

Abstract
Camurati‐Engelmann disease (CED) is a rare autosomal dominant type of bone dysplasia. This review is based on the unpublished and detailed clinical, radiological, and molecular findings in 14 CED families, comprising 41 patients, combined with data from 10 other previously reported CED families. For all 100 cases, molecular evidence for CED was available, as a mutation was detected in TGFB1, the gene encoding transforming growth factor (TGF) β1. Pain in the extremities was the most common clinical symptom, present in 68% of the patients. A waddling gait (48%), easy fatigability (44%), and muscle weakness (39%) were other important features. Radiological symptoms were not fully penetrant, with 94% of the patients showing the typical long bone involvement. A large percentage of the patients also showed involvement of the skull (54%) and pelvis (63%). The review provides an overview of possible treatments, diagnostic guidelines, and considerations for prenatal testing. The detailed description of such a large set of CED patients will be of value in establishing the correct diagnosis, genetic counselling, and treatment.
Keywords: Camurati‐Engelmann disease, progressive diaphyseal dysplasia, transforming growth factor β1, phenotypic variability
Camurati‐Engelmann disease (CED) or progressive diaphyseal dysplasia (MIM 131300) is an autosomal dominant condition belonging to the group of craniotubular hyperostoses. Initially described by Cockayne in 1920,1 Camurati was the first to suggest its hereditary nature in 1922.2 In 1929, Engelmann reported a single case with muscular wasting and marked bone involvement.3 The name progressive diaphyseal dysplasia emphasises the progressive nature of the hyperostosis and the ever present involvement of the diaphyses,4 but currently, the eponym Camurati‐Engelmann disease is widely accepted.
The hallmark of the disorder is the cortical thickening of the diaphyses of the long bones. Hyperostosis is bilateral and symmetrical and usually starts at the diaphyses of the femora and tibiae, expanding to the fibulae, humeri, ulnae, and radii. As the disease progresses, the metaphyses may become affected as well, but the epiphyses are spared.5 Sclerotic changes at the skull base may be present. The onset of the disease is usually during childhood and almost always before the age of 30. Most patients present with limb pain, muscular weakness, a waddling gait, and easy fatigability. Systemic manifestations—such as anaemia, leucopenia, and hepatosplenomegaly—occur occasionally.6 Abnormal values for several markers of bone formation and resorption have been reported in a few patients.7,8
In this review, clinical, radiological, and molecular data on 24 CED families were collected. Presentation of families from Europe, Asia, Africa, America, Australia, and Oceania shows that CED is spread worldwide. Fourteen of the families (41 patients) were examined by at least one of us. Data on 10 additional families (59 patients) were collected from published reports.9,10,11,12 Including the families presented in this paper, TGFB1 mutations in 45 CED families have been described worldwide.10,11,13,14,15,16,17,18 For the remaining 21 families, however, no published clinical or radiological information was available.
Radiological, scintigraphic, and clinical manifestations and phenotypic variability
Table 1 summarises clinical, radiological, scintigraphic, and molecular data on all the patients. Representative imaging studies and a clinical picture are presented in figs 1–4. From the data, several important conclusions can be drawn. In 94% of the patients—defined by the presence of a molecular defect in TGFB1—radiological symptoms are penetrant, with cortical thickening of the diaphyses of the long bones being the first manifestation. The skull (54%) and pelvis (63%) are other commonly involved sites. Scintigraphy detected increased osteoblastic activity in the affected regions (limbs, pelvis, skull, spine; see fig 2) in 74% of the investigated patients (17/22). As increased tracer uptake can be perceived even before sclerosis becomes radiologically visible, scintigraphy is a valuable technique for diagnosing CED in an early stage of disease.
Table 1 Overview of clinical, radiological, scintigraphic, and molecular data on patients from the 24 families.
| Family 1 | Family 2 | Family 3 | Family 4 | Family 5 | |
|---|---|---|---|---|---|
| (Belgium) | (Iraq) | (UK) | (Italy) | (Belgium) | |
| Mutation (DNA) | 673T→C | 653G→A | 652C→T | 28_36dup | 241T→C |
| Mutation (protein) | C225R | R218H | R218C | L10‐L12dup | Y81H |
| Number of patients | 3 | 5 | 3 | 2 | 3 |
| Clinical symptoms | 3/3 | 4/5 | 2/3 | 2/2 | 1/3 |
| Pain in extremities | 2/3 | 4/5 | 2/3 | 0/2 | 1/3 |
| Easy fatigability | 0/3 | 3/5 | 2/3 | 0/2 | 1/3 |
| Muscle weakness | 0/3 | 2/5 | 2/3 | 0/2 | 1/3 |
| Waddling gait | 1/3 | 3/5 | 1/3 | 0/2 | 1/3 |
| Hearing loss | 0/3 | 2/5 | 0/3 | 2/2 | 0/3 |
| Reduced subcutaneous fat | 1/3 | 2/5 | 0/3 | 2/2 | 1/3 |
| Other | Hyperthermia (1/3) | Hepatosplenomegaly (1/3) | ESR and CRP↑ (2/3) | – | Small stature (1/3) |
| Cranial nerve compression (1/3) | |||||
| Small stature (2/3) | |||||
| Headache (1/3) | |||||
| Radiological abnormalities | 3/3 | 5/5 | 3/3 | 2/2 | 1/3 |
| Cortical thickening diaphyses | 3/3 | 5/5 | 3/3 | 2/2 | 1/3 |
| Sclerosis of skull | 1/3 | 2/5 | ND | 2/2 | 0/3 |
| Other | – | – | Spine osteoporotic (2/3) | Spine, pelvis (1/2) | – |
| Coxa valga (2/3) | |||||
| Increased BMD | ND | ND | + (hip) (2/3) | 2/2 | ND |
| − (spine) (2/3) | |||||
| Scintigraphic abnormalities | ND | 4/4 | ND | 1/1 | 1/1 |
| Treatment | GC (1/3) | GC (1/5) | GC (2/3) | Calcitonin (1/2) | GC (1/3) |
| NSAIDs (1/3) | NSAIDs (2/3) | BP (2/2) | NSAIDs (1/3) | ||
| Analgesics (2/3) | Analgesics (1/3) | ||||
| BP (2/3) | |||||
| Tibial osteotomy (1/3) | |||||
| Femoral osteotomy (1/3) |
| Family 6 | Family 7 | Family 8 | Family 9 | Family 10 | |
|---|---|---|---|---|---|
| (Belgium) | (Italy) | (Germany) | (UK) | (Tonga‐Oceania) | |
| Mutation (DNA) | 241T→C | 653G→A | 653G→A | 653G→A | 653G→A |
| Mutation (protein) | Y81H | R218H | R218H | R218H | R218H |
| Number of patients | 6 | 2 | 3 | 2 | 2 |
| Clinical symptoms | 3/6 | 2/2 | 1/3 | 1/2 | 1/2 |
| Pain in extremities | 2/6 | 1/2 | 1/3 | 1/2 | 0/2 |
| Easy fatigability | 1/6 | 1/2 | 1/3 | 0/2 | 0/2 |
| Muscle weakness | 1/6 | 1/2 | 1/3 | 0/2 | 0/2 |
| Waddling gait | 2/6 | 2/2 | 1/3 | 0/2 | 0/2 |
| Hearing loss | 0/6 | 0/2 | 0/3 | 0/2 | 1/2 |
| Reduced subcutaneous fat | 0/6 | 1/2 | 0/3 | 0/2 | 0/2 |
| Other | – | – | – | – | Proptosis (1/2) |
| Radiological abnormalities | 4/6 | 2/2 | 1/1 | 2/2 | 2/2 |
| Cortical thickening diaphyses | 4/6 | 2/2 | 1/1 | 2/2 | 2/2 |
| Sclerosis of skull | 1/2 | 0/2 | ND | ND | 1/1 |
| Other | Pelvis (1/2) | – | – | ND | – |
| Increased BMD | ND | ND | ND | ND | 1/1 |
| Scintigraphic abnormalities | 1/6 | ND | 1/1 | ND | 1/1 |
| Treatment | GC (1/6) | – | Penicillin | Treated with vitamin D for presumed rickets (1/2) | Orbital decompression (1/1) |
| BP (1/6) | Gold salts | ||||
| NSAIDs |
| Family 11 | Family 12 | Family 13 | Family 14 | Family 15* | |
|---|---|---|---|---|---|
| (Morocco) | (Belgium) | (Spain) | (Germany) | (Israel) | |
| Mutation (DNA) | 463C→T | 673T→C | 652C→T | 664C→G | 652C→T |
| Mutation (protein) | R156C | C225R | R218C | H222D | R218C |
| Number of patients | 2 | 3 | 4 | 1 | 16 |
| Clinical symptoms | 2/2 | 2/3 | 4/4 | 1/1 | 10/16 |
| Pain in extremities | 2/2 | 2/3 | 2/4 | 1/1 | 8/8 |
| Easy fatigability | 1/2 | 1/3 | 0/4 | 1/1 | ND |
| Muscle weakness | 0/2 | 1/3 | 4/4 | 1/1 | 4/8 |
| Waddling gait | 1/2 | 1/3 | 1/4 | 1/1 | 6/8 |
| Hearing loss | 0/2 | 0/3 | 0/4 | 0/1 | 0/8 |
| Reduced subcutaneous fat | Obese (2/2) | Obese (1/3) | 0/4 | 1/1 | ND |
| Other | ESR and CRP↑ (1/2) | – | Vision ↓ (1/4) | Delayed puberty | Inability to run quickly (3/8) |
| Mild splenomegaly (2/4) | Small stature | ||||
| Recurrent facial paralysis (1/4) | |||||
| Hypertension (1/4) | |||||
| Radiological abnormalities | 2/2 | 3/3 | 4/4 | 1/1 | 11/11 |
| Cortical thickening diaphyses | 2/2 | 3/3 | 4/4 | 1/1 | 11/11 |
| Sclerosis of skull | 0/2 | 0/3 | 4/4 | 0/1 | ND |
| Other | Enlarged mandible (1/2) | Kyphoscoliosis (1/3) | Pelvis (3/4) | Genu valgum | ND |
| Coxa valga (1/3) | Spine (1/4) | Pes valgus | |||
| Vertebrae (1/4) | Coxa valga | ||||
| Increased BMD | ND | ND | ND | ND | ND |
| Scintigraphic abnormalities | 2/2 | 1/2 | 4/4 | ND | ND |
| Treatment | Analgesics (2/2) | NSAIDs (2/3) | GC (1/4) | – | ? |
| GC (2/2) |
| Family 16† | Family 17‡ | Family 18‡ | Family 19‡ | Family 20‡ | |
|---|---|---|---|---|---|
| (Japan) | (Portugal) | (France) | (Belgium) | (France) | |
| Mutation (DNA) | 673T→C | 653G→A | 652C→T | 673T→C | 673T→C |
| Mutation (protein) | C225R | R218H | R218C | C225R | C225R |
| Number of patients | 12 | 12 | 2 | 3 | 3 |
| Clinical symptoms | 10/12 | 10/12 | 2/2 | 2/3 | 2/3 |
| Pain in extremities | 10/12 | 10/12 | 2/2 | 2/3 | 2/3 |
| Easy fatigability | ND | 8/12 | 1/2 | 2/3 | 2/3 |
| Muscle weakness | 7/12 | 7/12 | 1/2 | 2/3 | 2/3 |
| Waddling gait | 5/12 | 7/12 | 1/1 | 1/3 | 2/3 |
| Hearing loss | 3/12 | ND | ND | ND | ND |
| Reduced subcutaneous fat | ND | ND | ND | ND | ND |
| Other | Marfanoid habitus (3/12) | Headache (2/12)Poor appetite (2/12) | Poor appetite (1/2) | Headache (3/3)Poor appetite (1/3) | |
| Facial nerve palsy (1/12) | Delayed puberty (?) | ||||
| Delayed puberty (1/1) | |||||
| Radiological abnormalities | 12/12 | 10/10 | ND | 3/3 | 3/3 |
| Cortical thickening diaphyses | 12/12 | 10/10 | ND | 3/3 | 3/3 |
| Sclerosis of skull | 3/12 | 9/11 | ND | ND | 1/2 |
| Other | ND | ND | ND | ND | ND |
| Increased BMD | ND | 8/10 | 2/2 | 3/3 | 3/3 |
| Scintigraphic abnormalities | 1/1 | ND | ND | ND | ND |
| Treatment | ? | GC (1/12) | ? | ? | ? |
| Family 21‡ | Family 22‡ | Family 23‡ | Family 24§ | ||
|---|---|---|---|---|---|
| (Australia) | (France) | (France) | (USA) | Summary | |
| Mutation (DNA) | 673T→C | 652C→T | 652C→T | 653G→A | |
| Mutation (protein) | C225R | R218C | R218C | R218H | |
| Number of patients | 2 | 1 | 2 | 6 | 100 |
| Clinical symptoms | 2/2 | 1/1 | 2/2 | 4/6 | 74/100 (74%) |
| Pain in extremities | 2/2 | 1/1 | 2/2 | 3/6 | 63/92 (68%) |
| Easy fatigability | 2/2 | 1/1 | 1/2 | 3/6 | 32/72 (44%) |
| Muscle weakness | 1/2 | 1/1 | 1/2 | 4/6 | 36/92 (39%) |
| Waddling gait | 1/2 | 1/1 | 1/2 | 4/6 | 44/92 (48%) |
| Hearing loss | ND | ND | ND | 2/6 | 10/67 (15%) |
| Reduced subcutaneous fat | ND | ND | ND | 2/6 | 10/47 (21%)–3/47 (6%) obese |
| Other | Headache (2/2)Poor appetite (2/2) | Poor appetite | Headache (1/2)Poor appetite (1/2) | Vertigo, tinnitus, balance problems (2/6) | |
| Delayed puberty (1/6) | |||||
| Poor appetite (1/6) | |||||
| Facial paralysis (1/6) | |||||
| Radiological abnormalities | 2/2 | 1/1 | 2/2 | 3/4 | 82/87 (94%) |
| Cortical thickening diaphyses | 2/2 | 1/1 | 2/2 | 3/4 | 82/87 (94%) |
| Sclerosis of skull | 2/2 | 1/1 | 2/2 | 3/4 | 32/59 (54%) |
| Other | ND | ND | ND | Enlarged mandible (1/6) | Pelvis 5/8 (63%) |
| Genu valgum (3/6) | |||||
| Pes planus (3/6) | |||||
| Increased BMD | 2/2 | 1/1 | 2/2 | ND | 26/29 (90%) |
| Scintigraphic abnormalities | ND | ND | ND | ND | 17/23 (74%) |
| Treatment | ? | ? | ? | GC (2/6) | |
| Hip surface replacement (1/6) | |||||
| Tibial, fibular, and femoral osteotomy (1/6) |
*This family has been described by Janssens et al.12
†This family has been described by Makita et al.9
‡These families have been described by Campos‐Xavier et al.10
§This family has been described by Wallace et al.11
?, Data not available; –, data absent; ↑, increased; ↓, decreased.
BMD, bone mineral density; BP, bisphophonates; CRP, C reactive protein; ESR, erythrocyte sedimentation rate; GC, glucocorticoids; ND, not determined; NSAIDs, non‐steroidal anti‐inflammatory drugs.

Figure 1 Typical radiographs of CED patients from different families. (A) AP radiographs of both lower legs of a patient from family 14. There is cortical thickening and severe modelling defect at the diaphysis of both tibiae and fibulae. (B) Full leg radiograph (AP view) of a patient from family 2. Note the cortical sclerosis and the modelling defect with symmetrical distribution at the diaphyses of the femora, tibiae, and fibulae, with sparing of the metaphyses and epiphyses. (C) Radiograph of the left distal femur (AP view) of a patient from family 11. Cortical thickening at the diaphysis of the femur—especially at the medial aspect—results in a modelling defect. Note sparing of the metaphysis and epiphysis. (D) Plain radiograph of the right forearm (AP view) from a patient from family 5. Cortical sclerosis and modelling defect can be seen at both radius and ulna, but are most striking at the proximal diaphysis of the ulna. (E) Standard radiograph of the forearm of a patient from family 10. Marked cortical thickening at the diaphysis of the ulna and radius can be observed, causing obliteration of the medullary cavity and hypertrophy of the long bones. Note extension of the cortical sclerosis towards the distal metaphysis of the radius. (F) Radiograph of the right arm (AP view) of a patient from family 14. Thickening of the cortex of the diaphyseal portion of the humerus, ulna, and radius is present, resulting in narrowing of the medullary canal. Note also the modelling defect of the long bones, which is most extensive at the diaphysis of the ulna. (G) Radiograph of the left hand (AP view) of a patient from family 10, showing cortical sclerosis, cortical thickening, and medullary cavity obliteration at the diaphysis of metacarpals 2 and 3. (H) Radiograph of the skull (lateral view) of a patient from family 1. Sclerosis of the calvaria, the tympanic portion of the skull base, and the ascending ramus of the mandible is visible. Note relatively small frontal and sphenoidal sinuses resulting from adjacent sclerosis of the frontal bone and upper part of the face. The maxillary sinuses are spared.
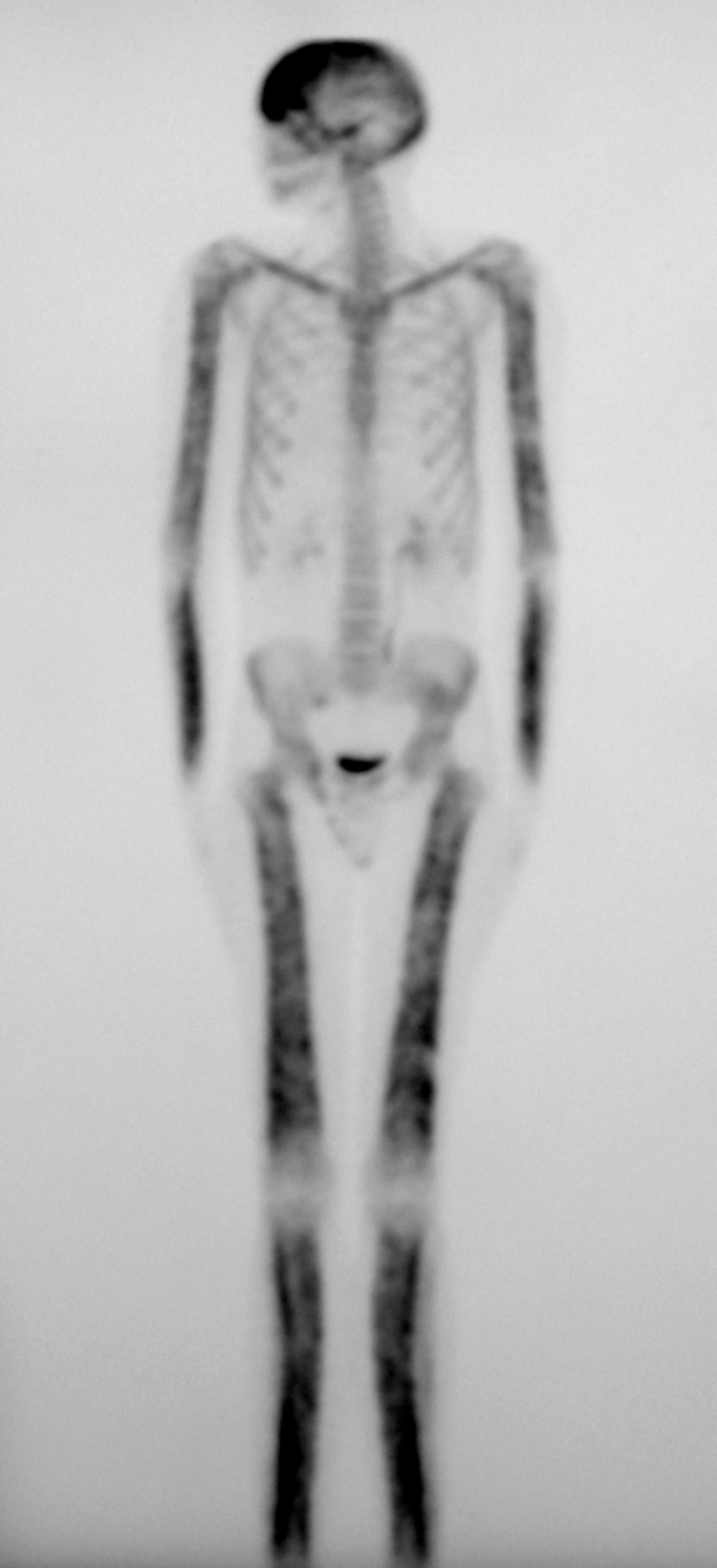
Figure 2 Whole body bone scintigraphy of a patient from family 13 showing the symmetrical distribution of the disease. Increased tracer uptake is visible in the diaphyseal portion of the long bones of the femora, lower legs, humeri and forearms, clavicles, and frontal bones. There is minor increased uptake at the parietal and occipital bones. Also note the slight valgus deformity of the knees.

Figure 3 Axial computed tomography (bone window) of the head of a patient from family 10, showing extensive sclerosis and thickening at the calvaria and petrous bones with loss of the diploe. Note also obliteration of the left frontal sinus.
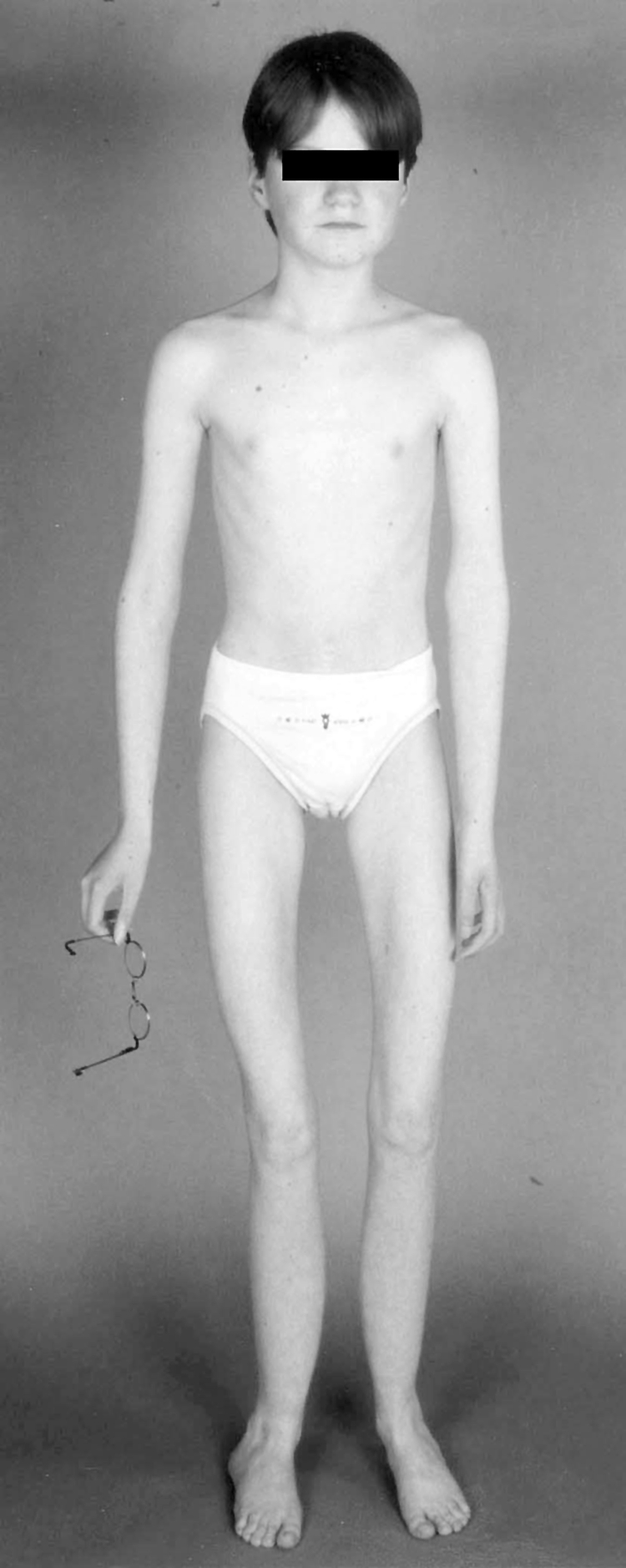
Figure 4 Clinical picture of the patient from family 14 at the age of 15. Note the absence of subcutaneous fat (weight 27 kg), muscle hypotrophy, and valgus deformity of the knees and feet. Muscle weakness restricts her maximum walking distance to 20 to 50 m. Secondary sex characteristics (breast development, menstruation) were delayed. Written permission of the patient for reproduction of this photograph was obtained.
Most of the patients also express clinical symptoms (74%). The most common symptoms are pain in the extremities (68%), a peculiar waddling gait (48%), easy fatigability (44%), and muscle weakness (39%). Reduced subcutaneous fat (21%) and hearing loss (15%) are less common.
The extreme variability in phenotypical expression, both between families sharing the same mutation and among members of the same family, makes it difficult to detect possible genotype–phenotype correlations. Irrespective of the nature of the mutation, the age of onset and disease progression appear highly unpredictable. As previously observed by others,5,19 there seems to be a tendency for an earlier age of onset or a more severe phenotype, or both, in successive generations, a phenomenon known as anticipation. A trend towards increased severity in successive generations was observed in at least seven families. However, in five of these families, diagnosis in the asymptomatic parent was made after giving birth to a severely affected child, creating the appearance of anticipation. Additionally, there was amelioration of disease outcome in successive generations in two families. Furthermore, the nature of the mutations is not in favour of anticipation. The Leu repeat expansion in family 4 forms an exception, but 60 Italian control individuals did not show evidence of instability in this repeat. It seems more plausible that additional genetic factors (for example, single nucleotide polymorphisms (SNPs) in TGFB1 or other genes) modulate the outcome of the principal mutation. A study by Campos‐Xavier et al10 detected no association between the promoter SNPs C‐509T and C‐800T or the coding SNPs T29C and G75C and disease severity in families 17 to 23. Likewise, no association was found between the same four and four additional TGFB1 polymorphisms and disease outcome in family 24.11 These results suggest that genes different from TGFB1 might influence the disease outcome.
Molecular analysis and pathogenesis
Mutation analysis in 46 CED families10,11,13,14,15,16,17,18,20 identified 10 different mutations in the TGFB1 gene in all but one family (table 2; fig 5). The absence of a mutation in a family described by Hecht et al15 raises the possibility of genetic heterogeneity in this disorder. This is further suggested by the absence of mutations in the coding region of TGFB1 in several isolated patients and small families investigated in our laboratory (unpublished data). However, the disease in the latter families might be caused by a mutation in a non‐coding position of TGFB1, affecting, for example, mRNA stability, protein expression level, or transcription factor binding. The possibility that CED is not the underlying disorder in these families should also not be overlooked: in a substantial subset of our patients lacking a TGFB1 mutation, we found indications that the diagnosis was incorrect (either because they had atypical radiological, clinical, or biochemical findings, or because of a different inheritance pattern). Thus far, we have not been able to find convincing evidence for genetic heterogeneity in our set of cases and families.
Table 2 Overview of mutations reported in Camurati‐Engelmann disease families thus far.
| Mutations (DNA): | Exon 1 | Exon 2 | Exon 4 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 28_36dup | 241T→C | 463C→T | 652C→T | 653G→A | 664C→G | ? | 667T→C | 667T→G | 673T→C | |
| Mutations (protein): | L10‐L12dup | Y81H | R156C | R218C | R218H | H222D | C223S | C223R | C223G | C225R |
| Number of families reported | 2 | 2 | 4 | 17 | 10 | 1 | 1 | 1 | 1 | 6 |
| Percentage | 4.4 | 4.4 | 8.9 | 37.8 | 22.2 | 2.2 | 2.2 | 2.2 | 2.2 | 13.3 |
| Percentage per exon | 8.9 | 8.9 | 82.2 | |||||||

Figure 5 Position of Camurati‐Engelmann disease (CED) mutations identified in TGFβ1 at DNA and protein level. Numbering of the mutations starts from the ATG start codon. Numbers indicate the TGFB1 exons. TGF, transforming growth factor.
Transforming growth factor β1 (TGFβ1) is formed as a pro‐precursor molecule, consisting of the signal peptide, the latency associated peptide, and the mature peptide. Post‐translational processing yields the small latent complex, a non‐covalent association between two latency associated peptides and two mature peptides. The majority (7/10) of the mutations detected in CED are missense mutations located in exon 4, coding for the region in the latency associated peptide surrounding the residues responsible for homodimerisation (Cys223 and Cys225)—making up 82.2% of all mutations reported so far. The arginine residue at position 218 is a mutation hotspot, representing 60% of the mutations. Mutations outside exon 4 include a nine base pair duplication in the part of exon 1 encoding the signal peptide, and two missense mutations in exon 1 and exon 2 at the N‐terminus of the latency associated peptide.
Functionally, the CED mutations have been classified into two groups.17 Exon 4 mutations destabilise disulphide bridging of the latency associated peptides, causing premature activation of the mature peptide. Exon 1 mutations rather affect secretion, leading to intracellular retention of the mutant protein. All mutations investigated so far increase TGFβ1 activity.17,21 In CED patients, the narrowing of the medullary cavity at the endosteal side and the modelling defect at the periosteal side of the diaphyses of the long bones suggest that the osteoclastic resorption capacity and the osteoblastic bone formation are both disturbed. This observation is in line with the presumed action of the mutant protein, as TGFβ1 has been shown to stimulate bone formation and suppress bone resorption under physiological conditions.22
Most clinical features of CED—such as bone pain in the limbs, waddling gait, and auditory impairment—are secondary to the hyperostosis and sclerosis of the skeleton. However, the reduction in fat and muscle mass, observed in a significant percentage of the patients (21% and 39%, respectively, in this population), seems to be unrelated to the affection of the skeleton. We sought to clarify these additional symptoms on the basis of the mutations detected. TGFβ1 is a known inhibitor of myogenesis, impairing fusion of myoblasts into multinucleated myotubes.23 Indeed, recent evidence points to a role for the TGFβ pathway in repressing the expression of two important myogenic transcription factors.24 TGFβ1 also inhibits adipogenesis,25 at least partly through the transcriptional repression of genes important in adipocyte differentiation.26 Increased TGFβ1 activity, as seen in CED patients, is therefore expected to inhibit muscle and fat development. It is of note that dexamethasone, a synthetic glucocorticoid and a known stimulator of adipogenesis, was recently shown to reverse TGFβ mediated inhibition of preadipocyte differentiation.27
How can it be explained that activating mutations in a protein like TGFβ1, whose receptors are ubiquitously expressed,28 cause the relatively mild CED phenotype? One possible explanation is the presence of modifier genes that modulate the outcome of the principal mutation (see above). However, in our opinion this is insufficient to account for the absence of symptoms during embryonic development, in which TGFβ1 is shown to play a crucial role,29,30,31 or in tissues like heart, pancreas, kidney, lung, and skin, where TGFβ1 is highly expressed during adult life.32,33 We propose the following hypothesis. TGFβ1 is post‐translationally processed to a non‐covalent small latent complex of the mature peptide and latency associated peptide. In most tissues, this complex covalently associates with a latency associated TGFβ binding protein to form a high molecular weight latent complex (large latent complex) that is directed for storage in the extracellular matrix.34 However, bone forms an exception, as bone cells produce predominantly the small latent complex,35,36,37 a form suggested to represent a pool of readily available TGFβ1—necessary in an environment where TGFβ1 plays such an important role throughout life.37 We raise the possibility that the capacity of a mutation to alter the structure of the latent complex depends on the presence of the latency associated TGFβ binding protein. Following this hypothesis, the conformational changes needed for premature activation of the mature peptide are quenched in the large latent complex. However, when secreted as the small latent complex, the latency associated peptide alters its conformation under the influence of the mutation, releasing some of the mature peptide or at least facilitating its activation. Experiments are under way to confirm or reject this hypothesis.
Treatment
Drug treatment
Glucocorticosteroids are anti‐inflammatory and immunosuppressive agents. In bone, they exert the unfavourable side effect of decreasing bone density, first by increasing the apoptosis rate of osteoblasts and osteocytes while suppressing osteoblast proliferation, differentiation, and bone matrix synthesis38,39; second, by enhancing proliferation and differentiation of osteoclast precursors40,41; and third, by decreasing intestinal calcium absorption.42 In CED patients, this “side effect” is turned into an advantage: glucocorticoids are applied to counteract the increased bone formation. Moreover, they exert a direct effect on TGFβ expression, activation, and signalling, although the exact mechanism needs further clarification. On the one hand, glucocorticoids are seen to stimulate TGFβ expression43 and increase latent TGFβ activation,44 which would imply that they could have adverse effects in CED patients, who show TGFβ1 overactivity. On the other hand, they have been found to induce a shift of TGFβ binding from the signalling‐capable receptor to the non‐signalling receptor,45,46,47 thereby downregulating signalling. Moreover, the glucocorticoid dexamethasone has been shown to interfere more downstream in the signalling pathway, thereby inhibiting TGFβ induced transcription of target genes.48
Several reports have described successful treatment of CED patients with the glucocorticoid prednisolone.49,50,51,52,53 In all cases, there was an improvement in clinical symptoms such as pain and fatigue. Correction of radiographic abnormalities has been documented as well. Of our patient set, 12 patients from nine different families are being treated with prednisolone or related drugs (table 1). For seven of them, information on the effect of treatment was made available. Suffering from lower limb pain, one of the patients from family 1 received high prednisone doses over a two year period. One month treatment courses kept her pain‐free for several months, but pain relapsed in the course of time. One of the affected children from family 2 was treated with prednisone for one year, starting on a dose of 30 mg/day, which was lowered to 10 mg every second day. Weight, appetite, and walking ability increased notably and he complained of skeletal pain much less. Treatment was discontinued because he developed aggressive behaviour which was thought to be related to prednisolone. Treatment of the two clinically affected patients from family 3 resulted in improved mobility and decreased bone pain. However, treatment had to be suspended as the patients became Cushingoid. The symptomatic patient from family 5 benefited from prednisolone treatment as pain and muscle weakness disappeared, while appetite and vigour increased. For the patients from family 11, low glucocorticoid doses helped to suppress pain. Although it is tempting to speculate that glucocorticoids improve bone pain by suppressing bone formation, the improvement in clinical symptoms to treatment can be very rapid and is therefore unlikely to be caused by the suppressive effect on osteoblast function.
Despite these positive reports, long term glucocorticoid treatment is not advisable, as unfavourable side effects can occur. For example long term prednisolone use in children will impair linear growth.54 Furthermore, spinal osteoporosis—as present in two patients from family 3—might be related to long term corticosteroid treatment, as spine bone mineral density (BMD) in two patients from family 4 who were not treated with glucocorticoids was increased. Thus it is important to define the minimum effective dose. A good starting dose is 1 mg/kg/day, but this can and should be lowered during long term treatment. Deflazacort, a derivative of prednisolone, was reported to have a comparable effect in improving clinical and radiological symptoms, but to have fewer adverse effects, and might therefore form a safer alternative.55
The value of bisphosphonates in the treatment of CED is disputed. Besides the five patients described here (table 1), there are very few reports of treatment with these drugs. One report mentions a worsening of bone pain on treatment with pamidronate,51 while in another, a patient profited by treatment with the same drug.56 A patient from family 6 underwent a five week course of treatment with weekly pamidronate infusions without amelioration of her symptoms. Likewise, two patients from family 3 did not benefit from a three month course of intravenous pamidronate treatment. Etidronate treatment in another patient (family 4) even had an adverse effect, as it augmented serum alkaline phosphatase levels above normal. Taking into account that bisphosphonates are widely used as antiresorptive drugs in the treatment of osteoporosis—increasing BMD and lowering fracture risk57—they do not seem likely to have promise for the treatment of CED.
The treatment of one of the patients with calcitonin (family 4) is the first report on the use of this drug in CED therapy. Besides functioning as an analgesic, capable of relieving bone pain,58 calcitonin is also known as a potent inhibitor of bone resorption, hence its application in osteoporosis and Paget's disease.59 Consequently, it is unlikely that this drug will be useful for treating CED, although recent in vivo findings point to an additional role of calcitonin as an inhibitor of bone formation.60 Application in the patient described here was discontinued as no improvement was apparent.
Other drugs used include non‐steroid anti‐inflammatory drugs (NSAIDs) such as aspirin. Although these drugs can alleviate pain, they are not effective at improving bone changes.
Surgery
An alternative to drug treatment is surgery. Bone overgrowth in the diaphyses, with concomitant narrowing of the medullary canal and modelling defects, can be alleviated by reaming of the medullary canal61,62 or osteotomy (this report; families 3 and 24). Orbital decompression to remove bone encroachment on the optic nerves has been used in one case (family 10). However, as the disorder is progressive the symptoms will recur in time.
Gene therapy
In the future, gene therapy might be considered as an additional way to cure CED patients. Based on their capacity for sequestering the mature peptide, decorin, biglycan, α2 macroglobulin, soluble β‐glycan, α2‐HS glycoprotein/fetuin, anti‐TGFβ monoclonal antibodies, or a soluble inactive type II receptor could be considered as possible drugs.63,64,65,66 Alternatively, inhibitors of downstream signalling molecules could be used. In all cases, it should be taken into account that TGFβ1 is implicated in a myriad of functions in the body, increasing the risk of unwanted side effects upon systemic administration of such a “quencher”. Consequently, local application, confined to bone and muscle tissue, would be is preferable.
Diagnosis and genetic counselling
As clinical and radiological variability is extensive, molecular analysis can provide an additional resource for making a correct diagnosis. Family 5 presents a good example of the complementary nature of the clinical, radiological, and molecular findings. In a previous publication on this family, dating from 1994, the mother and maternal grandfather of a severely affected girl had been diagnosed with CED.67 The mother did not show any radiological abnormalities, but scintigraphy demonstrated a focus of increased tracer uptake at the base of the skull. The grandfather was found to have marked fusiform enlargement and cortical thickening along the medial borders of the long bones, despite being symptom‐free. Linkage analysis in the 19q13 region—previously defined as containing the CED gene12—excluded this locus. Although this could point to genetic heterogeneity, mutation analysis of TGFB1 showed a Y81H missense mutation in the girl. The presence of the same mutation in family 6 confirmed this to be the disease causing mutation. The absence of the mutation in the mother of the girl and in other family members at the maternal side suggested that this was a de novo mutation. Alternatively, the mutation could be segregating in the paternal branch of the pedigree. Mutation analysis showed that the latter was the case, as the mutation was detected in the girl's father and paternal grandmother. Radiographic analyses of both individuals showed no signs of the disorder (though scintigraphy was not done). Although radiographs of the maternal grandfather were thought diagnostic of CED, bone scintigrams were considered normal. Unfortunately, no additional data on this individual were made available for further diagnosis. Furthermore, increased tracer uptake at the skull base, as seen in the mother, is a common phenomenon in the normal population and cannot be used as a diagnostic marker of CED. This example shows that a combination of clinical, radiological, scintigraphic, and molecular data may be mandatory for a definitive diagnosis of this disorder.
Interestingly, four of the five patients with radiological non‐penetrance belong to the two families (families 5 and 6) carrying the Y81H mutation. On comparison, it appears that the disease has a significantly lower penetrance in patients with the Y81H variant. Of nine patients with the Y81H mutation, only five (56%) show signs of the disorder. On the other hand, the genotype is penetrant in 77 of 78 patients with a mutation different from the Y81H variant (99%) (p<0.02). Although this could imply that the Y81H variant is not the disease causing mutation, earlier functional experiments provided evidence to the contrary. Overexpression of the mutant protein in a cell culture system showed that the protein is less efficiently secreted than the wild‐type protein, but far more capable of initiating the TGFβ signalling pathway.17
The extreme phenotypic variability of the disorder and occasional lack of penetrance render genetic counselling problematic, in particular dealing with the issue of prenatal diagnosis. A healthy carrier can give birth to a severely affected child. On the other hand, a severe affection status of the parent does not necessarily imply a negative disease course in the child. In the most severely affected patients, a normal way of life becomes difficult, as they are in constant pain and likely to be bedridden. As there is no possibility of predicting the disease outcome, even on the basis of the nature of the mutation, abortion upon prenatal testing can be contemplated. To our knowledge, only one affected parent from our series of patients considered prenatal diagnosis and obtained it following an in depth discussion with a genetic counsellor.
In conclusion, this survey of a large collection of families suffering from this rare bone disorder will aid the definition of the full spectrum and frequency of the various CED phenotypes, and may be of assistance to clinicians for both diagnosis and treatment.
Acknowledgements
This study is supported by the Fund for Scientific Research Flanders with a research project (G.0404.00), by an Interuniversity Attraction Poles programme P5/19 of the Belgian Federal Science Policy Office grant, and by a EU FP6 grant (Anabonos; LSHM‐CT‐2003‐503020) to WVH. KJ is a postdoctoral researcher of the Fund for Scientific Research Flanders.
Abbreviations
BMD - bone mineral density
CED - Camurati‐Engelmann disease
TGF - transforming growth factor
Footnotes
Conflicts of interest: none declared
References
- 1.Cockayne E A. Case for diagnosis. Proc R Soc Med 192013132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Camurati M. Di un raro caso di osteite simmetrica ereditaria degli arti inferiori. Chirurgia degli Organi di Movimento 19226662–665. [Google Scholar]
- 3.Engelmann G. Ein fall von ostheopathia hyperostotica (sclerotisans) multiplex infantilis. Forschritte auf dem Gebiete der Röntgenstrahlen und der Nuklearmedizin 1929391101–1106. [Google Scholar]
- 4.Neuhauser E B, Schwachman H, Wittenborg M, Cohen J. Progressive diaphyseal dysplasia. Radiology 19485111–22. [DOI] [PubMed] [Google Scholar]
- 5.Sparkes R S, Graham C B. Camurati‐Engelmann disease. Genetics and clinical manifestations with a review of the literature. J Med Genet 1972973–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crisp A J, Brenton D P. Engelmann's disease of bone – a systemic disorder? Ann Rheum Dis 198241183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernandez M V, Peris P, Guanabens N, Alvarez L, Monegal A, Pons F, Ponce A, Munoz‐Gomez J. Biochemical markers of bone turnover in Camurati‐Engelmann disease: a report on four cases in one family. Calcif Tissue Int 19976148–51. [DOI] [PubMed] [Google Scholar]
- 8.Smith R, Walton R J, Corner B D, Gordon I R. Clinical and biochemical studies in Engelmann's disease (progressive diaphyseal dysplasia). QJM 197746273–294. [PubMed] [Google Scholar]
- 9.Makita Y, Nishimura G, Ikegawa S, Ishii T, Ito Y, Okuno A. Intrafamilial phenotypic variability in Engelmann disease (ED): are ED and Ribbing disease the same entity? Am J Med Genet 200091153–156. [DOI] [PubMed] [Google Scholar]
- 10.Campos‐Xavier B, Saraiva J M, Savarirayan R, Verloes A, Feingold J, Faivre L, Munnich A, Le Merrer M, Cormier‐Daire V. Phenotypic variability at the TGF‐beta1 locus in Camurati‐Engelmann disease. Hum Genet 2001109653–658. [DOI] [PubMed] [Google Scholar]
- 11.Wallace S E, Lachman R S, Mekikian P B, Bui K K, Wilcox W R. Marked phenotypic variability in progressive diaphyseal dysplasia (Camurati‐Engelmann disease): report of a four‐generation pedigree, identification of a mutation in TGFB1, and review. Am J Med Genet 2004129A235–247. [DOI] [PubMed] [Google Scholar]
- 12.Janssens K, Gershoni‐Baruch R, Van Hul E, Brik R, Guanabens N, Migone N, Verbruggen L A, Ralston S H, Bonduelle M, Van Maldergem L, Vanhoenacker F, Van Hul W. Localisation of the gene causing diaphyseal dysplasia Camurati‐Engelmann to chromosome 19q13. J Med Genet 200037245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janssens K, Gershoni‐Baruch R, Guanabens N, Migone N, Ralston S, Bonduelle M, Lissens W, Van Maldergem L, Vanhoenacker F, Verbruggen L, Van Hul W. Mutations in the gene encoding the latency‐associated peptide of TGF‐beta 1 cause Camurati‐Engelmann disease. Nat Genet 200026273–275. [DOI] [PubMed] [Google Scholar]
- 14.Kinoshita A, Saito T, Tomita H, Makita Y, Yoshida K, Ghadami M, Yamada K, Kondo S, Ikegawa S, Nishimura G, Fukushima Y, Nakagomi T, Saito H, Sugimoto T, Kamegaya M, Hisa K, Murray J C, Taniguchi N, Niikawa N, Yoshiura K. Domain‐specific mutations in TGFB1 result in Camurati‐Engelmann disease. Nat Genet 20002619–20. [DOI] [PubMed] [Google Scholar]
- 15.Hecht J T, Blanton S H, Broussard S, Scott A, Rhoades Hall C, Milunsky J M. Evidence for locus heterogeneity in the Camurati‐Engelmann (DPD1) syndrome. Clin Genet 200159198–200. [DOI] [PubMed] [Google Scholar]
- 16.Mumm S R, Obrecht S, Podgornik M N, Whyte M P. Camurati‐Engelmann Disease: New mutations in the latency‐associated peptide of the transforming growth factor beta‐1 gene. J Bone Miner Res 200116(suppl 1)S223 [Google Scholar]
- 17.Janssens K, ten Dijke P, Ralston S H, Bergmann C, Van Hul W. Transforming growth factor‐beta 1 mutations in Camurati‐Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J Biol Chem 20032787718–7724. [DOI] [PubMed] [Google Scholar]
- 18.Kinoshita A, Fukumaki Y, Shirahama S, Miyahara A, Nishimura G, Haga N, Namba A, Ueda H, Hayashi H, Ikegawa S, Seidel J, Niikawa N, Yoshiura K. TGFB1 mutations in four new families with Camurati‐Engelmann disease: confirmation of independently arising LAP‐domain‐specific mutations. Am J Med Genet 2004127A104–107. [DOI] [PubMed] [Google Scholar]
- 19.Saraiva J M. Progressive diaphyseal dysplasia: a three‐generation family with markedly variable expressivity. Am J Med Genet 199771348–352. [DOI] [PubMed] [Google Scholar]
- 20.Simsek S, Janssens K, Kwee M L, Van Hul W, Veenstra J, Netelenbos J C. Camurati‐Engelmann Disease (progressive diaphyseal dysplasia) in a Moroccan family. Osteoporos Int 2005161167–1170. [DOI] [PubMed] [Google Scholar]
- 21.Saito T, Kinoshita A, Yoshiura K, Makita Y, Wakui K, Honke K, Niikawa N, Taniguchi N. Domain‐specific mutations of a transforming growth factor (TGF)‐beta 1 latency‐associated peptide cause Camurati‐Engelmann disease because of the formation of a constitutively active form of TGF‐beta 1. J Biol Chem 200127611469–11472. [DOI] [PubMed] [Google Scholar]
- 22.Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming growth factor‐beta1 to the bone. Endocr Rev 200526743–774. [DOI] [PubMed] [Google Scholar]
- 23.Massague J, Cheifetz S, Endo T, Nadal‐Ginard B. Type beta transforming growth factor is an inhibitor of myogenic differentiation. Proc Natl Acad Sci USA 1986838206–8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu S, Goldschmidt‐Clermont P J, Dong C. Transforming growth factor‐beta‐induced inhibition of myogenesis is mediated through Smad pathway and is modulated by microtubule dynamic stability. Circ Res 200494617–625. [DOI] [PubMed] [Google Scholar]
- 25.Ignotz R A, Massague J. Type beta transforming growth factor controls the adipogenic differentiation of 3T3 fibroblasts. Proc Natl Acad Sci USA 1985828530–8534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choy L, Derynck R. Transforming growth factor‐beta inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer‐binding protein (C/EBP) and repressing C/EBP transactivation function. J Biol Chem 20032789609–9619. [DOI] [PubMed] [Google Scholar]
- 27.Shin S M, Kim K, Kim J K, Yoon S R, Choi I, Yang Y. Dexamethasone reverses TGF‐beta‐mediated inhibition of primary rat preadipocyte differentiation. FEBS Lett 200354325–30. [DOI] [PubMed] [Google Scholar]
- 28.Roberts A B, Sporn M B. The transforming growth factor‐betas. In: Sporn MB, Roberts AB, eds. Peptide growth factors and their receptors. Heidelberg: Springer‐Verlag, 1990419–472.
- 29.Heine U, Munoz E F, Flanders K C, Ellingsworth L R, Lam H Y, Thompson N L, Roberts A B, Sporn M B. Role of transforming growth factor‐beta in the development of the mouse embryo. J Cell Biol 19871052861–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehnert S A, Akhurst R J. Embryonic expression pattern of TGF beta type‐1 RNA suggests both paracrine and autocrine mechanisms of action. Development 1988104263–273. [DOI] [PubMed] [Google Scholar]
- 31.Pelton R W, Saxena B, Jones M, Moses H L, Gold L I. Immunohistochemical localization of TGF beta 1, TGF beta 2, and TGF beta 3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J Cell Biol 19911151091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller D A, Lee A, Matsui Y, Chen E Y, Moses H L, Derynck R. Complementary DNA cloning of the murine transforming growth factor‐beta 3 (TGF beta 3) precursor and the comparative expression of TGF beta 3 and TGF beta 1 messenger RNA in murine embryos and adult tissues. Mol Endocrinol 198931926–1934. [DOI] [PubMed] [Google Scholar]
- 33.Thompson N L, Flanders K C, Smith J M, Ellingsworth L R, Roberts A B, Sporn M B. Expression of transforming growth factor‐beta 1 in specific cells and tissues of adult and neonatal mice. J Cell Biol 1989108(2)661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oklü R, Hesketh R. The latent transforming growth factor beta binding protein (LTBP) family. Biochem J 2000352 Pt 3601–610. [PMC free article] [PubMed] [Google Scholar]
- 35.Pedrozo H A, Schwartz Z, Mokeyev T, Ornoy A, Xin‐Sheng W, Bonewald L F, Dean D D, Boyan B D. Vitamin D3 metabolites regulate LTBP1 and latent TGF‐beta1 expression and latent TGF‐beta1 incorporation in the extracellular matrix of chondrocytes. J Cell Biochem 199972151–165. [PubMed] [Google Scholar]
- 36.Bonewald L F, Wakefield L, Oreffo R O, Escobedo A, Twardzik D R, Mundy G R. Latent forms of transforming growth factor‐beta (TGF beta) derived from bone cultures: identification of a naturally occurring 100‐kDa complex with similarity to recombinant latent TGF beta. Mol Endocrinol 19915741–751. [DOI] [PubMed] [Google Scholar]
- 37.Dallas S L, Park‐Snyder S, Miyazono K, Twardzik D, Mundy G R, Bonewald L F. Characterization and autoregulation of latent transforming growth factor beta (TGF beta) complexes in osteoblast‐like cell lines. Production of a latent complex lacking the latent TGF beta‐binding protein. J Biol Chem 19942696815–6821. [PubMed] [Google Scholar]
- 38.Cooper M S, Hewison M, Stewart P M. Glucocorticoid activity, inactivity and the osteoblast. J Endocrinol 1999163159–164. [DOI] [PubMed] [Google Scholar]
- 39.Weinstein R S, Jilka R L, Parfitt A M, Manolagas S C. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest 1998102274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takuma A, Kaneda T, Sato T, Ninomiya S, Kumegawa M, Hakeda Y. Dexamethasone enhances osteoclast formation synergistically with transforming growth factor‐beta by stimulating the priming of osteoclast progenitors for differentiation into osteoclasts. J Biol Chem 200327844667–44674. [DOI] [PubMed] [Google Scholar]
- 41.Hirayama T, Sabokbar A, Athanasou N A. Effect of corticosteroids on human osteoclast formation and activity. J Endocrinol 2002175155–163. [DOI] [PubMed] [Google Scholar]
- 42.Gennari C. Differential effect of glucocorticoids on calcium absorption and bone mass. Br J Rheumatol 199332(suppl 2)11–14. [DOI] [PubMed] [Google Scholar]
- 43.Almawi W Y, Abou Jaoude M M, Li X C. Transcriptional and post‐transcriptional mechanisms of glucocorticoid antiproliferative effects. Hematol Oncol 20022017–32. [DOI] [PubMed] [Google Scholar]
- 44.Oursler M J, Riggs B L, Spelsberg T C. Glucocorticoid‐induced activation of latent transforming growth factor‐beta by normal human osteoblast‐like cells. Endocrinology 19931332187–2196. [DOI] [PubMed] [Google Scholar]
- 45.Centrella M, McCarthy T L, Canalis E. Glucocorticoid regulation of transforming growth factor beta 1 activity and binding in osteoblast‐enriched cultures from fetal rat bone. Mol Cell Biol 1991114490–4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang D J, Ji C, Kim K K, Casinghino S, McCarthy T L, Centrella M. Reduction in transforming growth factor beta receptor I expression and transcription factor CBFa1 on bone cells by glucocorticoid. J Biol Chem 19982734892–4896. [DOI] [PubMed] [Google Scholar]
- 47.Nakayama H, Ichikawa F, Andres J L, Massague J, Noda M. Dexamethasone enhancement of betaglycan (TGF‐beta type III receptor) gene expression in osteoblast‐like cells. Exp Cell Res 1994211301–306. [DOI] [PubMed] [Google Scholar]
- 48.Song C Z, Tian X, Gelehrter T D. Glucocorticoid receptor inhibits transforming growth factor‐beta signaling by directly targeting the transcriptional activation function of Smad3. Proc Natl Acad Sci USA 19999611776–11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bourantas K, Tsiara S, Drosos A A. Successful treatment with corticosteroid in a patient with progressive diaphyseal dysplasia. Clin Rheumatol 199514485–486. [DOI] [PubMed] [Google Scholar]
- 50.Heymans O, Gebhart M, Alexiou J, Sokolow Y. Camurati‐Engelmann disease. Effects of corticosteroids. Acta Clin Belg 199853189–192. [PubMed] [Google Scholar]
- 51.Inaoka T, Shuke N, Sato J, Ishikawa Y, Takahashi K, Aburano T, Makita Y. Scintigraphic evaluation of pamidronate and corticosteroid therapy in a patient with progressive diaphyseal dysplasia (Camurati‐Engelmann disease). Clin Nucl Med 200126680–682. [DOI] [PubMed] [Google Scholar]
- 52.Naveh Y, Alon U, Kaftori J K, Berant M. Progressive diaphyseal dysplasia: evaluation of corticosteroid therapy. Pediatrics 198575321–323. [PubMed] [Google Scholar]
- 53.Verbruggen L A, Bossuyt A, Schreuer R, Somers G. Clinical and scintigraphic evaluation of corticosteroid treatment in a case of progressive diaphyseal dysplasia. J Rheumatol 198512809–813. [PubMed] [Google Scholar]
- 54.Emma F, Sesto A, Rizzoni G. Long‐term linear growth of children with severe steroid‐responsive nephrotic syndrome. Pediatr Nephrol 200318783–788. [DOI] [PubMed] [Google Scholar]
- 55.Bas F, Darendeliler F, Petorak I, Sadikoglu B, Bilir A, Bundak R, Saka N, Gunoz H. Deflazacort treatment in progressive diaphyseal dysplasia (Camurati‐Engelmann disease). J Paediatr Child Health 199935401–405. [DOI] [PubMed] [Google Scholar]
- 56.Cherie‐Ligniere G, Santalena G, Parafioriti A. Pamidronate in the treatment of progressive diaphyseal dysplasia (Camurati‐Engelmann disease). Clin Exp Rheumatol 199917264. [PubMed] [Google Scholar]
- 57.Watts N B. Bisphosphonate treatment of osteoporosis. Clin Geriatr Med 200319395–414. [DOI] [PubMed] [Google Scholar]
- 58.Silverman S L, Azria M. The analgesic role of calcitonin following osteoporotic fracture. Osteoporos Int 200213858–867. [DOI] [PubMed] [Google Scholar]
- 59.Zaidi M, Inzerillo A M, Moonga B S, Bevis P J, Huang C L. Forty years of calcitonin – where are we now? A tribute to the work of Iain Macintyre, FRS. Bone 200230655–663. [DOI] [PubMed] [Google Scholar]
- 60.Hoff A O, Catala‐Lehnen P, Thomas P M, Priemel M, Rueger J M, Nasonkin I, Bradley A, Hughes M R, Ordonez N, Cote G J, Amling M, Gagel R F. Increased bone mass is an unexpected phenotype associated with deletion of the calcitonin gene. J Clin Invest 20021101849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lazzarone C, Cartesegna M, Crova M, Calorio D. Progressive diaphyseal dysplasia: Camurati‐Engelmann's disease. Ital J Orthop Traumatol 19839109–114. [PubMed] [Google Scholar]
- 62.Raffaelli P, Ronzini M F. Camurati‐Engelmann's disease. A case report. Ital J Orthop Traumatol 198814267–271. [PubMed] [Google Scholar]
- 63.Yamaguchi Y, Mann D M, Ruoslahti E. Negative regulation of transforming growth factor‐beta by the proteoglycan decorin. Nature 1990346281–284. [DOI] [PubMed] [Google Scholar]
- 64.O'Connor‐McCourt M D, Wakefield L M. Latent transforming growth factor‐beta in serum. A specific complex with alpha 2‐macroglobulin. J Biol Chem 198726214090–14099. [PubMed] [Google Scholar]
- 65.Lopez‐Casillas F, Payne H M, Andres J L, Massague J. Betaglycan can act as a dual modulator of TGF‐beta access to signaling receptors: mapping of ligand binding and GAG attachment sites. J Cell Biol 1994124557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Szweras M, Liu D, Partridge E A, Pawling J, Sukhu B, Clokie C, Jahnen‐Dechent W, Tenenbaum H C, Swallow C J, Grynpas M D, Dennis J W. Alpha 2‐HS glycoprotein/fetuin, a transforming growth factor‐beta/bone morphogenetic protein antagonist, regulates postnatal bone growth and remodeling. J Biol Chem 200227719991–19997. [DOI] [PubMed] [Google Scholar]
- 67.Clybouw C, Desmyttere S, Bonduelle M, Piepsz A. Camurati‐Engelmann disease: contribution of bone scintigraphy to genetic counseling. Genet Couns 19945195–198. [PubMed] [Google Scholar]