

Abstract
Background
The apolipoprotein E (ApoE) polymorphism has been well studied in the adult human population, in part because the e4 allele is a known risk factor for Alzheimer's disease. Little is known of the distribution of ApoE alleles in newborns, and their association with perinatal brain damage has not been investigated.
Methods
ApoE genotyping was undertaken in a Scottish cohort of perinatal deaths (n = 261), some of whom had prenatal brain damage. The distribution of ApoE alleles in perinatal deaths was compared with that in healthy liveborn infants and in adults in Scotland.
Results
ApoE e2 was over‐represented in 251 perinatal deaths (13% v 8% in healthy newborns, odds ratio (OR) = 1.63, 95% confidence interval (CI) 1.13 to 2.36 and 13% v 8% in adults, OR = 1.67, 95% CI 1.16 to 2.41), both in liveborn and stillborn perinatal deaths. In contrast, the prevalence of ApoE e4 was raised in healthy liveborn infants (19%) compared with stillbirths (13%, OR = 1.59, 95% CI 1.11 to 2.26) and with adults (15%, OR = 1.35, 95% CI 1.04 to 1.76). However, no correlation was found between ApoE genotype and the presence or absence of perinatal brain damage.
Conclusions
This study shows a shift in ApoE allelic distribution in early life compared with adults. The raised prevalence of ApoE e2 associated with perinatal death suggests that this allele is detrimental to pregnancy outcome, whereas ApoE e4 may be less so. However, ApoE genotype did not appear to influence the vulnerability for perinatal hypoxic/ischaemic brain damage, in agreement with findings in adult brains and in animal models.
Keywords: perinatal death, stillbirth, apolipoprotein E, alleles, brain damage
Apolipoprotein E (ApoE) is a 299 amino acid protein primarily concerned with lipoprotein and cholesterol metabolism. The human ApoE gene is located on chromosome 19 and has three allelic forms, e2, e3, and e4, encoding different isoforms of the ApoE protein.1 ApoE is expressed at high levels in brain tissue where it is produced mainly by astrocytes, which package ApoE with cholesterol for presentation to neurones.2,3 ApoE e4 is a well known risk factor for sporadic and familial Alzheimer's disease.4,5 and is also associated with a poorer outcome following head injury.6 On the other hand, ApoE e2 has been linked with other central nervous system (CNS) disorders, including sporadic Parkinson's disease.7
There is growing evidence that ApoE, lipid metabolism and immune responses are related both systemically and in the CNS.8,9 Thus ApoE appears to suppress the production by glia of various pro‐inflammatory cytokines, including tumour necrosis factor‐α, interleukin‐1β, and interleukin‐6.10 Accumulating evidence suggests that ApoE e3 is more effective than e4 in neuronal repair mechanisms and the maintenance of synapses and dendrites.11
Much of what is known of the role of ApoE in the CNS relates to adult life and older age groups. The influence of ApoE alleles on human development has received little attention despite the importance attached to this polymorphism in relation to brain pathology in later life. We have recently established that there is a high prevalence of prenatal brain damage in Scottish perinatal deaths, both in liveborn and stillborn infants.12,13 In the present study, we hypothesised that the absence of one or more ApoE e3 alleles might predispose to brain injury in the perinatal period. It is known from population studies that the distribution of ApoE polymorphisms shows geographic and ethnic variations.14 For this reason we took as our standards for comparison both the data published previously for the adult Scottish population15 and our own study of the ApoE genotype distribution in Scottish healthy newborns.16 We report here the ApoE genotype distribution in a geographically matched cohort of perinatal deaths and examine a possible association between brain damage and particular ApoE alleles.
Materials and methods
In a Scotland wide study undertaken in the late 1990s, all perinatal deaths that were at least 24 weeks gestation at birth and <7 days at the time of death were identified over a 2 year period (n = 745). The initial purpose of the study was to investigate the epidemiology and neuropathological status of these infant and fetal deaths and to identify any factors that might predispose to brain damage, particularly of prenatal origin. Neonates and fetuses with malformations of the CNS (n = 12) and of the heart (n = 28), those with major chromosomal abnormalities (n = 28) or with evidence of CNS infection (n = 0), and extremely macerated stillbirths (n = 118) were all excluded from the study. Consent for the study was withheld in 229 cases and 69 deaths were notified only after the close of the study. After these exclusions, 70 liveborn and 191 stillborn infants qualified for this study. Two pairs of twins were enrolled in this study, one pair liveborn and one stillborn. For the purposes of this study, the ApoE result of the second twin was excluded from analysis. In other multiple pregnancies occurring in the study, only one infant of the siblings had died. No sequential sibling pregnancies were entered in this 2 year study. Therefore, this perinatal ApoE study investigated 261 perinatal deaths, each born within a different family in Scotland during a 2 year period, representing 35% of the total perinatal deaths in that time.
Frozen tissue samples were not available. A PCR method for ApoE genotyping, described by Hixson and Vernier,17 was adapted for analysis of formalin fixed, paraffin embedded tissue. DNA was extracted in each case from two or three 5 μm sections using a commercial kit (Qiagen DNeasy tissue kit, catalogue no 69506; Qiagem). The eluted DNA solution was added to a master mix solution containing customised primers (MWG Biotech; downstream primer: 5′‐ACAGAATTCGCCCCGGCCTGGTACACTGCCA‐3′, upstream primer: 5′‐TCCAAGGAGCTGCAGGCGGCGCA‐3′). The master mix also contained 5 μl of 10× PCR buffer, 8 μl of 5 mmol/l nucleotides, 1.5 μl 50 mmol/l MgCl, 1.25 μl Taq polymerase (Invitrogen), and 20 pmol of each primer. DNA cycling was run on a Techne Flexigene thermocycler at 95°C for 3 minutes followed by 45 cycles at 94°C for 1 minute, 62°C for 1 minute, and 72°C for 2 minutes, with a final extension of 72°C for 8 minutes. The PCR product of 227 bp was visualised with ethidium bromide on a 3% agarose gel. The remaining PCR product was mixed with 1 μl HhaI restriction digest enzyme (New England Biolabs) at 37°C overnight, run on a 4% Metaphor gel, and again visualised with ethidium bromide under ultraviolet light.
The genotype for each sample was determined from the migration pattern of bands in the gel. After digestion, fragments of different sizes allow ApoE genotyping (fig 1). ApoE e2/2 is characterised by the presence of two fragments, 91 bp and 81 bp in size. ApoE e4/4 is characterised by a unique 72 bp fragment, while ApoE e3/3 lacks the 81 bp and 72 bp fragments. Heterozygotes have a combination of different fragments. PCR was repeated for cases in which the band pattern was faint or equivocal until a definite result was obtained. In the small number of cases in which paraffin embedded brain tissue failed to yield a result despite repeated extraction, blocks of spleen sometimes gave a positive result, presumably due to shorter periods of formalin fixation.
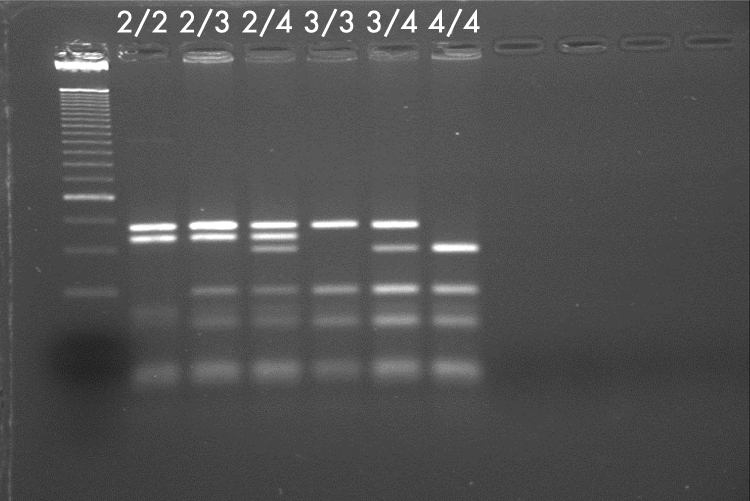
Figure 1 Metaphor gel showing the migration pattern for ApoE genotypes. Band 1 shows the control ladder.
When the ApoE analyses were complete for the whole cohort, the cases were classified according to whether they were liveborn or stillborn and analysed for association with evidence of brain damage. The ApoE allele distribution for these perinatal deaths was compared with published data for Scottish adults15 and healthy liveborn infants.16 The Scottish adults were healthy, late middle aged men and women randomly selected from General Practitioner lists in the mid 1980s. The healthy newborns were infants who were born in a single delivery unit within Edinburgh. Blood was collected randomly from the placentas following delivery and all infants were full term and had a birthweight >2 kg. No infant required medical care and all left hospital when due to do so.
Statistical analysis
Comparisons of ApoE distribution between populations were made using the χ2 test. Odds ratios (OR) and 95% confidence intervals (CI) were calculated for p<0.05. Similarly, χ2 analysis was used to determine differences between perinatal deaths showing neuropathological changes and those without such change.
This study was approved by the Lothian research ethics committee (LREC 1998/6/53). Additional to standard necropsy consent, written parental consent was obtained for extensive detailed neuropathological investigation.
Results
Unequivocal results with clear ApoE banding patterns were obtained for 67 liveborn infants who died at ⩽7 days of age, and 186 stillborn fetuses in this study. Both the stillborn and liveborn groups contained a pair of twins, and all four babies were found to have an ApoE 2/3 genotype. The second twin of each pair was excluded from further analysis. Spleen blocks were used in eight cases, all other results being from brain tissue.
The distribution of ApoE alleles and genotypes for the 251 perinatal deaths included in this study is shown in tables 1–3, which also includes previously published data for 400 Scottish adults15 and for 371 healthy newborn infants.16 Table 3 shows that departure from the Hardy‐Weinberg expectation is not significant for any of the three populations. Tables 4 and 5 compare ApoE results in the present study for infants who were liveborn but who died subsequently, and those for stillbirths.
Table 1 Comparison of adults, healthy newborns, and perinatal deaths for apolipoprotein E genotype: distribution of ApoE alleles .
| Cohort | e2 | e3 | E4 | Total |
|---|---|---|---|---|
| Adults (n = 400)* | 66 (8%) | 616 (77%) | 118 (15%) | 800 |
| HN (n = 371)† | 63 (8%) | 538 (72%) | 141 (19%) | 742 |
| PD(n = 251) | 64 (13%) | 365 (72%) | 73 (15%) | 502 |
Table 2 Comparison of adults, healthy newborns, and perinatal deaths for apolipoprotein E genotype: distribution of ApoE genotypes.
| Cohort | 2/2 | 3/3 | 4/4 | 2/3 | 2/4 | 3/4 |
|---|---|---|---|---|---|---|
| Adults(n = 400)* | 2 (0.5%) | 233 (58.2%) | 4 (1%) | 51 (12.8%) | 11 (2.7%) | 99 (24.8%) |
| HN (n = 371)† | 2 (0.5%) | 199 (53.6%) | 15 (4%) | 44 (11.9%) | 15 (4%) | 96 (25.9%) |
| PD (n = 251) | 5 (2%) | 138 (54.9%) | 3 (1.2%) | 38 (15.1%) | 16 (6.4%) | 51 (20.3%) |
Table 3 Comparison of adults, healthy newborns, and perinatal deaths for apolipoprotein E genotype: distribution n of alleles with Hardy‐Weinberg expectation.
| e3/3 | e3/− | Others | Total | p | |
|---|---|---|---|---|---|
| Adults | |||||
| Observed | 232 | 152 | 16 | 400 | 0.63 |
| Expected | 237 | 142 | 21 | 400 | |
| HN | |||||
| Observed | 199 | 140 | 32 | 371 | 0.58 |
| Expected | 195 | 148 | 28 | 371 | |
| PD | |||||
| Observed | 138 | 89 | 24 | 251 | 0.55 |
| Expected | 133 | 99 | 19 | 251 |
HN, healthy newborns; PD, perinatal deaths.
Table 4 ApoE genotype in perinatal deaths, liveborn versus stillborn: distribution of ApoE alleles.
| Cohort | e2 | e3 | e4 | Total |
|---|---|---|---|---|
| Liveborn PD (n = 66) | 16 (12%) | 91 (69%) | 25 (19%) | 132 |
| Stillbirths (n = 185) | 48 (13%) | 274 (74%) | 48 (13%) | 370 |
PD, perinatal deaths.
Table 5 ApoE genotype in perinatal deaths, liveborn versus stillborn: distribution of ApoE genotypes.
| Cohort | 2/2 | 3/3 | 4/4 | 2/3 | 2/4 | 3/4 |
|---|---|---|---|---|---|---|
| Liveborn PD (n = 66) | 2 (3%) | 28 (42.4%) | 0 (0%) | 11 (16.7%) | 1 (1.5%) | 24 (34.4%) |
| Stillbirths (n = 185) | 3 (1.6%) | 110 (59.5%) | 3 (1.6%) | 27 (14.6%) | 15 (8.1%) | 27 (14.6%) |
PD, perinatal deaths.
We found over‐representation of ApoE e2 in perinatal deaths (13%) compared with healthy newborns (8%, OR = 1.63, 95% CI 1.13 to 2.36) and with adults (8%, OR = 1.67, 95% CI 1.16 to 2.41) (table 1). The prevalence of all genotypes possessing one or more e2 alleles (2/2, 2/3 and 2/4) was higher among perinatal deaths (24%) than among healthy liveborn infants (16%, OR = 1.56, 95% CI 1.43 to 1.68) and adults (16%, OR = 1.64, 95% CI 1.51 to 1.77) (table 2). The higher prevalence of ApoE e2 was found both in liveborn and stillborn perinatal deaths (tables 4 and 5). The prevalence of ApoE e4 was higher in healthy liveborn infants (19%) than in both normal adults (15%, OR = 1.35, 95% CI 1.04 to 1.76) and stillbirths (13%, OR = 1.59, 95% CI 1.11 to 2.26). Review of tables 4 and 5 suggests that liveborn infants who die in the perinatal period have over‐representation of both e2 and e4 alleles compared with normal adults, but the number of cases (n = 66) is too small to place reliance on statistical analysis.
Table 6 shows the variation in allele distribution across gestational groups. Although there appears to be an increase in e2 in extremely preterm gestations, this difference is not statistically significant.
Table 6 Allele distributions by gestational group in all perinatal deaths.
| Total alleles (n = 502) | e2 | e3 | e4 |
|---|---|---|---|
| 24–26 weeks (n = 92) | 17 (18%) | 63 (68%) | 12 (13%) |
| 27–29 weeks (n = 62) | 7 (11%) | 48 (77%) | 7 (11%) |
| 30–32 weeks (n = 56) | 6 (11%) | 37 (66%) | 13 (23%) |
| 33–35 weeks( n = 76) | 12 (16%) | 57 (75%) | 7 (9%) |
| 36or >wks (n = 216) | 22 (10%) | 160 (74%) | 34 (16%) |
| HN (n = 742 alleles) | 63 (8%) | 538 (72%) | 141 (19%) |
HN, healthy newborns.
Tables 7 and 8 show the distribution of ApoE genotype according to the presence or absence of brain damage in this cohort. No correlation was detected between ApoE status and the neuropathological findings in liveborn or stillborn babies. Similarly there was no link between ApoE and the presence of haemorrhage in the brain (table 9).
Table 7 Correlation of ApoE alleles with prenatal brain damage in liveborn babies aged ⩽3 days at death* (n = 57).
| Prenatal brain damage present | No prenatal brain damage* | |
|---|---|---|
| Infants with ApoE 3/3 (25) | 12 (48%) | 13 (52%) |
| Infants with ⩾1 ApoE 2 (12) | 5 (42%) | 7 (58%) |
| Infants with ⩾1 ApoE 4 (19) | 10 (53%) | 9(47%) |
| Infants with ApoE 2/4 (1) | 0 | 1 (100%) |
*The presence of prenatal damage cannot be determined reliably in infants aged more than three days (Becher et al12).
Table 8 Correlation of ApoE alleles with brain damage in stillborn babies (n = 185).
| Established brain damage | Recent brain damage | No brain damage | |
|---|---|---|---|
| Infants with ApoE 3/3 (110) | 42 (38%) | 34 (31%) | 34 (31%) |
| Infants with ⩾1 ApoE 2 (30) | 10 (33%) | 7 (23%) | 13 (43%) |
| Infants with ⩾1 ApoE 4 (30) | 12 (40%) | 11 (37%) | 7 (23%) |
| Infants with ApoE 2/4 (15) | 2 (13%) | 5 (33%) | 8 (53%) |
Table 9 Correlation of ApoE alleles with brain haemorrhage in liveborn babies who died aged ⩽7 days (n = 66), and in stillborn babies (n = 185) .
| Haem. | No haem. | |
|---|---|---|
| Liveborn infants | ||
| Infants with ApoE 3/3 (28) | 14 (50%) | 14 (50%) |
| Infants with ⩾1 ApoE 2 (13) | 7 (54%) | 6 (46%) |
| Infants with ⩾1 ApoE 4 (24) | 11 (46%) | 13 (54%) |
| Infants with ApoE 2/4 (1) | 1 (100%) | 0 |
| Stillbirths | ||
| Infants with ApoE 3/3 (110) | 48 (44%) | 62 (56%) |
| Infants with ⩾1 ApoE 2 (30) | 8 (27%) | 22 (73%) |
| Infants with ⩾1 ApoE 4 (30) | 7 (23%) | 23 (77%) |
| Infants with ApoE 2/4 (15) | 4 (27%) | 11 (73%) |
Taken together, the results of this study suggest that the absence of one or more ApoE e3 predisposes to perinatal death but not to perinatal brain injury.
Discussion
This study reveals significant differences in ApoE allele distribution when comparing perinatal deaths with healthy liveborn infants and with previously published data for the adult population, all in Scotland. Cumming and Robertson15 showed that ApoE e2 was present in 8%, e3 in 77% ,and e4 in 15% of 400 adults aged 45–60 years, chosen randomly from north Scotland. Although this is a historical cohort, the distribution of allele frequencies is similar to that of other white populations in the literature. In the review by Davignon et al14 of all published data, it was found that in 5805 white subjects, the ApoE allele frequency was 8% for e2, 77% for e3, and 15% for e4, which is precisely the same distribution as that reported for the Scottish adult population used in this study. The exceptions in this large survey were adults in France and New Zealand, who have a raised prevalence of e2, at 13% and 12%, respectively, and the Finnish adult population, who have a reduced e2 (4%) and higher e4 (22%) prevalence.14 These three studies comprised 223, 426, and 615 subjects, respectively, and are therefore comparable in size with our study, although the adults were younger than those in the Scottish adult study.
We have shown in 371 healthy newborn infants that the corresponding ApoE distribution was 8% for e2, 72% for e3, and 19% for e4 (p = 0.024 for over‐representation of e4 in healthy newborns compared to healthy adults).16 The present study of 251 perinatal deaths shows that ApoE e2 is significantly over‐represented in both stillborn and liveborn perinatal deaths, compared with the lower prevalence in both healthy newborn and late middle aged adults in Scotland. Infants who are born alive but who die in the first week of life also show over‐representation of e4 and resemble healthy newborns in this respect. While it is possible that these shifts in ApoE distribution have occurred by chance, the numbers in our study are not small. The shift in genotype is also borne out by examination of individual genotypes. However, the limitations in comparing newborn with adult populations should be noted, in that both ApoE e2 and e4 are associated with diseases that have their onset in middle life and carry a significant mortality risk. In addition, data from the French, Finnish, and New Zealand studies quoted above14 indicate that the prevalence of non‐e3 alleles is not entirely consistent in adults.
Support in the literature for a developmental association with different ApoE alleles comes from the small number of studies already available in human infants and pregnancy loss. Zetterberg et al18 found a decreased prevalence of e4 in spontaneous abortions in Crete, 87% of which were earlier than 12 weeks gestation, and proposed that e4 may have a protective effect in pregnancy, which would be consistent with our findings in healthy newborns. The contrast between ApoE e4 prevalence in liveborn and in stillborn infants (e4 being higher in the former group), also suggests that e4 may have a protective effect. Wright et al19 showed that e4 infants have a better mental developmental index at 2 years of age, suggesting that the effects of e4 at an early age may be beneficial rather than detrimental. Similar results have been demonstrated in Brazilian children.20 Other studies suggest that the e4 allele may be associated with improved cognitive performance and higher educational achievement in early adulthood.21,22 In contrast, possession of one or more ApoE e2 alleles appears to be detrimental to pregnancy outcome. Relatively little attention has been paid to this, the rarest ApoE allele, perhaps because it is regarded as the “good” gene with respect to Alzheimer's disease and ischaemic heart disease. ApoE e2 was reported in an early study to be associated with pre‐eclampsia,23 but this was not upheld in subsequent studies.24,25 ApoE e2 is less often transmitted to babies who are small for gestational age26 and perinatally growth restricted babies are more likely to develop cardiovascular disease later in life.27 According to the antagonistic pleiotropic theory of ageing, natural selection favours genes that confer short term benefits at the expense of deterioration in later life. Such opposing effects of the e4 allele may result in beneficial effects in early development but detrimental effects in adulthood.
We found no evidence in this study for an association between ApoE genotype and vulnerability for brain damage or haemorrhage in the perinatal period. The relationship between ApoE genotype and hypoxic/ischaemic injury in the brain is complex, and conflicting results have been reported in the literature. It seems that ApoE e2 is associated with cerebral haemorrhage only in the context of amyloid angiopathy28 while e4 is linked to poorer outcome after haemorrhagic stroke but to better survival following ischaemic damage in the adult.3 However, no effect of ApoE allele variations was noted in neonatal mouse models transgenic for human ApoE e3 and e4 and subjected to ischaemic/hypoxic or excitotoxic insults and neuronal survival appeared to be similar in the two animal groups.29 Our findings for human infants would be in keeping with these animal experiments, and extend the results to the e2 allele.
The role of ApoE in brain tissue is not fully understood, but appears to be neuroprotective and immunosuppressive. Protein expression of ApoE is reported to be increased in neurones following a global ischaemic insult in the human brain, but this effect is not influenced by the host genotype in respect of ApoE e3 or e4.30 ApoE e2 was not included in this study due to the scarcity of this allele. Our findings suggest that ApoE does not display allele specific neuroprotective variations in the perinatal period.
In summary, this study demonstrates that ApoE e2 is linked to a poor gestational outcome, while e4 may confer benefits during pregnancy which make live birth more likely than stillbirth. However, we have found no evidence that ApoE status influences the likelihood of sustaining pregnancy associated brain damage. The absence of one of more ApoE e3 alleles did not predispose to brain injury or to haemorrhage. More thorough investigation of the effects of ApoE on the outcome of pregnancy may require paired parent/fetus analysis,26 as it is possible that fetal wellbeing may vary according to the parental source of the e2 or e4 allele.
Acknowledgements
The authors would like to thank the Chief Scientist's Office of the Scottish Home and Health Department for funding for the study. I Croy and I Anthony gave considerable help with the ApoE methodology, Dr M Lo provided the gel photograph, and Drs R Elton and W Lam advised on the statistical analysis.
Footnotes
Competing interests: J Bell, J Keeling, and N McIntosh are consulted regularly by both defence and prosecution teams with regard to legal proceedings in infant deaths. There are no financial competing interests for any of the authors.
References
- 1.Utermann G, Langenbeck U, Beisiegel U, Weber W. Genetics of the apolipoprotein E system in man. Am J Hum Gen 198032339–347. [PMC free article] [PubMed] [Google Scholar]
- 2.Boyles J K, Pitas R E, Wilson E, Mahley R W, Taylor J M. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest 1985761501–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCarron M O, Muir K W, Weir C J, Dyker A G, Bone I, Nicoll J A, Lees K R. The apolipoprotein E ε4 allele and outcome in cerebrovascular disease. Stroke 1998291882–1887. [DOI] [PubMed] [Google Scholar]
- 4.Saunders A M, Strittmatter W J, Schmechel D, George‐Hyslop P H, Pericak‐Vance M A, Joo S H, Rosi B L, Gusella J F, Crapper‐MacLachlan D R, Alberts M J, Hulette C, Crain B, Goldgaber D, Roses A D. Association of apolipoprotein E allele epsilon 4 with late‐onset familial and sporadic Alzheimer's disease. Neurology 1993431467–1472. [DOI] [PubMed] [Google Scholar]
- 5.Farrer L A, Cupples L A, Haines J L, Hyman B, Kukull W A, Mayeux R, Myers R H, Pericak‐Vance M A, Risch N, van Duijn C M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. ApoE and Alzheimer disease meta analysis consortium. JAMA 19972781349–1356. [PubMed] [Google Scholar]
- 6.Graham D I, Horsburgh K, Nicoll J A, Teasdale G M. Apolipoprotein E and the response of the brain to injury. Acta Neurochir Suppl 19997389–92. [DOI] [PubMed] [Google Scholar]
- 7.The French Parkinson's Disease Genetics Study Group Apolipoprotein E genotype in familial Parkinson's disease. J Neurol Neurosurg Psych 199763394–395. [PMC free article] [PubMed] [Google Scholar]
- 8.Posse de Chaves E I, Rusinol A E, Vance D E, Campenot R B, Vance J E. Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J Biol Chem 199727230766–30773. [DOI] [PubMed] [Google Scholar]
- 9.Mahley R W, Rall S C. Apolipoprotein E: Far more than a lipid transport protein. Annu Rev Genomics Hum Genet 20001507–537. [DOI] [PubMed] [Google Scholar]
- 10.Laskowitz D T, Sheng H, Bart R D, Joyner K A, Roses A D, Warner D S. Apolipoprotein E‐deficient mice have increased susceptibility to focal cerebral ischaemia. J Cereb Blood Flow Metab 199717753–758. [DOI] [PubMed] [Google Scholar]
- 11.Nathan B P, Bellosta S, Sanan D A, Weisgraber K H, Mahley R W, Pitas R E. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science 1994264850–852. [DOI] [PubMed] [Google Scholar]
- 12.Becher J‐C, Bell J E, Keeling J W, McIntosh N, Wyatt B. The Scottish National Perinatal Neuropathology Study. Clinicopathological correlation in early neonatal deaths. Arch Dis Child 200489F399–F407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becher J‐C, Bell J E, Keeling J W, McIntosh N, Wyatt B. The Scottish Perinatal Neuropathology Study – clinico‐pathological correlation in stillbirths. BJOG 2006113310–317. [DOI] [PubMed] [Google Scholar]
- 14.Davignon J, Gregg R E, Sing C F. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis 198881–21. [DOI] [PubMed] [Google Scholar]
- 15.Cumming A M, Robertson F W. Polymorphism at the Apoprotein‐E locus in relation to risk of coronary disease. Clin Genet 198425310–313. [DOI] [PubMed] [Google Scholar]
- 16.Becher J‐C, Bell J E, McIntosh N, Keeling J W. The distribution of apolipoprotein E alleles in a Scottish healthy newborn population. Biol Neonate 200588164–167. [DOI] [PubMed] [Google Scholar]
- 17.Hixson J E, Vernier D T. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 199031545–548. [PubMed] [Google Scholar]
- 18.Zetterberg H, Palmer M, Ricksten A, Poirier J, Palmqvist L, Rymo L, Zafiropoulos A, Arvanitis D A, Spandidos D A, Blennow K. Influence of the apolipoprotein E ε4 allele on human embryonic development. Neurosci Lett 2002324189–192. [DOI] [PubMed] [Google Scholar]
- 19.Wright R O, Hu H, Silverman E K, Tsaih S W, Schwartz J, Bellinger D, Palazuelos E, Weiss S T, Hernandez‐Avila M. Apolipoprotein E genotype predicts 24‐month bayley scales infant development score. Pediatr Res 200354819–825. [DOI] [PubMed] [Google Scholar]
- 20.Oria R B, Patrick P D, Zhang H, Lorntz B, de Castro Costa C M, Brito G A, Barrett L J, Lima A A, Guerrant R L. ApoE4 protects the cognitive development in children with heavy diarrhea burdens in Northeast Brazil. Pediatr Res 200557310–316. [DOI] [PubMed] [Google Scholar]
- 21.Hubacek J A, Pitha J, Skodova Z, Adamkova V, Lanska V, Poledne R. A possible role of apolipoprotein E polymorphism in predisposition to higher education. Neuropsychobiology 200143200–203. [DOI] [PubMed] [Google Scholar]
- 22.Puttonen S, Elovainio M, Kivimaki M, Lehtimaki T, Keltikangas‐Jarvinen L. The combined effects of apolipoprotein E polymorphism and low‐density lipoprotein cholesterol on cognitive performance in young adults. Neuropsychobiology 20034835–40. [DOI] [PubMed] [Google Scholar]
- 23.Nagy B, Rigo J, Fintor L, Karadi I, Toth T. Apolipoprotein E alleles in women with severe pre‐eclampsia. J Clin Pathol 199851324–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagy B, Rigo J, Fintor L, Romics L, Papp Z, Karadi I. Distribution of apolipoprotein(a) isoforms in normotensive a severe preeclamptic women. J Matern Fetal Med 19998270–274. [DOI] [PubMed] [Google Scholar]
- 25.Makkonen N, Heinonen S, Hiltunen M, Helisalmi S, Mannermaa A, Kirkinen P. Apolipoprotein E alleles in women with pre‐eclampsia. J Clin Pathol 200154652–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Infante‐Rivard C, Levy E, Rivard G E, Guiguet M, Feoli‐Fonseca J C. Small babies receive the cardiovascular protective apolipoprotein epsilon 2 allele less frequently than expected. J Med Genet 200340626–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barker D J, Eriksson J G, Forsen T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol 2002311235–1239. [DOI] [PubMed] [Google Scholar]
- 28.Nicoll J A R, Burnett C, Love S, Graham D I, Dewar D, Ironside J W, Stewart J, Vinters H V. High frequency of Apolipoprotein E ε2 allele in haemorrhage due to cerebral amyloid angiopathy. Ann Neurol 199741716–721. [DOI] [PubMed] [Google Scholar]
- 29.Lendon C L, Han B H, Salimi K, Fagan A M, Behrens M I, Muller M C, Holtzman D M. No effect of apoliprotein E on neuronal cell death due to excitotoxic and apoptotic agents in vitro and neonatal hypoxia ischaemia in vivo. Eur J Neurosci 2000122235–2242. [DOI] [PubMed] [Google Scholar]
- 30.Horsburgh K, Graham D I, Stewart J, Nicoll J A R. Influence of Apolipoprotein E genotype on neuronal damage and ApoE immunoreactivity in human hippocampus following global ischaemia. J Neuropath Exp Neurol 199958227–234. [DOI] [PubMed] [Google Scholar]