using the StatView program (version 5.0; SAS Institute, Cary, NC). Data are presented as means ± SEM. Statistical significances were determined by comparing means of different groups using ANOVA, followed by Fisher's PLSD test.
Results
LRRK2 forms a complex with Hsp90 in vivo
To investigate how the G2019S mutation in LRRK2 causes neuronal dysfunction in vivo, we generated a conditional transgenic mouse model in which the expression of C-terminal HA-tagged human LRRK2 was under the control of a tetracycline-responsive regulator. The expression of mutant LRRK2 protein was detected in the forebrain after having been crossed with CaMKII–tTA transgenic mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Using a HA affinity column, we purified LRRK2-associated protein complexes from transgenic mouse brains to identify any proteins that might interact with mutant LRRK2 in vivo. Proteins specifically present in mutant mouse brains, including LRRK2 and a pair of proteins with an apparent molecular weight of ∼90 kDa, were identified by silver staining (Fig. 1A). Mass spectrometry revealed that these 90 kDa proteins were two splice variants of Hsp90: Hsp90α and Hsp90β. The appearance of Hsp90 in the LRRK2-containing protein complex was further confirmed by immunoblot analysis with an Hsp90-specific antibody after immunoprecipitation of HA-tagged LRRK2 from transgenic mouse brains (Fig. 1B). This observation that LRRK2 forms a complex with Hsp90 in vivo is consistent with previous in vitro studies (Gloeckner et al., 2006; Dachsel et al., 2007).
Figure 1.

LRRK2 complexes with Hsp90 in LRRK2 G2019S mutant transgenic mouse brains. A, Silver staining of purified LRRK2-containing protein complexes from LRRK2 G2019S transgenic (G2019S) mouse brains. Protein bands seen in LRRK2 G2019S but not in ntg mouse brain samples were subjected to mass spectrometry analysis. LRRK2 and Hsp90 chaperone proteins were selectively identified in LRRK2 immunoprecipitates. B–D, LRRK2 was immunoprecipitated with HA matrix from G2019S and ntg mouse brains, and the immunoprecipitates (IP) were blotted with antibodies against Hsp90, Cdc37, and Hsp70. Hsp90 and Cdc37, but not Hsp70, were coimmunoprecipitated with LRRK2 from mouse brain extracts. Mr, Molecular weight; WB, Western blots.
Hsp90 functions in concert with a set of cochaperones that link Hsp90 to distinct classes of client proteins (Pearl and Prodromou, 2006). The mammalian homolog of the yeast cell cycle control protein Cdc37 acts as a kinase-specific target subunit for Hsp90. Cdc37 was also coimmunoprecipitated with LRRK2 (Fig. 1C). Other cochaperones, including Hsp70, Hsp70/Hsp90-organizing protein (HOP), and p23, however, did not show the same interaction with LRRK2 (Fig. 1D, 2A).
Figure 2.

Hsp90 inhibitor disrupts the LRRK2/Hsp90 complex. A, HEK-293 cells transfected with either WT or G2019S mutant LRRK2 were treated with Hsp90 inhibitor GA (+) or vehicle (−) for 1 h, lysed, immunoprecipitated with HA matrix, and then subjected to Western blot analysis with antibodies against Hsp90, LRRK2, Cdc37, p23, HOP, and Hsc70. B, The expression of Hsp90, LRRK2, Cdc37, p23, HOP, and Hsc70 in cell extracts was revealed by Western blot analysis with antibodies against Hsp90, Cdc37, p23, HOP, and Hsc70. LRRK2 was detected by the anti-HA antibody. IP, Immunoprecipitates; Mr, molecular weight.
Hsp90 inhibitor disrupts the formation of the LRRK2/Hsp90 complex
The chaperone activities of Hsp90 are ATP dependent. GA, which interferes with the association of Hsp90 and ATP, specifically inhibits Hsp90 chaperone activity in cells (Chiosis et al., 2004). To examine whether the formation of the LRRK2/Hsp90 complex depends on Hsp90 chaperone activity, we immunoprecipitated LRRK2 from LRRK2-transfected HEK-293 cells after a short treatment with GA (1 μm). We found that treatment of GA effectively disrupted the coimmunoprecipitation of both WT and G2019S mutant LRRK2 with endogenous Hsp90 and Cdc37 in HEK-293 cells (Fig. 2). These data demonstrate that the formation of the LRRK2/Hsp90 complex is dependent on the chaperone activity of Hsp90. Interestingly, more constitutive heat shock protein 70 (Hsc70) was pulled down with LRRK2 after treatment of GA. Because Hsc70 is involved in protein folding, the dissociation of LRRK2 with the Hsp90-mediated chaperone complex likely results in misfolding of LRRK2 and more binding of LRRK2 with Hsc70-mediated chaperone complex.
Inhibition of Hsp90 reduces the steady level of LRRK2 expression in cell lines
Hsp90 is known to regulate the function and stability of many protein kinases (Sreedhar et al., 2004). To investigate whether the activity of Hsp90 is required for the stability of LRRK2, we transfected WT and G2019S mutant LRRK2 into HEK-293 cells and treated the transfected cells with Hsp90 specific inhibitor PU-H71 at different doses. Similar to GA, purine-scaffold derivative PU-H71 also disrupts the association of Hsp90 with ATP, resulting in inhibition of Hsp90 chaperone activity (Rodina et al., 2007). Furthermore, whereas GA activity could be modulated by a reductive environment in the cell, resulting in effects that are not a direct consequence of Hsp90 inhibition (Kelland et al., 1999), PU-H71 is not metabolically sensitive to the cellular environment (Rodina et al., 2007). Both G2019S (Fig. 3A) and WT LRRK2 (Fig. 3B) displayed a comparable sensitivity to PU-H71-mediated Hsp90 inhibition in HEK-293 cells (Fig. 3C) (IC50-G2019S of 130.0 nm vs IC50-WT of 112.0 nm, n = 4; p = 0.86). Similar results were also obtained when WT and G2019S LRRK2-transfected HEK-293 cells were treated with GA and another Hsp90 inhibitor, PU-DZ8 (data not shown).
Figure 3.

Hsp90 inhibitors suppress the steady level of LRRK2 but not α-synuclein expression in HEK-293 cells. A, B, Western blot analysis of LRRK2 expression in HEK-293 cells transfected with G2019S mutant (G2019S) (A) and WT LRRK2 after treatment with the indicated doses of Hsp90 inhibitor PU-H71 for 24 h (B). C, Dose–response curve of overexpressed wild-type (filled triangles) and G2019S mutant (open triangles) LRRK2 protein treated with PU-H71. D, Western blot analysis of α-synuclein A53T expression in transfected HEK-293 cells treated with vehicle (DMSO) or 500 nm GA for 24 h. Mr, Molecular weight.
To investigate whether Hsp90 inhibitors generally suppress the overall steady level of protein expression in cells, we examined the expression of human α-synuclein A53T in transfected HEK-293 cells after treatment with 500 nm GA. Consistent with previous observations (Auluck and Bonini, 2002; Klucken et al., 2004; Auluck et al., 2005; Flower et al., 2005), the steady level of α-synuclein expressed in transfected HEK-293 cells was not affected by Hsp90 inhibition (Fig. 3D).
Inhibition of Hsp90 reduces the steady level of mutant and endogenous LRRK2 in cultured neurons
To investigate whether the activity of Hsp90 is required for the stability of LRRK2 in neurons, we examined the expression of LRRK2 in primary cultured cortical neurons derived from nontransgenic (ntg) and LRRK2 G2019S mutant pups after treatment with Hsp90-specific inhibitors. Treatment of neurons with GA suppressed the steady levels of both mutant and endogenous LRRK2 expression in both a dose-dependent (Fig. 4A) and time-dependent (Fig. 4B) manner. It appears that the exogenous G2019S LRRK2 was more sensitive to GA treatment than endogenous protein (Fig. 4C) (IC50-G2019S of 4.7 nm, 95% CI of 1.6–13.6 nm vs IC50-endogenous of 37.2 nm, 95% CI of 11.8–117.5 nm, n = 4; p < 0.05). Similarly, the suppression of LRRK2 expression was also observed when neurons were treated with PU-H71 (Fig. 4D) (IC50-G2019S of 26.9 nm, 95% CI of 8.3–87.2 nm vs IC50-endogenous of 387.0 nm, 95% CI of 144.9–1000 nm, n = 4; p < 0.0001). The increased sensitivity of G2019S mutation to Hsp90 inhibition is likely a result from the greater expression of mutant LRRK2 in neurons (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). In line with this notion, heterologous expression of WT and G2019S LRRK2 in HEK-293 cells had a comparable sensitivity to Hsp90 inhibition (Fig. 3C). These observations suggest that the alteration of the apparent IC50 of Hsp90 inhibitors is attributable to increased expression levels rather than an LRRK2 mutation. Nonetheless, lower concentrations of GA and PU-H71 could be used to selectively suppress the expression of G2019S mutant but not endogenous LRRK2 protein in neurons derived from LRRK2 transgenic mice.
Figure 4.

Hsp90 inhibitors suppress the steady level of mutant and endogenous LRRK2 in neurons. A, B, Western blot analysis of LRRK2 expression in primary cultured cortical neurons derived from LRRK2 G2019S transgenic (G2019S) and littermate nontransgenic pups (endogenous) after treatment of indicated doses of Hsp90 inhibitor GA for 24 h (A) or 10 nm GA for indicated time points (B). C, D, Dose–response curves of endogenous (filled triangles) and G2019S mutant (open triangles) LRRK2 protein treated with GA (C) or PU-H71 (D). LRRK2 was detected by a LRRK2 polyclonal antibody, JH5514 (Biskup et al., 2006). Mr, Molecular weight.
To investigate whether Hsp90 inhibitors interfere with the transcription of the LRRK2 gene, the expression of mutant and endogenous LRRK2 messenger RNA was quantified by real-time RT-PCR in vehicle- and GA-treated neurons. No significant difference between expression of mutant and endogenous LRRK2 messenger RNA was found (data not shown), suggesting that Hsp90 inhibitors suppress the LRRK2 expression via inhibition of the chaperone activities of Hsp90 rather than interference with the transcription of LRRK2.
Hsp90 regulates the stability of LRRK2
To investigate whether Hsp90 plays a role in maintaining the stability of LRRK2 in cells, we examined the t1/2 of both WT and G2019S mutant LRRK2 proteins in transfected HEK-293 cells after treatment with 1 μm PU-H71 (Fig. 5A). The t1/2 for untreated WT and G2019S mutant LRRK2 were comparable (Fig. 5B,C) (t1/2-WT of 8.66 h, 95% CI of 5.22–25.30 h vs t1/2-G2019S of 8.51 h, 95% CI of 4.94–30.81 h; r2 = 0.96; n = 2). Meanwhile, the t1/2 of WT and G2019S mutant LRRK2 proteins in PU-H71-treated cells were significantly reduced compared with vehicle-treated cells (Fig. 5B,C) (t1/2-WT of 0.75 h, 95% CI of 0.49–1.59 h vs t1/2-G2019S of 0.83 h, 95% CI of 0.54–1.73 h; r2 = 0.95; n = 2). These data demonstrate that Hsp90 is critical for maintaining the stability of both WT and mutant LRRK2 in cells.
Figure 5.
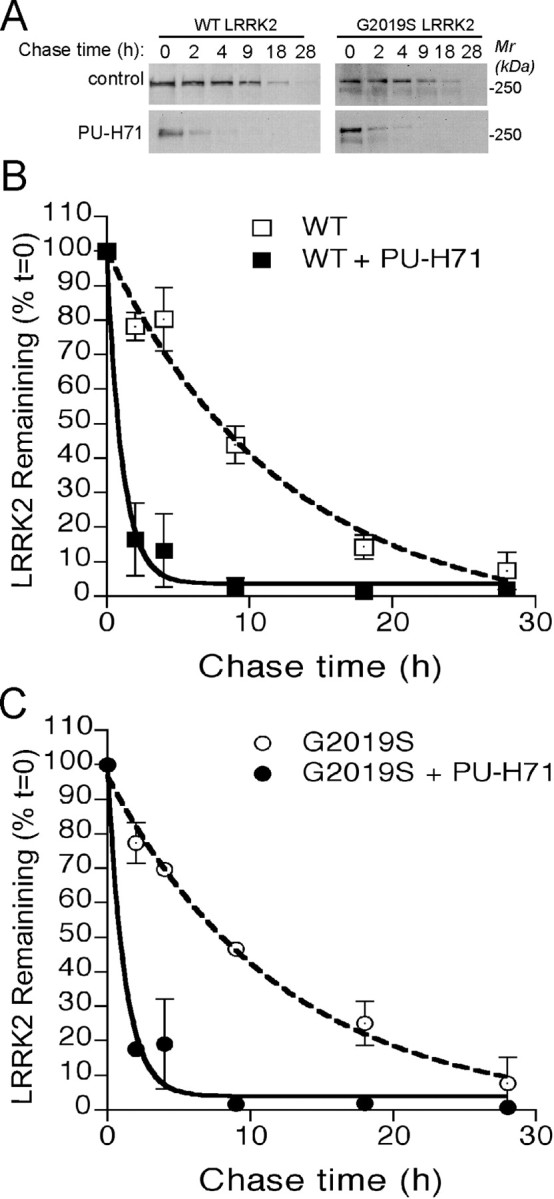
Hsp90 regulates the stability of LRRK2. A, WT and G2019S mutant LRRK2 proteins were pulse labeled with biotinylated Halo ligand and chased at indicated time intervals in the presence or absence (control) of 1 μm PU-H71. B, C, Time courses of the decay of biotinylated WT (B) and G2019S mutant (C) LRRK2 proteins in the presence or absence (control) of Hsp90 inhibitor PU-H71. Mr, Molecular weight.
The Hsp90 inhibitor-induced suppression of LRRK2 expression is mediated by the proteasome-dependent protein degradation pathway
Hsp90 client proteins tend to be degraded via the proteasome-dependent pathway during chaperone inhibition (Goryunov and Liem, 2007). Concordantly, the GA-induced suppression of mutant and endogenous LRRK2 was selectively blocked by a proteasome inhibitor, MG132, but not the lysosome inhibitor chloroquine in neurons (Fig. 6A,B). These observations demonstrate that LRRK2, like other Hsp90 client proteins, are stabilized by Hsp90 against proteasome-mediated protein degradation.
Figure 6.

Hsp90 inhibitor-induced suppression of LRRK2 expression is mediated by the proteasome-dependent protein degradation pathway. A, B, Western blot analyses of LRRK2 protein in primary cultured cortical neurons derived from LRRK2 G2019S mutant (A) and ntg (B) pups after treatment with Hsp90 inhibitor GA in the presence or absence of the proteasome inhibitor MG132 (MG) or the lysosome inhibitor chloroquine (Ch). LRRK2 was detected by an LRRK2 polyclonal antibody, JH5514. Mr, Molecular weight.
Hsp90 inhibitor rescues the axonal outgrowth deficits of neurons expressing the LRRK2 G2019S mutation
A previous report indicates that LRRK2 is involved in maintaining the extension and branching of neurites (MacLeod et al., 2006). Mutant LRRK2, particularly the G2019S mutation but not wild-type LRRK2, causes neurite shortening via a potential autophagy-mediated pathway (Plowey et al., 2008). Consistent with this observation, we recently found that neurons derived from LRRK2 G2019S transgenic mouse brains also displayed retarded axonal outgrowth during neuron morphogenesis (C. Xie, unpublished data). To determine whether Hsp90 inhibitors are capable of rescuing the axonal growth deficit in LRRK2 G2019S transgenic neurons by suppressing mutant LRRK2 expression, we treated the neurons with PU-H71 immediately after they were plated onto culture dishes. We used PU-H71 at 40 nm, which significantly suppressed the expression of mutant LRRK2 but had a minimal effect on endogenous LRRK2 (Fig. 4D). The length of axons, the longest neurites, of vehicle-treated mutant neurons (G2019S) was significantly shorter than that of ntg littermate controls after 2d in culture (Fig. 7A,B,E) (lengthG2019S of 68.30 ± 2.73 μm, n = 79 vs lengthntg of 93.75 ± 4.87 μm, n = 48; p < 0.001). PU-H71 significantly increased the length of axons of mutant neurons compared with treatment with vehicle (Fig. 7D,E) (lengthG2019S/H71 of 112.47 ± 7.04 μm, n = 45; p < 0.001). As a positive control, treatment with Dox, which suppresses the expression of mutant LRRK2 through the tetracycline responsible regulator, also rescued the axon growth defect of mutant neurons (Fig. 7C,E) (lengthG2019S/Dox of 96.58 ± 4.09 μm, n = 62; p < 0.001). In contrast, no significant alteration of axon length was detected in vehicle-, Dox-, and PU-H71-treated nontransgenic neurons (Fig. 7F). These results demonstrate that Hsp90 inhibitors can ameliorate mutant LRRK2-elicited loss of axonal outgrowth.
Figure 7.

PU-H71 rescues the axonal growth deficit of neurons derived from LRRK2 G2019S mutant mice. A–D, Representative images of hippocampal neurons derived from nontransgenic (A) and LRRK2 G2019S transgenic (C, D) pups treated with vehicle (Veh), doxycycline (Dox), or PU-H71 for 2 d and visualized by immunostaining of βIII-tubulin antibody. Scale bar, 100 μm. E, F, Bar graphs of axon length of LRRK2 G2019S (E) and nontransgenic (F) neurons after 2 d in culture. Data are presented as means ± SEM. *p < 0.0001.
Discussion
Mutations in LRRK2 are causative for both familial and apparently sporadic forms of PD (Farrer et al., 2005; Hardy et al., 2006). Because mutations in LRRK2 are likely associated with a toxic gain of function, destabilization of LRRK2 presents an attractive way to limit its detrimental effects in neurons. Here, we have shown that inhibition of Hsp90 chaperone function dramatically decreases the stability of LRRK2 in cell lines and primary neuronal cultures via a proteasome-mediated protein degradation mechanism, suggesting that Hsp90 may serve as a useful target for suppressing the accumulation and pathogenic activity of PD-related LRRK2 mutations in neurons. Indeed, treatment of neurons with an Hsp90 inhibitor rescued the axon growth retardation defect caused by overexpression of the LRRK2 G2019S mutation.
Hsp90 and Cdc37 contribute to the stabilization, activation, and/or translocation of client protein kinases, such as Src family tyrosine kinase and the serine/threonine kinase Raf and MEK (mitogen-activated protein kinase kinase) (Pearl and Prodromou, 2006). Although its physiological substrates remain to be determined, LRRK2 is likely an active serine/threonine protein kinase (Greggio et al., 2006; Smith et al., 2006; Guo et al., 2007; Li et al., 2007; West et al., 2007). The G2019S substitution at the kinase domain (Goldwurm et al., 2005; Bonifati, 2006) likely increases the kinase activity of LRRK2 (Greggio et al., 2006; Smith et al., 2006; Jaleel et al., 2007; Luzon-Toro et al., 2007). In line with previous in vitro observations (Gloeckner et al., 2006; Dachsel et al., 2007), we found that Hsp90 and Cdc37 formed a complex with LRRK2 in vivo. Furthermore, we demonstrated that inhibition of Hsp90 chaperone activities caused the dissociation of LRRK2 with Hsp90/Cdc37 chaperone complex and shifted LRRK2 to Hsc70 and other chaperone proteins linked to the proteasome degradation pathway. Therefore, the chaperone activity of Hsp90 is critical for maintaining the stability of LRRK2 in cells. Because of the existence of sequence polymorphism between mouse and human LRRK2 (Paisan-Ruiz et al., 2004; Zimprich et al., 2004), mouse and human LRRK2 may potentially function differently. In our study, we tested the role of Hsp90 on the stability of both human LRRK2 (wild-type or the G2019S mutation overexpressed in cell lines and neurons) and mouse LRRK2 (endogenous LRRK2 in neurons). We found that inhibition of Hsp90 activity led to degradation of both human and mouse LRRK2, which is independent of the origin of LRRK2 protein.
Because inhibition of Hsp90 chaperone function dramatically decreases the stability of LRRK2 in cells, Hsp90 may serve as a valid therapeutic target for treatment of PD via suppression of mutant LRRK2 accumulation and related pathogenic activities in neurons. In fact, a large number of natural and synthetic Hsp90 inhibitors have been developed for cancer therapy, and some are already in clinical trials (Solit and Rosen, 2006). In tumor cells, inhibition of Hsp90 chaperone activity leads to a selective degradation of signaling molecules, including protein kinases that are involved in cell proliferation, cell cycle regulation, and apoptosis (McDonald et al., 2006). Additionally, several Hsp90 inhibitors have also been shown to successfully ameliorate neuropathological abnormalities in mouse models of neurodegenerative diseases (Waza et al., 2006; Dickey et al., 2007; Luo et al., 2007). Moreover, the Hsp90 inhibitor GA prevents α-synuclein-mediated toxicity in several animal models through upregulation of Hsp70 chaperone activity, which prevents the formation of α-synuclein aggregates (Auluck and Bonini, 2002; Klucken et al., 2004; Auluck et al., 2005; Flower et al., 2005). Therefore, Hsp90 inhibitors may ameliorate the cytotoxicity induced by these two PD-related dominant mutations.
A main concern for Hsp90 inhibition is the lack of specificity for the affected proteins or cells. Indeed, among the three Hsp90 inhibitors we tested, none of them showed specificity for the LRRK2 G2019S mutation, although we have shown that exogenous LRRK2 was more susceptible to Hsp90 inhibition than the endogenous LRRK2 in neurons. However, progress has been made to develop Hsp90 inhibitors that display selectivity to aberrant proteins and cells. For example, PU-H71 has been shown to be more effective in suppressing Hsp90 activities in tumor cells compared with normal cells (Rodina et al., 2007). Therefore, it is of interest to screen more Hsp90 inhibitors that display selectivity to mutant LRRK2. In addition, it will be interesting to postulate whether Hsp90 inhibitors can prevent any pathological or behavioral phenotypes in LRRK2 G2019S mutant transgenic mice. We, however, have not detected any obvious neuropathological abnormalities or motor dysfunctions in LRRK2 mutant mice to date (12 months of age) (X. Lin, unpublished data). Therefore, it is premature to test any efficacy of Hsp90 inhibition in these mice. Nonetheless, because of the drastic effect of Hsp90 inhibition on the expression of LRRK2, Hsp90 inhibitors are likely to be able to rescue the pathological and behavioral phenotypes caused by overexpression of mutant LRRK2 in mice. Together, our findings establish a novel function of Hsp90 in regulating the stability of LRRK2, suggesting that Hsp90 inhibitors are valid therapeutic candidates for treatment of both α-synuclein and LRRK2-related PD.
Footnotes
This work was supported by the intramural research programs of the National Institute on Aging and the National Human Genome Research Institute and in part by the National Institute of Aging Grant 1R21AG028811 (G.C.). We thank Amy Chen of the Mouse Embryonic Stem Cell and Transgenic Core Facility, National Human Genome Research Institute, National Institutes of Health for pronuclear injection, and Dr. David Borchelt (University or Florida, Gainesville, FL) for providing the Prp-tet gene expression vector.
The authors declare no competing financial interests.
References
- Auluck PK, Bonini NM. Pharmacological prevention of Parkinson disease in Drosophila. Nat Med. 2002;8:1185–1186. doi: 10.1038/nm1102-1185. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Meulener MC, Bonini NM. Mechanisms of suppression of α-synuclein neurotoxicity by geldanamycin in Drosophila. J Biol Chem. 2005;280:2873–2878. doi: 10.1074/jbc.M412106200. [DOI] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- Bonifati V. Parkinson's disease: the LRRK2–G2019S mutation: opening a novel era in Parkinson's disease genetics. Eur J Hum Genet. 2006;14:1061–1062. doi: 10.1038/sj.ejhg.5201695. [DOI] [PubMed] [Google Scholar]
- Cai H, Lin X, Xie C, Laird FM, Lai C, Wen H, Chiang HC, Shim H, Farah MH, Hoke A, Price DL, Wong PC. Loss of ALS2 function is insufficient to trigger motor neuron degeneration in knock-out mice but predisposes neurons to oxidative stress. J Neurosci. 2005;25:7567–7574. doi: 10.1523/JNEUROSCI.1645-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiosis G, Vilenchik M, Kim J, Solit D. Hsp90: the vulnerable chaperone. Drug Discov Today. 2004;9:881–888. doi: 10.1016/S1359-6446(04)03245-3. [DOI] [PubMed] [Google Scholar]
- Dachsel JC, Taylor JP, Mok SS, Ross OA, Hinkle KM, Bailey RM, Hines JH, Szutu J, Madden B, Petrucelli L, Farrer MJ. Identification of potential protein interactors of Lrrk2. Parkinsonism Relat Disord. 2007;13:382–385. doi: 10.1016/j.parkreldis.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Jr, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M, Stone J, Mata IF, Lincoln S, Kachergus J, Hulihan M, Strain KJ, Maraganore DM. LRRK2 mutations in Parkinson disease. Neurology. 2005;65:738–740. doi: 10.1212/01.wnl.0000169023.51764.b0. [DOI] [PubMed] [Google Scholar]
- Flower TR, Chesnokova LS, Froelich CA, Dixon C, Witt SN. Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson's disease. J Mol Biol. 2005;351:1081–1100. doi: 10.1016/j.jmb.2005.06.060. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- Goldwurm S, Di Fonzo A, Simons EJ, Rohe CF, Zini M, Canesi M, Tesei S, Zecchinelli A, Antonini A, Mariani C, Meucci N, Sacilotto G, Sironi F, Salani G, Ferreira J, Chien HF, Fabrizio E, Vanacore N, Dalla LA, Stocchi F, et al. The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson's disease and originates from a common ancestor. J Med Genet. 2005;42:e65. doi: 10.1136/jmg.2005.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryunov D, Liem RK. CHIP-ping away at tau. J Clin Invest. 2007;117:590–592. doi: 10.1172/JCI31505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Singleton A. Kinase signaling pathways as potential targets in the treatment of Parkinson's disease. Expert Rev Proteomics. 2007;4:783–792. doi: 10.1586/14789450.4.6.783. [DOI] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG. The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. 2007;313:3658–3670. doi: 10.1016/j.yexcr.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson's disease and parkinsonism. Ann Neurol. 2006;60:389–398. doi: 10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Gonzales V, Savonenko AV, Wen JC, Jenkins NA, Copeland NG, Younkin LH, Lester HA, Younkin SG, Borchelt DR. Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS Med. 2005;2:e355. doi: 10.1371/journal.pmed.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelland LR, Sharp SY, Rogers PM, Myers TG, Workman P. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst. 1999;91:1940–1949. doi: 10.1093/jnci/91.22.1940. [DOI] [PubMed] [Google Scholar]
- Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 reduces alpha-synuclein aggregation and toxicity. J Biol Chem. 2004;279:25497–25502. doi: 10.1074/jbc.M400255200. [DOI] [PubMed] [Google Scholar]
- Lai C, Xie C, McCormack SG, Chiang HC, Michalak MK, Lin X, Chandran J, Shim H, Shimoji M, Cookson MR, Huganir RL, Rothstein JD, Price DL, Wong PC, Martin LJ, Zhu JJ, Cai H. Amyotrophic lateral sclerosis 2-deficiency leads to neuronal degeneration in amyotrophic lateral sclerosis through altered AMPA receptor trafficking. J Neurosci. 2006;26:11798–11806. doi: 10.1523/JNEUROSCI.2084-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, Moulick K, Aguirre J, Wu N, Greengard P, Chiosis G. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc Natl Acad Sci USA. 2007;104:9511–9516. doi: 10.1073/pnas.0701055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzon-Toro B, de la Torre ER, Delgado A, Perez-Tur J, Hilfiker S. Mechanistic insight into the dominant mode of the Parkinson's disease-associated G2019S LRRK2 mutation. Hum Mol Genet. 2007;16:2031–2039. doi: 10.1093/hmg/ddm151. [DOI] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McDonald E, Workman P, Jones K. Inhibitors of the HSP90 molecular chaperone: attacking the master regulator in cancer. Curr Top Med Chem. 2006;6:1091–1107. doi: 10.2174/156802606777812004. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der BM, Lopez dM, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ, III, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodina A, Vilenchik M, Moulick K, Aguirre J, Kim J, Chiang A, Litz J, Clement CC, Kang Y, She Y, Wu N, Felts S, Wipf P, Massague J, Jiang X, Brodsky JL, Krystal GW, Chiosis G. Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat Chem Biol. 2007;3:498–507. doi: 10.1038/nchembio.2007.10. [DOI] [PubMed] [Google Scholar]
- Skipper L, Li Y, Bonnard C, Pavanni R, Yih Y, Chua E, Sung WK, Tan L, Wong MC, Tan EK, Liu J. Comprehensive evaluation of common genetic variation within LRRK2 reveals evidence for association with sporadic Parkinson's disease. Hum Mol Genet. 2005;14:3549–3556. doi: 10.1093/hmg/ddi376. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Solit DB, Rosen N. Hsp90: a novel target for cancer therapy. Curr Top Med Chem. 2006;6:1205–1214. doi: 10.2174/156802606777812068. [DOI] [PubMed] [Google Scholar]
- Sreedhar AS, Soti C, Csermely P. Inhibition of Hsp90: a new strategy for inhibiting protein kinases. Biochim Biophys Acta. 2004;1697:233–242. doi: 10.1016/j.bbapap.2003.11.027. [DOI] [PubMed] [Google Scholar]
- Wang L, Shim H, Xie C, Cai H. Activation of protein kinase C modulates BACE1-mediated beta-secretase activity. Neurobiol Aging. 2008;29:357–367. doi: 10.1016/j.neurobiolaging.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waza M, Adachi H, Katsuno M, Minamiyama M, Tanaka F, Doyu M, Sobue G. Modulation of Hsp90 function in neurodegenerative disorders: a molecular-targeted therapy against disease-causing protein. J Mol Med. 2006;84:635–646. doi: 10.1007/s00109-006-0066-0. [DOI] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]