

Abstract
Background
Heritable alterations in CDKN2A account for a subset of familial melanoma cases although no robust method exists to identify those at risk of being a mutation carrier.
Methods
We set out to construct a model for estimating CDKN2A mutation carrier probability using a cohort of 116 consecutive familial cutaneous melanoma patients evaluated at Massachusetts General Hospital Pigmented Lesion Center between April 2001 and September 2004. Germline CDKN2A and CDK4 status on the familial melanoma cases and clinical features associated with mutational status were then used to build a multiple logistic regression model to predict carrier probability and performance of model on external validation.
Results
From the 116 kindreds prone to melanoma in the Boston area, 13 CDKN2A mutation carriers were identified and 12 were subsequently used in the modeling. Proband age at diagnosis, number of proband primaries, and number of additional family primaries were most closely associated with germline mutations. The estimated probability of the proband being a mutation carrier based on the logistic regression model (MELPREDICT) is given by
where L = 1.99+[0.92×(no. of proband primaries)]+[0.74×(no. of additional family primaries)]−[2.11×ln(age)]. The mean estimated probabilities for subjects in the Boston dataset were 55.4% and 5.1% for the mutation carriers and non‐carriers respectively. In a receiver operator characteristic analysis, the area under the curve was 0.881 (95% confidence interval 0.739 to 1.000) for the Boston model set (n = 116) and 0.803 (0.729 to 0.877) for an external Toronto hereditary melanoma cohort (n = 143).
Conclusions
These results represent the first‐iteration logistic regression model to approximate CDKN2A carrier probability. Validation of this model with an external dataset revealed relatively robust performance.
Keywords: melanoma, genetics, model, carrier, probability
Germline mutations in CDKN2A have been implicated in a significant subset of melanoma prone families and patients with multiple melanomas.1,2,3 The mutation frequency among families with 2 affected members is <5% while the prevalence for kindreds with ⩾3 members range from 20–40%.2 Moreover, the mutation frequency among individuals with multiple primary melanomas is approximately 15%.2,4 Thus, the number of affected family members and the number of cases of melanoma seem to govern carrier probability.
As other factors, such as sun exposure, may modulate the penetrance of mutant alleles, the Melanoma Genetics Consortium (Genomel; http://www.genomel.org) suggests that issues such as indications for and interpretation of genetic results should remain within the research context.5 Although genetic testing for CDKN2A is commercially available but not routinely recommended at the current time, individuals identified to be at significant risk for harbouring germline CDKN2A mutations can be enrolled in ongoing genetic studies with commercial testing used in a confirmatory capacity. Moreover, patients with a low carrier probability can be appropriately counselled against testing as a negative result may not mitigate melanoma risk significantly. For general physicians, dermatologists, and oncologists involved in the care of familial melanoma patients, a simple tool to estimate carrier probability based on clinical parameters would be a useful instrument to direct patients into cancer risk counselling and proper research channels, and potentially away from inappropriate CDKN2A testing. As such, we have devised a logistic regression model, designated MELPREDICT, to estimate CDKN2A carrier probability based on number of primary proband melanomas, number of primary melanomas in the family, and age. We have also tested the performance of MELPREDICT on an independent group of 143 families derived from a melanoma registry in Toronto, Canada.
PATIENTS AND METHODS
Boston patient population
This study was performed in accordance with a protocol approved by our institutional review board. Between April 2001 to September 2004, all patients with invasive or in situ melanoma, who were seen either in initial consultation or in follow up at the Massachusetts General Hospital Pigmented Lesion Center were screened for eligibility based on the following: (a) one or more first degree relatives with melanoma, or (b) two or more affected relatives with melanoma on one side of the family, or (c) three or more primary cutaneous melanomas irrespective of family history. The presence and number of melanomas for probands were confirmed via pathology reports for all but a small number of cases (<10%, data not shown). As per our protocol, we were permitted to pursue medical record confirmation of reported family histories only if probands' relatives provided prior consent to participate in our study.
Toronto patient population
Patients in the Toronto registry were enrolled in accordance with a protocol approved by the institutional review board at the University of Toronto. The 143 probands were consecutive referrals to the familial melanoma clinic specifically for genetic assessment. These referrals were both internal (via the general melanoma clinic and pigmented lesion clinic) and external (from counsellors/dermatologists from other centres in Ontario). The latter group was only seen for genetic consultations or testing then referred back to their primary care physician or dermatologist for subsequent follow up. The probands from Toronto were selected based on the same criteria as those for the Boston families, except that patients were also eligible if they had two or more primary melanomas, or had a personal or family history of pancreatic carcinoma in addition to melanoma.
Mutation analysis
For the Boston cohort, DNA from either peripheral blood leukocytes (PBLs) or PBLs immortalised with Epstein‐Barr virus was extracted with the Qiagen DNEasy kit (Qiagen; Valencia, CA, USA). CDKN2A exons 1α and 2 were amplified and sequenced using published primers and conditions.6 Any mutation detected in immortalised PBLs was subsequently confirmed using frozen blood from the patient. CDKN2A exon 1β was screened for sequence variants using PCR single strand conformation polymorphism (SSCP) analysis as previously described,7 except that this exon was amplified using two overlapping sets of primers (p14ARF‐96F:5′GCTCAGGGAAGGCGGGTGC3′; p14ARF‐320R: 5′AACCCTCACTCGCGGCGG3′; p14ARF‐253F: 5′ACATGGTGCGCAGGTTCTTGGT3′; and p14ARF‐473R: 5′CCGGACTTTTCGAGGGCCTTT3′). Exon 2 of CDK4 was screened for mutations by PCR‐SSCP as previously described.8 The IVS‐105 mutation was screened for by PCR‐PCR using two primers (IV2‐105F: 5′ACCAGGGAGGTGTGGGAGAG3′ and IV2‐105R: 5′TGGTTCTTTCAATCGGGGATG3′).
For the Toronto cohort, all probands were genotyped for CDKN2A exons 1α and 2 using published primers and conditions.9 In addition, an allele specific PCR assay that detects the CDK4 R24C and R24H mutations were also performed on the probands.
Statistical analysis
All analyses were performed using SPSS (version 11.5) and SAS (version 8.2). Variables that were tested for association with the presence of CDKN2A mutations included sex, age at first diagnosis of melanoma in proband (natural logarithm transformed), number of proband melanoma primaries, number of additional family members affected by melanoma, and number of cases of melanoma in the family (other than proband). These last three variables were modelled as ordinal. Univariate associations were tested using Fisher's exact test for the dichotomous variables, and the exact version of the Cochran‐Armitage test for trend for the ordinal variables. Multiple logistic regression analysis was used to estimate adjusted odds ratios, the associated 95% confidence intervals, and p values. A backwards stepwise procedure with a cutoff p value of 0.10 was used to select the final model. The fit of the multiple logistic regression model was evaluated using the Hosmer‐Lemeshow goodness of fit test.
Receiver operator characteristic (ROC) analysis is frequently used to determine the threshold values for a test that provides the best discrimination between “normal” and “abnormal.” We plotted the sensitivity of a particular threshold value for detecting CDKN2A carriers on the y axis and 1 minus the specificity for that threshold value on the x axis. The area under the curve (AUC) for the ROC curves was then calculated as a measure of the overall discrimination that a given test can provide between the individuals with our condition of interest: a CDKN2A gene mutation. In our study, the AUC corresponds to the probability that any given family with a CDKN2A mutation will have a mutation probability greater than any family without a mutation chosen at random. An AUC of 1 represents a perfect test; an area of 0.5 represents a test that discriminates no better than random chance.
RESULTS
Boston cohort
In total, 169 patients enrolled and donated blood for genotyping (fig 1). This represents approximately 10% of all patients seen in the Pigmented Lesion Center over the study period (data not shown). Of the patients who enrolled, seven were excluded because of incorrect medical histories or ineligible pathological diagnoses. The 162 confirmed subjects comprised 128 probands from unique families and 34 relatives of these probands. Of the 128 probands, five had a family history of a non‐cutaneous melanoma (three ocular, one gastric and one vaginal) and six individuals had at least three melanomas but no family history of melanomas. All 162 patients whose pathology reports were confirmed were subsequently subjected to molecular analysis. Six probands had a family history of pancreatic cancer.

Figure 1 Flow chart of study patients ‐ Boston. The grey boxes indicate the cohort that was used to further model MELPREDICT.
Mutation analysis
We analysed the CDKN2A locus for mutations in exons 1α and 2 by direct sequencing, for mutations in exon 1β and CDK4 exon 2 by PCR‐SSCP, and for the intronic IVS2‐105 A/G mutation by allele specific bidirectional PCR. In total, 17 sequence variants were detected in exons 1α and 2 of CDKN2A; no exon 1β or CDK4 exon 2 variants and no IVS2‐105 A/G mutations were detected.
Of the 18 evident variants, 13 are known mutations that have been previously described (fig 2).3 In exon 1α, we found three unrelated probands with 1–8dup8 mutations (9–32dup24 by nucleotide); two of the three probands with 1–8dup8 mutations were of English descent while one individual was of French‐Canadian descent. One of the patients with the 1–8dup8 mutation had subcutaneous metastasis and was thus excluded from the subsequent modelling. There were also two unrelated probands with the Trp15X mutation, of Irish and Scottish backgrounds, respectively.

Figure 2 p16 Mutations detected in the Boston (top) and Toronto (bottom) cohorts. Although 13 mutations were detected in the Boston cohort, the training set used only 12. Each vertical tick represents 10 amino acids along the coding region of p16. Numbers in parentheses reflect number of independent families with the designated mutation.
In exon 2, we detected two unrelated probands with the 240–253del14 deletion and three unrelated probands with the Met53Ile mutation. At position 53, we recently reported a Met53Val missense mutation10 that resulted from an A→G transition at position +157. This p16 INK4A mutation is probably disruptive, as another change at this codon, Met53Ile, is known to be disease associated and does not bind CDK4 normally.4,11 We also identified individuals with the Gly101Trp founder mutation and the Val126Asp mutation. One of the five families with pancreatic cancer had a p16 INK4A mutation (Val126Asp). None of the six individuals with multiple melanomas and without a positive family history had a germline CDKN2A mutation.
Five variants were probably polymorphisms. A G→C transversion at −33 of the 5′UTR was found in one proband and has been previously reported in two French families12 even though it does not appear to be a common polymorphism in the general population.12 Four alanine to threonine alterations (one at codon 60 and three at codon 148) were also detected and probably represent polymorphisms.13
There is some precedence for a direct relationship between the number of affected members in a family and the prevalence of CDKN2A mutations. Individuals with multiple primary melanomas but no family history have been shown to carry CDKN2A mutations at varying rates.14 Some studies also suggest that the family history is extant, but just undisclosed.4 Similarly, there is also some evidence that the rare cutaneous/ocular melanoma families may be due to a locus on chromosome 1p22 distinct from CDKN2A.15 Taken together, we decided to build the first iteration of the regression model using only familial cutaneous melanoma probands.
Associations between CDKN2A mutations and clinical features
For the training set, the mean ages of onset for the 51 men and 65 women were 45.8 and 41.3 years, respectively. There were 386 melanomas in the 116 families (median 3 per kindred; mean 3.3 per kindred).
The mean numbers of primary melanomas reported in the family by CDKN2A carriers and non‐carriers were 7.2 (95% confidence interval (CI) 4.9 to 9.4) and 2.9 (5% CI 2.7 to 3.1), respectively. The mean ages of first diagnosis were 33.2 years (95% CI 25.9 to 40.5) for the CDKN2A carriers and 44.4 years (95% CI 42.0 to 46.9) for non‐carriers. One of the probands developed melanoma at 74 years of age, which was significantly older than any other patient in the training set. Although this single patient was a carrier (1–8dup8), his late diagnosis had a strong destabilising influence on the rest of the model and thus he was dropped from the final analysis.
The results of the univariate and multivariate analyses are given in table 1. In the univariate analysis, ln(age) at proband diagnosis, and higher numbers of proband primaries, other affected family members, and other family primaries were significantly associated with having a CDKN2A mutation. Female sex was non‐significantly associated with mutation status. All variables were tested in an initial multiple logistic regression model because all were significant or close to significance in the univariate analysis. The results are presented in table 1. The number of proband primaries (p = 0.008) and the number of additional family primaries (p = 0.002) remained significant in the multiple logistic regression model while the proband age of diagnosis (ln transformed) was very close to significance (p = 0.059). We chose to log transform age because the effect of increasing age at diagnosis on the probability of having the mutation appeared to lessen with increasing age, and the log transformed covariate resulted in a lower p value than using age directly. The Hosmer‐Lemeshow goodness of fit test indicated an adequate fit for the model (p = 0.138). As presence of ancestry in the British Isles was not available for the validation dataset we did not include this variable in the final multiple logistic regression model. However when we tried it in the model along with the three covariates selected for the final model, it was significant (adjusted odds ratio 207.5 (95% CI 1.05 to 41170.55, p = 0.048).
Table 1 Distribution of p16 mutations in the 101 Boston model set and associations .
| Variable | No. (%) with mutation | Univariate | Multivariate | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | p | Adjusted OR(95% CI) | p* | |||||||
| Sex | ||||||||||
| Male (ref) | 2/51 (3.9%) | 4.46 (0.93 to 21.33) | 0.064† | Dropped | — | |||||
| Female | 10/65 (15.4%) | |||||||||
| Ln(proband age at diagnosis) | — | 0.094 (0.017 to 0.53)§ | 0.008* | 0.12 (0.013 to 1.09)§ | 0.059 | |||||
| British/Irish ancestry | ||||||||||
| Yes | 11/85 (12.9%) | 4.31 (0.53 to 34.91) | 0.18† | Not included | — | |||||
| Unknown | 0/1 (0%) | |||||||||
| No. (ref %) | 1/30 (3.3%) | |||||||||
| No. of proband melanoma cases | ||||||||||
| 1 | 2/93 (2.2%) | 3.03 (1.67 to 5.47) | 0.0002‡ | 2.51 (1.27 to 4.97) | 0.0085 | |||||
| 2 | 4/12 (33.3%) | |||||||||
| ⩾3 | 5/11 (45.5%) | |||||||||
| No. of additional affected family members | ||||||||||
| 1 | 4/75 (5.3%)¶ | 2.82 (1.57 to 5.07) | <0.0001‡ | Dropped | — | |||||
| 2 | 2/24 (8.3%) | |||||||||
| 3 | 2/12 (16.7%) | |||||||||
| ⩾4 | 4/5 (80.0%) | |||||||||
| No. of melanoma cases in other family members | ||||||||||
| 1 | 3/64 (4.7%) | 2.33 (1.56 to 3.50) | <0.0001‡ | 2.09 (1.30 to 3.36) | 0.0023 | |||||
| 2 | 1/25 (4.0%) | |||||||||
| 3 | 1/15 (6.7%) | |||||||||
| ⩾4 | 7/12 (58.3%) | |||||||||
*From multiple logistic regression; †Fisher's exact test; ‡exact version of the Cochran‐Armitage test for trend; §corresponds to a 1 unit increase in ln(age). For reference the range of ln(age) in the data was 2.48 (12 years old) to 4.30 (74 years old). A ln(age) of 3.0 corresponds to a 20 year old, a ln(age) of 3.5 to a 33 year old, and a ln(age) of 4.0 to a 54 year old.
There is also evidence that the risk of pancreatic cancer is elevated among CDKN2A mutation carriers.16,17,18,19,20 In our registry, six probands report a family history of pancreatic cancer, and one of these probands had a CDKN2A mutation. As this frequency is low, an association could not be detected (data not shown).
Carrier probability model:MELPREDICT
The estimated probability of the proband being a mutation carrier based on the logistic regression model is shown by:
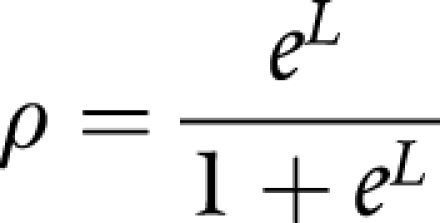 |
where ρ = the probability of being a carrier, L = 1.99+[0.92×(CM)]+0.74×(FM)]−[2.11×ln(age)], PM = number of proband primaries, FM = number of additional family primaries, and ln(age) = natural logarithm of age at diagnosis.
The mean estimated probabilities for subjects in our dataset were 55.4% and 5.1% for the mutation carriers and non‐carriers respectively (p<0.00001). Seven of the 12 mutation carriers had predicted probabilities >83%, while the highest predicted probability for a non‐carrier was 68.9% and the second and third highest were 31.8% and 29.5%. For MELPREDICT, a predicted probability of 50% as a cutoff gives a sensitivity of 54.5% and a specificity of 98.9, a predicted probability of 20% as a cutoff yields a sensitivity of 63.6% and a specificity of 94.4%, and a predicted probability of 10% gives a sensitivity of 81.8% and a specificity of 85.6%. Nine of the 11 carriers had probabilities greater than 10%.
Two mutation carriers had predicted probabilities less than 5%; both individuals (one man, one woman) had late onset melanoma (agd 52 and 60 years) and only one affected first degree relative with a single primary lesion. One of the individuals had the 1–8dup8 mutation, which is one of the few variants that appear to be fully functional in biochemical assays,4 although it is not necessarily “benign” as another 1–8dup8 carrier had a 98.9% probability estimate.
External validation set
We then proceeded to apply MELPREDICT to a cohort of 143 unique familial melanoma probands from the Toronto area. The mutations identified in the Toronto cohort are shown in fig 2. Overall, the prevalence of CDKN2A mutations was 10.3% and 27.2% among the Boston and Toronto families, respectively (table 2; stratified by tumour number). Some substrata of families show high rates of CDKN2A mutations, albeit in the context of only a few families. Prevalence estimates in these larger pedigrees will clearly stabilise as we enrol more families. In families with 3 or more affected members, which is a commonly used cutoff,2 we detected eight mutations out of 41 families (20%). Smaller kindreds with only two primary melanoma cases accounted for 64.7% of the Boston cohort but only 35.0% of the Toronto set.
Table 2 Comparison of Boston model and Toronto test sets.
| No. of melanomas | Boston | Toronto | ||||||
|---|---|---|---|---|---|---|---|---|
| No. of families (%) | No. of p16 mutations (%) | No. of families (%) | No. of p16 mutations (%) | |||||
| 2 | 54(46.6%) | 1 (1.9%) | 50 (35.0%) | 5 (10.0%) | ||||
| 3 | 26 (22.4%) | 0 (0%) | 41 (28.7%) | 10 (24.4%) | ||||
| 4 | 20 (17.2%) | 4 (20.0%) | 22 (15.4%) | 9 (40.9%) | ||||
| >4 | 16 (13.8%) | 7 (43.8%) | 30 (21.0%) | 15 (50.0%) | ||||
| Total | 116 (100%) | 12 (10.3%) | 143 (100%) | 39 (27.2%) | ||||
The mean estimated probabilities for mutation carriers and non‐carriers in the Toronto test set were 20.1% (95% CI 11.6% to 28.6%) and 5.2% (93.7% to 6.8%), respectively. In the Toronto test set, the highest predicted probabilities for a carrier and a non‐carrier were >99.9% and 44.7%, respectively, while the lowest predicted probabilities for carriers and non‐carriers were 0.36% and 1.20%, respectively (both were probands with one primary melanoma and one family member with a single melanoma; the carrier was younger than the non‐carrier).
In order to compare the performance of MELPREDICT on the two datasets independently, we generated ROC curves separately for the two groups (fig 3). In this analysis, we tested whether the predicted probability of mutation has at least one tie between the positive actual state group and the negative actual state group using nonparametric assumptions and a null hypothetical true area of 0.5. Both datasets were significantly different from the null hypothesis. The AUC was 0.881 (95% CI 0.739 to 1.000) for the Boston model set and 0.803 (95% CI 0.728 to 0.877) for the Toronto test set. While the discrimination ability of MELPREDICT for the Toronto data was very good, the predicted probabilities underestimated the actual rate of mutations for this data. Of the 88 subjects with predicted probabilities between 0 and 5%, 13 (14.4%) actually had mutations; of those with probabilities between 5 and 10%, 37.5% had mutations; for those with probabilities between 10 and 20%, the proportion with mutations was 40.0%; for those with predicted probabilities between 20 and 50%, the observed mutation rate was 61.5%; and for those with probabilities over 50%, 100% harboured mutations. This underestimation is due to the fact that a much greater percentage of the Toronto subjects were mutation carriers.

Figure 3 Receiver operator characteristic (ROC) curves for MELPREDICT. Analysis using (A) the internal Boston dataset and (B) the external Toronto data set. AUC, area under the curve.
DISCUSSION
In this study, we (a) screened and enrolled consecutive melanoma patients into a newly established familial melanoma registry embedded within a New England based melanoma clinic, (b) screened and genotyped our probands and participating family members for mutations in CDKN2A and CDK4, (c) performed univariate and multivariate analyses to identify proband features most closely aligned with CDKN2A mutational status, (d) constructed a logistic regression model based on the multivariate analysis, (e) applied our model to an external test set of randomly selected families from a familial melanoma registry in the Toronto area, and (f) compared the performance of our model between the two datasets using ROC analysis.
Data from GenoMEL estimate a CDKN2A prevalence of up to 40% in families with three or more affected individuals.2 However, the prevalence varies geographically. For instance, the prevalence of CDKN2A mutations is approximately 10% in Queensland, Australia21 and 25% in Toronto, Canada.6 Overall, 11% of the Boston familial melanoma cases harboured a CDKN2A mutation; these rates are more akin to the estimates from Australia. In regions with a high population rate of melanoma, the apparent frequency of CDKN2A mutations may be diminished because sporadic melanoma cases could cluster within large families and produce phenocopies that dilute the true hereditary cases. In addition, if family history is assessed in the community and healthcare providers refer individuals to a genetic clinic for counselling and possible testing, the composition of the referred population would be enriched for hereditary melanoma cases. In this situation, the apparent mutation frequency may be higher compared with a clinic population where patients are simply referred for melanoma treatment. Alternatively, modifier genes may be selectively more prevalent in different geographical locations because of the ethnic composition of the area.
The method of mutation detection will determine the sensitivity and specificity and therefore the mutation frequency. We directly sequenced exons 1α and 2 of CDKN2A and screened for the most common IVS‐105 mutation by bidirectional PCR and mutations in CDKN2A exon 1β by PCR‐SSCP analysis. We recognise that our approach may fail to detect other less common alterations such as large deletions, deep intronic mutations, and potential distant mutations that affect transcription of p16 and/or p14ARF. The best strategy for genotyping CDKN2A is the subject of ongoing studies through Genomel.
Deviations from MELPREDICT's estimates include low probability individuals who are carriers (false negatives) and high probability probands who do not harbour germline mutations (false positives). With false negatives, modifier genes may attenuate the action of the specific mutant allele or environmental influences may modulate the expression of the phenotype. In this regard, the geographic variation described for CDKN2A penetrance3 most likely reflects a combination of these two inputs. With false positives, the most likely explanation is that mutations at other loci, such as the melanoma susceptibility locus on chromosome 1p22,15 may be responsible for the family history. Alternatively, undetected intronic or distant promoter CDKN2A mutations may play a role.
Not unexpectedly, the performance of our model in a ROC analysis was superior with the internal Boston set compared with the external Toronto test set (AUC 0.881; 95% CI 0.739 to 1.00 for Boston versus 0.803; 95% CI 0.728 to 0.877 for Toronto). As other predictive models for hereditary melanoma do not currently exist, we cannot compare this performance with the performance of other extant models. However, one recent study applied the BRCAPRO model to a set of 272 breast cancer families at eight cancer genetics clinics22 and reported a median AUC of 0.712 (range 0.709 to 0.720 at the eight centres),22 which is actually slightly less than the AUC for our external test set. Based on the ROC analysis, we found the performance of our model highly encouraging as it appears to be in line with other more mature cancer probability models, such as BRCAPRO.
At the current time, Genomel does not endorse CDKN2A genetic testing for clinical use, citing variance in its penetrance estimates and also lack of clinical utility.2,5 However, it does support the use of clinical testing in order to confirm research results. In addition, it is likely that patients are or will become aware of the commercial availability of CDKN2A testing and will thus query healthcare providers regarding its use. In the past, cancer predisposition testing had been recommended for individuals whose pretest mutation probability exceeded 10%.22,23 With MELPREDICT, a probability of 10% as a cutoff gave a sensitivity of 83.3% and a specificity of 85.6%. Thus, MELPREDICT will assign a probability of less than 10% for some mutation harbouring families. Thus, calculating the probability of a germline CDKN2A mutation should not be viewed as the primary method for selecting patients to undergo genetic testing. Rather, MELPREDICT is only a tool for the cancer risk counsellor to quantify carrier probability. Moreover, these estimates may also change. As personal and/or family histories evolve, the probability calculation may increase because family members often enter surveillance after a diagnosis in the family and additional melanomas become detected. In addition, as individuals are followed over time, new melanomas can develop. Our model provides information that can help counsellors educate patients regarding the usefulness of testing and stratify individuals into categories of hereditary risk for potential research applications.
CONCLUSION
We have constructed a logistic regression model to estimate CDKN2A carrier probability and have documented relatively robust performance when validated on an external cohort of hereditary melanoma kindreds. We are encouraged by this initial iteration of MELPREDICT and its potential usefulness in the field of melanoma genetics. Having a logistic regression model to quantitatively estimate carrier probability will encourage research by providing investigators with a common risk assessment tool and empower healthcare providers with more accurate counselling information. Moreover, cancer risk counsellors can use these concrete estimates of carrier probability in order to discourage patients who have an exaggerated perception of risk away from uninformed genetic testing.
ACKNOWLEDGEMENTS
H Tsao was partly supported by grants from the Dermatology Foundation, the National Institutes of Health and the American Skin Association. D Hogg was supported by a grant from the National Cancer Institute of Canada, and by the Michael Young Melanoma Foundation through the Ontario Cancer Research Network.
Abbreviations
AUC - area under the curve
PBL - peripheral blood leukocyte
ROC - receiver operating characteristic
SSCP - single strand conformation polymorphism
Footnotes
Competing interests: there are no competing interests
References
- 1.Hussussian C J, Struewing J P, Goldstein A M, Higgins P A, Ally D S, Sheahan M D, Clark W H, Jr, Tucker M A, Dracopoli N C. Germline p16 mutations in familial melanoma. Nat Genet 1994815–21. [DOI] [PubMed] [Google Scholar]
- 2.Kefford R F, Newton Bishop J A, Bergman W, Tucker M A. Counseling and DNA testing for individuals perceived to be genetically predisposed to melanoma: A consensus statement of the Melanoma Genetics Consortium. J Clin Oncol 1999173245–3251. [DOI] [PubMed] [Google Scholar]
- 3.Bishop D T, Demenais F, Goldstein A M, Bergman W, Bishop J N, Bressac‐de Paillerets B, Chompret A, Ghiorzo P, Gruis N, Hansson J, Harland M, Hayward N, Holland E A, Mann G J, Mantelli M, Nancarrow D, Platz A, Tucker MA; Melanoma Genetics Consortium Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst 200294894–903. [DOI] [PubMed] [Google Scholar]
- 4.Monzon J, Liu L, Brill H, Goldstein A M, Tucker M A, From L, McLaughlin J, Hogg D, Lassam N J. CDKN2A mutations in multiple primary melanomas. N Engl J Med 1998338879–887. [DOI] [PubMed] [Google Scholar]
- 5.Kefford R, Bishop J N, Tucker M, Bressac‐de Paillerets B, Bianchi‐Scarra G, Bergman W, Goldstein A, Puig S, Mackie R, Elder D, Hansson J, Hayward N, Hogg D, Olsson H; Melanoma Genetics Consortium Genetic testing for melanoma. Lancet Oncol 20023653–654. [DOI] [PubMed] [Google Scholar]
- 6.Hogg D, Liu L, Lassam N. Genetic testing in familial melanoma. In: Nickoloff BJ, ed. Melanoma: methods and protocols. Totowa, NJ: Humana Press Inc, 2001;61, [DOI] [PubMed]
- 7.Tsao H, Zhang X, Kwitkiwski K, Finkelstein D M, Sober A J, Haluska F G. Low prevalence of germline CDKN2A and CDK4 mutations in patients with early‐onset melanoma. Arch Dermatol 20001361118–1122. [DOI] [PubMed] [Google Scholar]
- 8.Tsao H, Benoit E, Sober A J, Thiele C, Haluska F G. Novel mutations in the p16/CDKN2A binding region of the cyclin‐dependent kinase‐4 gene. Cancer Res 199858109–113. [PubMed] [Google Scholar]
- 9.Lynch H T, Brand R E, Hogg D, Deters C A, Fusaro R M, Lynch J F, Liu L, Knezetic J, Lassam N J, Goggins M, Kern S. Phenotypic variation in eight extended CDKN2A germline mutation familial atypical multiple mole melanoma‐pancreatic carcinoma‐prone families: the familial atypical mole melanoma‐pancreatic carcinoma syndrome. Cancer 20029484–96. [DOI] [PubMed] [Google Scholar]
- 10.Yang G, Niendorf K B, Tsao H. A novel methionine‐53‐valine mutation of p16 in a hereditary melanoma kindred. J Invest Dermatol 2004123574–575. [DOI] [PubMed] [Google Scholar]
- 11.Pollock P M, Spurr N, Bishop T, Newton‐Bishop J, Gruis N, van der Velden P A, Goldstein A M, Tucker M A, Foulkes W D, Barnhill R, Haber D, Fountain J, Hayward N K. Haplotype analysis of two recurrent CDKN2A mutations in 10 melanoma families: evidence for common founders and independent mutations. Hum Mutat 199811424–431. [DOI] [PubMed] [Google Scholar]
- 12.Soufir N, Avril M F, Chompret A, Demenais F, Bombled J, Spatz A, Stoppa‐Lyonnet D, Benard J, Bressac‐de Paillerets B. Prevalence of p16 and CDK4 germline mutations in 48 melanoma‐prone families in France. Hum Mol Genet 19987209–216. [DOI] [PubMed] [Google Scholar]
- 13.Cairns P, Mao L, Merlo A, Lee D J, Schwab D, Eby Y, Tokino K, van der Riet P, Blaugrund J E, Sidransky D. Rates of p16 (MTS1) mutations in primary tumors with 9p loss. Science 1994265415–417. [DOI] [PubMed] [Google Scholar]
- 14.Hayward N K. Genetics of melanoma predisposition. Oncogene 2003223053–3062. [DOI] [PubMed] [Google Scholar]
- 15.Gillanders E, Juo S H, Holland E A, Jones M, Nancarrow D, Freas‐Lutz D, Sood R, Park N, Faruque M, Markey C, Kefford R F, Palmer J, Bergman W, Bishop D T, Tucker M A, Bressac‐de Paillerets B, Hansson J, Stark M, Gruis N, Bishop J N, Goldstein A M, Bailey‐Wilson J E, Mann G J, Hayward N, Trent J; Lund Melanoma Study Group; Melanoma Genetics Consortium Localization of a novel melanoma susceptibility locus to 1p22. Am J Hum Genet 200373301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartsch D K, Sina‐Frey M, Lang S, Wild A, Gerdes B, Barth P, Kress R, Grutzmann R, Colombo‐Benkmann M, Ziegler A, Hahn S A, Rothmund M, Rieder H. CDKN2A germline mutations in familial pancreatic cancer. Ann Surg 2002236730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lynch H T, Shaw T G, Lynch J F. Inherited predisposition to cancer: A historical overview. Am J Med Genet 2004129C5–22. [DOI] [PubMed] [Google Scholar]
- 18.Goldstein A M, Struewing J P, Fraser M C, Smith M W, Tucker M A. Prospective risk of cancer in CDKN2A germline mutation carriers. J Med Genet 200441421–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldstein A M. Familial melanoma, pancreatic cancer and germline CDKN2A mutations. Hum Mutat 200423630. [DOI] [PubMed] [Google Scholar]
- 20.Goldstein A M, Fraser M C, Struewing J P, Hussussian C J, Ranade K, Zametkin D P, Fontaine L S, Organic S M, Dracopoli N C, Clark W H, Jr, Tucker M A. Increased risk of pancreatic cancer in melanoma‐prone kindreds with p16INK4 mutations. N Engl J Med 1995333970–974. [DOI] [PubMed] [Google Scholar]
- 21.Aitken J, Welch J, Duffy D, Milligan A, Green A, Martin N, Hayward N. CDKN2A variants in a population‐based sample of Queensland families with melanoma. J Nat Cancer Inst 199991446–452. [DOI] [PubMed] [Google Scholar]
- 22.Euhus D M, Smith K C, Robinson L, Stucky A, Olopade O I, Cummings S, Garber J E, Chittenden A, Mills G B, Rieger P, Esserman L, Crawford B, Hughes K S, Roche C A, Ganz P A, Seldon J, Fabian C J, Klemp J, Tomlinson G. Pretest prediction of BRCA1 or BRCA2 mutation by risk counselors and the computer model BRCAPRO. J Natl Cancer Inst 200294844–851. [DOI] [PubMed] [Google Scholar]
- 23.American Society of Clinical Oncology Statement of the American Society of Clinical Oncology: genetic testing for cancer susceptibility. Adopted on February 20, 1996. J Clin Oncol 996141730–6discussion 173740. [DOI] [PubMed] [Google Scholar]