

Abstract
Purpose
To identify the gene responsible for autosomal dominant lamellar pulverulent cataract in a four‐generation British family and characterise the functional and cellular consequences of the mutation.
Methods
Linkage analysis was used to identify the disease locus. The GJA8 gene was sequenced directly. Functional behaviour and cellular trafficking of connexins were examined by expression in Xenopus oocytes and HeLa cells.
Results
A 262C>A transition that resulted in the replacement of proline by glutamine (P88Q) in the coding region of connexin50 (Cx50) was identified. hCx50P88Q did not induce intercellular conductance and significantly inhibited gap junctional activity of co‐expressed wild type hCx50 RNA in paired Xenopus oocytes. In transfected cells, immunoreactive hCx50P88Q was confined to the cytoplasm but showed a temperature sensitive localisation at gap junctional plaques.
Conclusions
The pulverulent cataract described in this family is associated with a novel GJA8 mutation and has a different clinical phenotype from previously described GJA8 mutants. The cataract likely results from lack of gap junction function. The lack of function was associated with improper targeting to the plasma membrane, most probably due to protein misfolding.
Keywords: congenital cataract, connexin50, intercellular communication
Cataract is the leading cause of blindness, accounting for almost half of all cases worldwide.1 Congenital cataract alone accounts for 10% of childhood blindness and is the most common treatable cause of childhood visual impairment.2 Inherited cataracts account for a third of all childhood cataracts and are genetically and phenotypically heterogeneous. Inheritance is most commonly autosomal dominant although autosomal recessive and X‐linked forms have also been reported. To date, 27 loci including 13 independent genes have been reported in hereditary isolated congenital cataracts.3 These genes encode transcription factors, crystallins, membrane transport proteins, and cytoskeletal proteins.3
Several mutations in connexin46 and connexin50 (Cx50) have been identified in autosomal dominant isolated congenital cataract.4,5,6,7,8,9,10,11,12,13,14 Here, we report a novel GJA8 mutation, hCx50P88Q, in a family with lamellar pulverulent cataract and describe the consequences of the mutation.
Methods
Patient ascertainment and collection of genetic material
The pedigree was identified through the database of Birmingham Children's Hospital, Birmingham, UK. Ethical approval for this study was obtained from the Moorfields Eye Hospital research ethics committee. Written informed consent was obtained from all individuals and the parents of children under 16 years of age. Full ophthalmic assessment was performed. Genomic DNA was extracted from venous blood leukocytes using the Nucleon II DNA extraction kit (Tepnel Life Sciences, Manchester, UK).
Linkage analysis
All individuals were genotyped with ABI microsatellite markers according to the manufacturer's instructions (Human Linkage Set V2.5; Applied Biosystems, Foster City, CA). Markers (5 and 10 cM) were chosen to flank known congenital cataract genes or loci.
The resulting fluorescently tagged PCR products were analysed on an ABI PRISM 3100 Genetic Analyser (Applied Biosystems). Two point LOD scores were calculated using MLINK (version 5.1) through the Genetic Linkage User Environment (GLUE; http://portal.litbio.org/Registered/Webapp/glue/demo). A fully penetrant autosomal dominant inheritance model was applied with a disease frequency of 6 per 10 000.
Sequencing
The entire coding region of the GJA8 gene (exon 2) was amplified from genomic DNA by PCR using primers 1F: 5′‐ATTCAGATCATGTTGGCACC‐3′, and 1R 5′‐AGAGGTACCCCG CGTTAGC‐3′. PCR products were purified from agarose gels using the QIA‐Quick Gel Extraction Kit (Qiagen, Valencia, CA) and then subcloned into pGEM‐T Easy (Promega, Madison, WI). Plasmid DNA was purified using the GenElute Plasmid Miniprep Kit (Sigma, St Louis, MO). The DNA insert was cycle sequenced with BigDye Terminator Ready Reaction Mix (Applied Biosystems) and analysed on an ABI PRISM 3100 Genetic Analyser (Applied Biosystems).
Restriction analysis
Segregation of the mutation in affected family members was determined by restriction digestion of DNA with BmrI (New England Biolabs, Ipswich, MA). The resulting restriction fragments were separated in 1% agarose gels and stained with ethidium bromide.
Subcloning of wild type and mutant human Cx50 DNA
Wild type (wt hCx50) and mutant (hCx50P88Q) human Cx50 alleles were PCR amplified from genomic DNA of affected individuals using primers (sense: 5′‐ATTGAATTCGCCGCCACCATGGGCGACTGGAGTTTCCTGGGGAACATCTTG‐3′, and anti‐sense: 5′‐GTCGAATTCTCATACGGTTAGATCGTCTGACCTGGCTCGGCTGCT‐3′) and products were sequenced to exclude introduction of random mutations. The PCR products were subcloned into pcDNA3.1/Hygro (+) (Invitrogen Life Technologies, Carlsbad, CA) or the RNA expression vector, SP64TII.15
Cell culture and stable transfection
HeLa cells were grown in DMEM supplemented with non‐essential amino acids, 10% fetal bovine serum (FBS), and 2 mM glutamine. Cell transfections with the pcDNA3.1/Hygro constructs were performed using lipofectin (Invitrogen Life Technologies). Stably transfected clones were selected by their resistance to 150 μg/ml hygromycin. For the temperature‐shift experiments, cells were grown at 37°C until they reached 40–50% confluence. They were then either transferred to 30°C or maintained at 37°C and incubated until they reached 80–90% confluence.
Immunofluorescence
Cells transfected with wt hCx50 or hCx50P88Q were plated on four‐well chamber slides (Lab‐Tek; Nalge Nunc International, Rochester, NY) and allowed to reach 80–90% confluence. Cells were rinsed with phosphate buffered saline (PBS) and fixed for 15 min in 4% paraformaldehyde at room temperature and permeabilised in 1% Triton X‐100 in PBS. Immunofluorescence microscopy was performed using affinity purified rabbit polyclonal anti‐Cx50 antibodies and/or mouse monoclonal anti‐protein disulphide isomerase (PDI; Affinity BioReagents, Golden, CO) or anti‐Golgi 58K protein (Sigma) antibody.16
Immunoblotting
Cells at about 90% confluence were harvested in PBS, 4 mM EDTA, and 2 mM PMSF and centrifuged at 13 800 g for 5 min. Pellets were sonicated in PBS, 4 mM EDTA, and 2 mM PMSF. Aliquots from cell homogenates were resolved on 9% SDS‐containing polyacrylamide gels, transferred to Immobilon P membranes (Millipore, Billerica, MA) and subjected to immunoblotting using anti‐Cx50 antibodies.16
Expression of connexins in Xenopus oocytes
Plasmids were linearised with SalI. Capped RNA was synthesised using the mMessage mMachine SP6 in vitro transcription kit (Ambion, Austin, TX). RNA was quantified by measuring absorbance at 260 nm and purity was further assessed by agarose gel electrophoresis.
Adult female Xenopus laevis were anaesthetised on ice and a partial ovariectomy was performed. The animals were maintained and treated in accordance with National Institutes of Health guidelines. The oocytes were defolliculated and microinjected with 14.5 ng of connexin cRNAs and an oligonucleotide antisense (AS) to endogenous Cx38 as previously described.17
Electrophysiological measurements
Connexin cRNA‐injected oocytes were devitellinised and paired.18 The oocyte pairs were allowed to incubate at room temperature for 4–6 h (or overnight) before electrophysiological recording. Double two‐microelectrode voltage clamp experiments were performed using Geneclamp 500 and an Axoclamp 2A voltage‐clamp amplifier (Axon Instruments, Union City, CA). The microelectrodes were filled with 3 M KCl and had a resistance between 0.1 and 0.6 MΩ. To prevent electrode leakage, the tips of the electrodes were backfilled with 1% agar in 3 M KCl. For measurements of gap junctional coupling, both cell of a pair were held initially at −40 mV. Increments (5 mV to 10 mV) were applied to one cell whilst holding the second cell at −40 mV. The junctional conductance (gj) was calculated as gj = Ij/Vj, where Vj = Vcell2−Vcell1. Pulse generation and data acquisition were performed using a PC computer equipped with pClamp 6 software and a TL‐1 acquisition system (Axon Instruments). Currents were filtered at 20–50 Hz and digitised using pClamp 6 software and a Digidata 1200 (Axon Instruments). All experiments were performed at room temperature (20–22°C).
Results
Linkage analysis and sequencing
A pulverulent cataract of novel phenotype was observed in members of a four‐generation English family. Nineteen members (nine affected, five unaffected, and five married‐in members) (fig 1) underwent a full ophthalmic assessment which revealed bilateral lamellar pulverulent cataract in all affected individuals (fig 2). Hospital records indicated that the cataract was present at birth. Severity was variable; six affected family members required surgery in their first or second decade. No other ocular or systemic defects were noted.

Figure 1 Pedigree and haplotype analysis of the family demonstrating segregation of seven microsatellite markers on chromosome 1q, listed in descending order from the centromere. Black symbols indicate affected individuals and white symbols indicate unaffected individuals. The disease haplotype is indicated as a black bar with alleles.
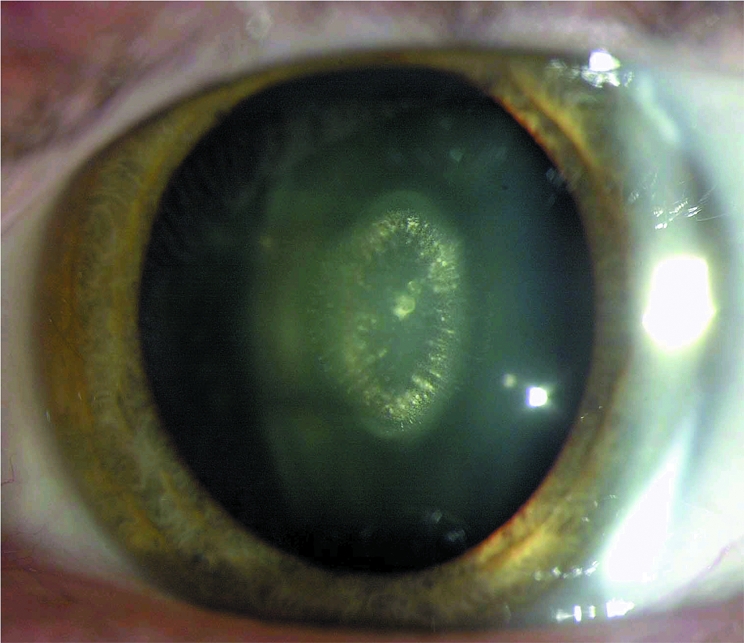
Figure 2 Slit lamp photograph demonstrating pulverulent opacities in the fetal nucleus. The embryonal nucleus is clear.
Linkage analysis was performed using markers from chromosomal regions known to contain relevant cataract candidate genes. Maximum LOD scores of 2.056 and 1.505 without recombination were obtained for markers D1S498 and D1S252, respectively. Linkage analysis was performed for a further five neighbouring markers (table 1). The disease interval contained the candidate gene GJA8.
Table 1 Two point z values for cataract phenotype and chromosome 1 markers.
| Distance (cM) | Marker | LOD score (z) at recombination (θ) of 0 |
|---|---|---|
| 110.4 | D1S2726 | 0.9031 |
| 116.3 | D1S252 | 1.505 |
| 144.1 | CX50 | 1.81 |
| 147.0 | D1S498 | 2.056 |
| 159.4 | D1S2635 | −∞ |
| 156.5 | D1S484 | −∞ |
| 161.0 | D1S2878 | −∞ |
| 164.7 | D1S196 | −∞ |
Subsequent sequence analysis of GJA8 revealed a nucleotide transition, 262C>A, within the coding region (fig 3); this results in the replacement of proline by glutamine (P88Q). This transition introduced a BmrI restriction site. Restriction analysis with BmrI confirmed segregation of the mutation with disease status in the family (fig 4). This sequence change was not found in 156 Caucasian chromosomal controls.

Figure 3 Mutation analysis of the GJA8 gene. Sequence chromatograms of the wild type allele (A) demonstrate a nucleotide sequence encoding a proline (CCG) at codon 88 and of the mutant allele (B) demonstrate a C to A transition resulting in the substitution of Pro by Gln at amino acid residue 88.

Figure 4 Confirmation of segregation of the P88Q mutation by restriction digestion. The mutation results in the gain of a BmrI site (TGACCC) producing digested fragments of 1.3 and 0.3 Kb (not shown) unlike wt Cx50, which remains undigested (1.6 Kb).
Altered trafficking/targeting of hCx50P88Q
To study the cellular behavioural consequences of this mutation, HeLa cells were stably transfected with wt hCx50 or hCx50P88Q. Expression of the protein in stable clones was confirmed by immunoblotting. A band of Mr ∼62 kDa was detected in homogenates of cells transfected with either wt hCx50 or hCx50P88Q; it was absent from homogenates of untransfected cells (fig 5).

Figure 5 Proteins from whole cell homogenates of untransfected HeLa cells or cells transfected with hCx50 or hCx50P88Q were resolved by SDS‐PAGE and subjected to immunoblotting using rabbit polyclonal anti‐human Cx50 antibodies. The positions of the 66, 45, and 30 kDa molecular mass standards are indicated on the right.
The subcellular distribution of Cx50 in stably transfected HeLa cells was assessed by immunofluorescence (fig 6). As expected for a gap junction protein, cells transfected with wt hCx50 demonstrated staining at appositional membranes. Cells transfected with hCx50P88Q showed anti‐Cx50 immunoreactivity in the cytoplasm; its appearance varied from diffuse to particulate and, in some cases, to distinct blobs (fig 5B–D). To determine the localisation of Cx50 within the cytoplasm, double labelling immunofluorescence was performed using antibodies directed against proteins that are resident in the ER (PDI) or Golgi (58K protein) compartments. Human Cx50P88Q showed partial co‐localisation with PDI and Golgi 58K (fig 7).

Figure 6 Photomicrographs showing the distribution of anti‐Cx50 immunoreactivity in HeLa cells transfected with wt hCx50 (A) or hCx50P88Q (B, C, D).

Figure 7 Photomicrographs of double immunofluorescence staining of HeLa cells stably transfected with wt hCx50 (A, C) or hCx50P88Q (B, D) using rabbit polyclonal anti‐Cx50 antibodies and mouse monoclonal antibody against PDI (A, B) or Golgi 58K protein (C, D). Anti‐Cx50 immunoreactivity is shown in red and immunoreactivity from subcellular compartment proteins is shown in green.
Misfolding of membrane proteins and consequent trafficking defects can be overcome by decreasing the temperature at which cells are incubated.19 To determine whether intracellular localisation of hCx50P88Q was due to protein misfolding, cells were incubated at 30°C and the Cx50 distribution was assessed by immunofluorescence. Cells shifted to 30°C showed increased immunoreactivity at appositional membranes compared to cells incubated at 37°C (fig 8).

Figure 8 Confocal images of anti‐Cx50 immunoreactivity from HeLa cells transfected with hCx50P88Q and incubated at 37°C and kept at 37°C (A) or transferred to 30°C (B). There is an increase in immunoreactivity at appositional membranes after incubation at lower temperatures (B, arrows).
Functional characterisation of wild type and mutant hCx50
Oocyte pairs injected with wt hCx50 cRNA developed gap junctional conductances with a mean junctional conductance of 4.02±0.368 μS (table 2). In contrast, pairs of oocytes injected with hCx50P88Q cRNA failed to demonstrate significant gap junctional coupling compared with control oocytes injected only with the Xenopus Cx38 antisense oligonucleotide (table 2).
Table 2 Wild type and mutant Cx50 gap junction coupling.
| Oocyte injection | Gap junction coupling | Duration of pairing prior to testing | ||
|---|---|---|---|---|
| Cell 1 | Cell 2 | Mean conductance (μS) | n | |
| wt hCx50 | wt hCx50 | 4.02±0.368* | 4 | 4–6 h |
| hCx50P88Q | hCx50P88Q | 0.127±0.030* | 6 | 4–6 h |
| wt hCx50:hCx50P88Q (1:1) | wt hCx50:hCx50P88Q (1:1) | 1.37±0.655* | 15 | 4–6 h |
| AS | AS | 0.078±0.18* | 4 | 4–6 h |
| wt hCx50 | wt hCx50 | 35.28±6.64 | 7 | Overnight |
| wt hCx50:hCx50P88Q (1:1) | wt hCx50:Cx50P88Q (1:1) | 8.7±12.23* | 7 | Overnight |
Values are mean±SE; n, number of cell pairs. To facilitate comparison of the homotypic and heteromeric pair data, the amount of wt hCx50 cRNA injected into each cell was always held constant.
*p<0.01 as compared to wt hCx50 by Student's t test.
To determine whether hCx50P88Q exerted a dominant negative effect on wt hCx50 channel activity, gap junction conductance was recorded from oocyte pairs co‐injected with equal amounts of wt hCx50 and hCx50P88Q cRNAs. Oocytes co‐injected with wt hCx50 and hCx50P88Q and paired for 4–6 h showed a 66% reduction in gap junction conductance compared to oocytes injected with wt hCx50 alone. This reduction in gap junction conductance was more pronounced (76%) in oocytes paired overnight (table 2). These results suggest that this mutation acts as a dominant negative inhibitor of wt hCx50.
Discussion
The lamellar pulverulent cataract characterised in this study was associated with a mutation in the GJA8 gene encoding Cx50, a gap junction protein. Gap junctions are membrane specialisations containing clusters of channels that allow intercellular passage of ions and molecules up to 1 kDa.20 The avascular nature of the lens and the intercellular permeability properties of gap junction channels have led to the hypothesis that gap junctions might play an important role in lens cell homeostasis and maintenance of transparency.21 This hypothesis found support when cataracts were observed in mice with targeted ablation of Cx4622 or Cx5023 or in mice carrying mutations in Cx50.24,25 Furthermore, mutations in the genes encoding Cx46 and Cx50 have been reported in autosomal dominant congenital cataract in man.4,5,6,7,8,9,10,11,12,13,14 Each of these mutations is associated with a nuclear pulverulent phenotype. In contrast, hCx50P88Q results in a lamellar pulverulent cataract; the opacity appears confined to the fetal nucleus with the embryonic nucleus remaining clear.
Connexins are integral membrane proteins containing four transmembrane domains, two extracellular loops, and an intracellular loop with both the amino and carboxyl termini located in the cytoplasm. The 262C>A transition results in the substitution of hydrophobic proline for non‐polar glutamine at position 88 in the second transmembrane domain (M2), an α‐helical region on the cytoplasmic face of the protein. Proline residues often introduce kinks, so substitution with another residue can lead to conformational changes. Another cataract associated Cx50 mutant in which this proline is substituted for a polar amino acid, serine (hCx50P88S), has been described.10 Like hCx50P88Q, hCx50P88S is unable to form functional channels and acts as a dominant negative inhibitor of wt hCx50.26 However hCx50P88S is associated with a nuclear pulverulent cataract.10
The proline at position 88 is conserved among all connexins and appears to be important in voltage gating of gap junction channels; substitution of the corresponding proline (P87) in Cx26 prevents the formation of functional homotypic gap junction channels and causes a reversal of voltage gating polarity when paired heterotypically with wild type Cx26.27 Studies of Cx32 site directed mutants suggest that P87 in the M2 α‐helix may act as a hinge for voltage gated opening/closing of the channel.28 Mutations at this residue in Cx32 result in the X‐linked form of Charcot‐Marie Tooth disease, an inherited peripheral neuropathy.29
It is remarkable that this mutation has occurred at a previously reported site.10 We can only speculate about why this may have occurred. Graw suggests that a frequently occurring mutation in CRYGD, Pro23Thr, may be sequence dependent.30 Interestingly, there is a similarity in the wild type sequence of the GJA8 and CRYGD genes at the site of the mutation, CCAACCCC. This wild type sequence changes to CCAACCCA in hCx50P88Q and CCAACCTC in hCx50P88S.10 Unlike the reported CRYGD mutations,30 only one of the GJA8 mutations is a C to A base pair change and it is therefore also unlikely that this mutation is the result of slippage during DNA replication. Furthermore, it is unlikely that this site is a hot spot for mutations as the substituted nucleotide is different for the two mutations. However, there may be a functional constraint where substitutions at this site in the protein are more likely to cause congenital cataract, or this amino acid may be more sensitive to mutation.
In the current study, we have shown that the replacement of a proline with glutamine in Cx50 led to a lack of intercellular communication. This loss of intercellular communication resulted, at least in part, from impaired trafficking of Cx50 to the plasma membrane as hCx50P88Q was not incorporated into gap junction plaques at 37°C. The distinct hCx50P88Q accumulations observed resembled those found in cells transfected with hCx50P88S,16 but the diffuse and rather particulate staining that was spread throughout the cytoplasm was not observed for the hCx50P88S mutant. The temperature‐shift experiments (that is, when cells were incubated at lower temperatures) in which hCx50P88Q could be observed at gap junctional plaques suggest that the impaired trafficking originates from protein misfolding.
We are unable to demonstrate a clear phenotype‐genotype correlation between the cataract phenotype (lamellar v nuclear pulverulent) observed in individuals carrying the hCx50P88Q compared to the hCx50P88S allele. We have however hypothesised several possible reasons as follows. (i) This difference could result from differences in the functional behaviour of the two mutations. Even though both mutants were non‐functional and acted as dominant negative inhibitors of wt hCx50, hCx50P88Q appeared to be a less potent inhibitor than hCx50P88S which causes a complete inhibition of function of wt hCx50 as previously reported.26 Thus, it is possible that the remaining intercellular communication was sufficient to maintain transparency of the embryonic nucleus but not of the fetal nucleus suggesting that the fetal nucleus is more critically dependent on Cx50 based intercellular communication. (ii) The differences in cataract phenotype might also reflect differences in accumulation of the mutant proteins. The hCx50P88Q mutant showed a more variable intracellular distribution than hCx50P88S, and did not always form the blobs consistently seen in cells expressing hCx50P88S. In this scenario, the interactions of hCx50P88Q or hCx50P88S with other lens proteins may differ or the cellular mechanisms for removal of the mutant protein may differ. The lamellar appearance of the hCx50P88Q cataract may reflect a decreased capability of cells in the fetal nucleus to handle the mutant protein. (iii) Epigenetic factors may be involved. The severity of the cataracts observed in Cx46‐null mice31 and Cx50‐null mice32 can be influenced by genetic background.
Conclusion
hCx50P88Q is a novel mutation in the GJA8 gene that was identified in an English family with autosomal dominant lamellar pulverulent congenital cataract. The hCx50P88Q protein does not traffic properly, is not incorporated into gap junctional plaques, and acts as a dominant negative inhibitor of wt hCx50 leading to a significant decrease in gap junction function. Identification and characterisation of this mutation confirms the importance of the proline in the second transmembrane domain in connexin function.
Electronic‐database information
Information on GLUE can be found at http://portal.litbio.org/Registered/Webapp/glue/demo.
Acknowledgments
We would like to thank the family for participating in this study.
Abbreviations
Cx50 - connexin50
PBS - phosphate buffered saline
PDI - protein disulphide isomerase
wt - wild type
Footnotes
Anita Arora is supported by the Wellcome Trust (Grant 068083/Z/02/Z), and Eric Beyer and Lisa Ebihara by the National Institutes of Health (Grants EY08368 and EY10589, respectively).
Competing interests: none declared
Information on GLUE can be found at http://portal.litbio.org/Registered/Webapp/glue/demo.
References
- 1.Resnikoff S, Pascolini D, Etya'ale D, Kocur I, Pararajasegaram R, Pokharel G P, Mariotti S P. Global data on visual impairment in the year 2002. Bull World Health Organ 200482844–851. [PMC free article] [PubMed] [Google Scholar]
- 2.Gilbert C E, Canovas R, Hagan M, Rao S, Foster A. Causes of childhood blindness: results from West Africa, South India and Chile. Eye 19937184–188. [DOI] [PubMed] [Google Scholar]
- 3.Reddy M A, Francis P J, Berry V, Bhattacharya S S, Moore A T. Molecular genetic basis of inherited cataract and associated phenotypes. Surv Ophthalmol 200449300–315. [DOI] [PubMed] [Google Scholar]
- 4.Mackay D, Ionides A, Kibar Z, Rouleau G, Berry V, Moore A, Shiels A, Bhattacharya S. Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet 1999641357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rees M I, Watts P, Fenton I, Clarke A, Snell R G, Owen M J, Gray J. Further evidence of autosomal dominant congenital zonular pulverulent cataracts linked to 13q11 (CZP3) and a novel mutation in connexin 46 (GJA3). Hum Genet 2000106206–209. [DOI] [PubMed] [Google Scholar]
- 6.Jiang H, Jin Y, Bu L, Zhang W, Liu J, Cui B, Kong X, Hu L. A novel mutation in GJA3 (connexin46) for autosomal dominant congenital nuclear pulverulent cataract. Mol Vis 20039579–583. [PubMed] [Google Scholar]
- 7.Bennett T M, Mackay D S, Knopf H L, Shiels A. A novel missense mutation in the gene for gap‐junction protein alpha3 (GJA3) associated with autosomal dominant “nuclear punctuate” cataracts linked to chromosome 13q. Mol Vis 200410376–382. [PubMed] [Google Scholar]
- 8.Li Y, Wang J, Dong B, Man H. A novel connexin46 (GJA3) mutation in autosomal dominant congenital nuclear pulverulent cataract. Mol Vis 200410668–671. [PubMed] [Google Scholar]
- 9.Burdon K P, Wirth M G, Mackey D A, Russell‐Eggitt I M, Craig J E, Elder J E, Dickinson J L, Sale M M. A novel mutation in the Connexin 46 gene causes autosomal dominant congenital cataract with incomplete penetrance. J Med Genet 200441106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shiels A, Mackay D, Ionides A, Berry V, Moore A, Bhattacharya S. A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant “zonular pulverulent” cataract, on chromosome 1q. Am J Hum Genet 199862526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berry V, Mackay D, Khaliq S, Francis P J, Hameed A, Anwar K, Mehdi S Q, Newbold R J, Ionides A, Shiels A, Moore T, Bhattacharya S S. Connexin 50 mutation in a family with congenital “zonular nuclear” pulverulent cataract of Pakistani origin. Hum Genet 1999105168–170. [DOI] [PubMed] [Google Scholar]
- 12.Polyakov A V, Shagina I A, Khlebnikova O V, Evgrafov O V. Mutation in the connexin 50 gene (GJA8) in a Russian family with zonular pulverulent cataract. Clin Genet 200160476–478. [DOI] [PubMed] [Google Scholar]
- 13.Willoughby C E, Arab S, Gandhi R, Zeinali S, Arab S, Luk D, Billingsley G, Munier F L, Heon E. A novel GJA8 mutation in an Iranian family with progressive autosomal dominant congenital nuclear cataract. J Med Genet 200340e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng J Q, Ma Z W, Sun H M. A heterozygous transversion of connexin 50 in a family with congenital nuclear cataract in the northeast of China. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 20052276–78. [PubMed] [Google Scholar]
- 15.Ebihara L, Berthoud V M, Beyer E C. Distinct behavior of connexin 56 and connexin 46 gap junction channels can be predicted from the behavior of their hemi‐gap junction channels. Biophys J 1995681796–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berthoud V M, Minogue P J, Guo J, Williamson E K, Xu X, Ebihara L, Beyer E C. Loss of function and impaired degradation of a cataract‐associated mutant connexin50. Eur J Cell Biol 200382209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tong J J, Liu X, Dong L, Ebihara L. Exchange of gating properties between rat Cx46 and chicken Cx45.6. Biophys J 200487(4)2397–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ebihara L. Expression of gap junction proteins in Xenopus oocyte pairs. In: Rudy B, Iverson LE, eds. Ion channels. San Diego, CA: Academic Press, 1992376–380. [DOI] [PubMed]
- 19.Denning G M, Anderson M P, Amara J F, Marshall J, Smith A E, Welsh M J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature sensitive. Nature 1992358761–764. [DOI] [PubMed] [Google Scholar]
- 20.Sáez J C, Berthoud V M, Branes M C, Martínez A D, Beyer E C. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev 2003831359–1400. [DOI] [PubMed] [Google Scholar]
- 21.Goodenough D A. Lens gap junctions: a structural hypothesis for nonregulated low‐resistance intercellular pathways. Invest Ophthalmol Vis Sci 1979181104–1122. [PubMed] [Google Scholar]
- 22.Gong X, Li E, Klier G, Huang Q, Wu Y, Lei H, Kumar N M, Horwitz J, Gilula N B. Disruption of α3 connexin gene leads to proteolysis and cataractogenesis in mice. Cell 199791833–843. [DOI] [PubMed] [Google Scholar]
- 23.White T W, Goodenough D A, Paul D L. Targeted ablation of connexin50 in mice results in microphthalmia and zonular pulverulent cataracts. J Cell Biol 1998143815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steele E C, Jr, Lyon M F, Favor J, Guillot P V, Boyd Y, Church R L. A mutation in the connexin 50 (Cx50) gene is a candidate for the No2 mouse cataract. Curr Eye Res 199817883–889. [DOI] [PubMed] [Google Scholar]
- 25.Chang B, Wang X, Hawes N L, Ojakian R, Davisson M T, Lo W K, Gong X. A Gja8 (Cx50) point mutation causes an alteration of alpha 3 connexin (Cx46) in semi‐dominant cataracts of Lop10 mice. Hum Mol Genet 200211507–513. [DOI] [PubMed] [Google Scholar]
- 26.Pal J D, Berthoud V M, Beyer E C, Mackay D, Shiels A, Ebihara L. Molecular mechanism underlying a Cx50‐linked congenital cataract. Am J Physiol 1999276C1443–C1446. [DOI] [PubMed] [Google Scholar]
- 27.Suchyna T M, Xu L X, Gao F, Fourtner C R, Nicholson B J. Identification of a proline residue as a transduction element involved in voltage gating of gap junctions. Nature 1993365847–849. [DOI] [PubMed] [Google Scholar]
- 28.Ri Y, Ballesteros J A, Abrams C K, Oh S, Verselis V K, Weinstein H, Bargiello T A. The role of a conserved proline residue in mediating conformational changes associated with voltage gating of Cx32 channels. Biophys J 1999762887–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krutovskikh V, Yamasaki H. Connexin gene mutations in human genetic diseases. Mutat Res 2000462197–207. [DOI] [PubMed] [Google Scholar]
- 30.Graw J. Congenital hereditary cataracts. Int J Dev Biol 2004481031–1044. [DOI] [PubMed] [Google Scholar]
- 31.Gong X, Agopian K, Kumar N M, Gilula N B. Genetic factors influence cataract formation in α3 connexin knockout mice. Dev Genet 19992427–32. [DOI] [PubMed] [Google Scholar]
- 32.Gerido D A, Sellitto C, Li L, White T W. Genetic background influences cataractogenesis, but not lens growth deficiency, in Cx50‐knockout mice. Invest Ophthalmol Vis Sci 2003442669–2674. [DOI] [PubMed] [Google Scholar]