

Abstract
Background
Golabi, Ito, and Hall reported a family with X linked mental retardation (XLMR), microcephaly, postnatal growth deficiency, and other anomalies, including atrial septal defect, in 1984.
Methods
This family was restudied as part of our ongoing study of XLMR, but significant linkage to X chromosome markers could not be found. Extreme short stature and microcephaly as well as other new clinical findings were observed. Mutations in the polyglutamine tract binding protein 1 gene (PQBP1) have recently been reported in four XLMR disorders (Renpenning, Hamel cerebro‐palato‐cardiac, Sutherland‐Haan, and Porteous syndromes) as well as in several other families. The clinical similarity of our family to these patients with mutations in PQBP1, particularly the presence of microcephaly, short stature, and atrial septal defect, prompted examination of this gene.
Results
A missense mutation in PQBP1 was identified which changed the conserved tyrosine residue in the WW domain at position 65 to a cysteine (p.Y65C).
Conclusions
This is the first missense mutation identified in PQBP1 and the first mutation in the WW domain of the gene. The WW domain has been shown to play an important role in the regulation of transcription by interacting with the PPxY motif found in transcription factors. The p.Y65C mutation may affect the proper functioning of the PQBP1 protein as a transcriptional co‐activator.
Keywords: Golabi‐Ito‐Hall syndrome, microcephaly, PQBP1 , Renpenning syndrome, X linked mental retardation
Knowledge of X chromosome gene mutations in families with X linked mental retardation (XLMR) continues to increase at a remarkable rate. The Greenwood‐Miami XLMR database now contains over 200 entries.1 Currently, 57 genes have been identified which encompass 77 syndromes and at least 37 non‐syndromic XLMR (MRX) families. Mutations in 36 of the 57 known genes result only in a syndromic phenotype, and mutations in 13 result only in a non‐syndromal phenotype. Both phenotypes have been associated with mutations in eight genes. In 53 other disorders and 50 MRX families, regional localisation of the gene loci has been accomplished, but the responsible genes have not been identified. Only 37 recognised XLMR syndromes still await localisation or identification of a mutation.1,2,3 The present report documents one more instance in the process of gene identification among the named XLMR syndromes with the finding that a missense mutation in the polyglutamine tract binding protein 1 gene (PQBP1) (p.Y65C) segregates with the clinical disorder known as Golabi‐Ito‐Hall syndrome. This syndrome was reported in 19844 and restudied as part of the Greenwood‐Miami XLMR project. The missense mutation in the Golabi‐Ito‐Hall syndrome is unique among the PQBP1 mutations reported to date.
Methods
Clinical and laboratory studies
Patients 1–3 and their pedigree numbers are the same in the present paper and in the report by Golabi, Ito, and Hall.4 Patient 1 was examined in both studies; patient 2 was examined only in the first study, but records from both the facility where he was institutionalised and from autopsy were available for the present study; patient 3 was seen only for the present study. The findings reported by Golabi, Ito, and Hall4 were specifically checked in the present study. Most significant findings were consistent except for the ectodermal abnormalities originally observed in patient 1 (dry, brittle, sparse hair and slight nail hypoplasia). His family reported that these were transient manifestations and they were not observed in either affected male examined in the present study. It is assumed they were incidental findings. A few other reported minor facial variations were also not observed and are not included in the summary of clinical data. Intelligence was normal in carrier females. The present studies were reviewed and approved by Human Subjects Review Committees at Self Regional Healthcare and the University of Miami.
Mutation screening
Mutation analysis of the PQBP1 gene was done by incorporation PCR SSCP (IPS) and by denaturing high performance liquid chromatography (DHPLC). Exons 1, 2, 3, 5, and 6 were screened by IPS and exon 4 by DHPLC.
IPS and sequence analysis
A 100 ng sample of genomic DNA was amplified in a total volume of 10 μl containing 1× GoTaq buffer (Promega, Madison, WI), 1 μM of each primer, 50 μM of DNTPs, and 0.05 μCi of α32P dCTP (3000 Ci/mmol; PerkinElmer Life and Analytical Sciences, Wellesley, MA).5 GoTaq DNA polymerase (1 U; Promega) and 0.2 μg of TaqStart antibody (Clontech, Palo Alto, CA) were incubated at 22°C for 5 min before being added to the PCR reaction mixture. Amplification was carried out in a PTC‐200 thermocycler (MJ Research, Waltham, MA) under the following conditions: 95°C for 5 min; 30 cycles of 95°C for 30s, 65°C for 30 s, and 72°C for 30s; 7 min extension at 72°C. Following PCR, 8 μl of the IPS loading dye (95% formamide, 10 mM NaOH, 0.25% bromophenol blue, 0.025% xylene cyanol) was added. The samples were denatured at 96°C for 5 min and resolved on a 0.5× MDE gel (FMC, Philadelphia, PA) prepared in 0.6× TBE. The gel was run at 8 W for 16 h at room temperature. After the run the gel was dried and the radioactive signal was visualised using BIOMAX MS films (Eastman Kodak, Rochester, NY).
DNA fragments exhibiting an abnormal pattern on the IPS gel were sequenced in both directions on the MegaBACE (Amersham Biosciences, Piscataway, NJ) using the DYEnamic dye terminator kit (Amersham Biosciences) according to the manufacturer's protocol. Alignment and analysis of the sequence was done by the Lasergene program (DNASTAR, Madison, WI).
Results
Clinical studies
A partial pedigree of the K8275 family is given in fig 1. The complete five generation pedigree includes 55 individuals, seven of whom were males affected with XLMR. Several died of accidental deaths at a relatively early age; only patient 3 is currently alive. Growth and clinical findings in three affected males are summarised in tables 1 and 2. The term “spastic diplegia” is used to indicate a predominance of spasticity in the lower limbs, but is often accompanied by variable spasticity in the upper limbs.

Figure 1 Partial pedigree of the K8275 family and segregation of the A>G mutation. A 2% agarose gel electrophoresis of the BsrI restriction endonuclease digestion of amplified DNA fragments from members of the Golabi‐Ito‐Hall family showing the segregation of the c.194A>G alteration. The numbers below the females indicate X inactivation status. The normal males have bands of 86 and 186 bp, whereas the affected males have a single 272 bp band. A 19 bp band which is present in all individuals has run off the gel.
Table 1 Growth in Golabi‐Ito‐Hall syndrome.
| Patient | IV‐2 | III‐7 | IV‐4 | ||||
|---|---|---|---|---|---|---|---|
| Age (years) | Birth | 1.25 | 14 | Birth | 16 | 42* | 21 |
| Head circumference, cm (centile) | – | 42 (<<3rd) | 48 (<<3rd) | – | 51 (<3rd) | – | 54 (5th) |
| Height, cm (centile) | 48.5 (20) | 70 (<3rd) | 129.5 (<<3rd) | – | 145 (<<3rd) | 157 (<3rd) | 144 (<<3rd) |
| Weight, kg (centile) | 2.8 (10) | 7.6 (<<3rd) | 21 (<<3rd) | 2.7 (10th) | 35 (<3rd) | 64 (25th) | – |
*At autopsy.
Table 2 Summary of clinical findings in three males with Golabi‐Ito‐Hall syndrome.
| Clinical findings | Frequency in patients with truncating mutations* | |
|---|---|---|
| Postnatal growth deficiency | 3/3 | |
| Lean body build | 3/3 | 85% |
| Face | ||
| Elongation with triangular lower face | 3/3 | 55% |
| Asymmetry | 2/3 | |
| Upslanting palpebral fissures | 3/3 | 39% |
| Cupped/prominent ears | 3/3 | 50% |
| Large/prominent tongue | 2/2 | |
| Micrognathia | 1/3 | |
| Atrial septal defect | 2/3 | 12.5% |
| Contractures and spastic diplegia | 3/3 |
*Percentages based on summary of Stevenson et al.11
Patient IV‐2
An atrial septal defect had been diagnosed at birth. The patient was originally examined at 16 months. Examination at 14 years of age revealed an extremely small, thin male with minimal body fat who stood with his legs flexed. He had no speech, but understood a few simple commands. Positive findings included slightly upslanting palpebral fissures (fig 2A), narrow face and triangular lower face, slightly large mouth, a grade IV/VI systolic murmur, limited movement in the arms, hyperactive deep tendon reflexes in the legs, and positive Babinski signs. Scoliosis, kyphosis, and the dry, brittle hair and nail hypoplasia described in the original report were not observed.
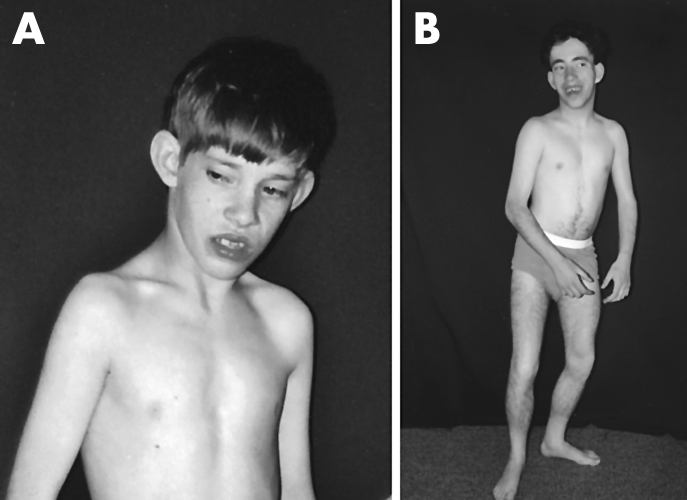
Figure 2 (A) Patient IV‐2 at 14 years of age. (B) Patient IV‐4 at 21 years of age. The lean habitus and cupped ears are evident in both, as is the crouched stance, which was due to a combination of spasticity and contractures. Both are markedly below the 3rd centile in height. (Photographs are published with consent.)
At 21 years of age, because of a tremor, an MRI was obtained, which was normal. Currently 26 years of age, the patient is in an assisted living home and has not developed any speech or additional neurological problems. Chromosome studies were normal.
Patient III‐7
The original report of this patient at 28 years of age noted growth deficiency, severe mental retardation (estimated IQ 23), mild sensorineural hearing loss, spastic diplegia, possible congenital heart diseases, hypospadias with normal testes, and chronic haematuria. Subsequently, biopsy proven chronic glomerulonephritis was documented. He was institutionalised nearly all of his life and was not examined by the current investigators. He had a long history of regurgitation and aspiration and died following aspiration at 42 years of age. Medical records documented short stature, severe acne, a very large tongue, no murmur, hyperreflexia, and flexion contractures of the right leg. Autopsy findings included aspiration pneumonia and minimal musculature in the legs; no cardiac defect was found. The gyral pattern and gross external structure of the brain were normal.
Patient IV‐4
The history of this patient was notable for a repaired atrial septal defect and severe mental retardation. At 15 years of age, a CT scan was performed because of seizures. This showed bilateral calcifications of the globus pallidus and periventricular frontal white matter. The cause of the calcifications was not clear. At 18 years of age, other radiological findings included mild rotatory scoliosis, mildly thickened ribs with short clavicles, and cardiomegaly. Normal laboratory findings included EEG, urinary mucopolysaccharide excretion, plasma amino acid, and urinary organic acid screens.
At 21 years of age, he was said to have a few words and no history of hearing defect. On examination, he was extremely small overall, had flexed arms, and stood with flexed legs (fig 2B). His elbows were contracted by 15°, but his flexed knees could be passively straightened. The head was small and the face thin with a triangular lower face and upslanting palpebral fissures. There was extensive dark hair on his legs and genital region as well as similar patches elsewhere. Brittle hair and nail hypoplasia were not noted. Testicular size (2.5×3.5 cm) was normal. No intelligible speech was heard and he did not follow simple commands.
He died at 25 years of age in a swimming pool accident.
Molecular studies
The entire PQBP1 coding region (exons 1–6) was screened for mutations in the proband by a combination of IPS6 and DHPLC.7 Screening of exon 3 by IPS resulted in an abnormal pattern (data not shown). Sequencing of exon 3 from the patient revealed that he was carrying an A to G substitution at position 194 (c.194A>G) (data not shown). This alteration changed the conserved tyrosine residue in the WW domain at position 65 to a cysteine (p.Y65C). The A>G alteration eliminated a BsrI site, making it possible to confirm co‐segregation of the c.194A>G alteration with the mental retardation in the Golabi‐Ito‐Hall family (fig 1). The segregation studies also revealed that individual IV‐5 was a carrier of the mutation (fig 1). Furthermore, the c.194A>G mutation was not found in 334 X chromosomes from normal males. X inactivation studies using the AR gene polymorphic repeated sequence (ACG) showed that the four carrier females were randomly or moderately skewed (fig 1).
Discussion
Mutations in PQBP1 have recently been reported in four other named XLMR disorders (Renpenning, Hamel cerebro‐palato‐cardiac, Sutherland‐Haan, and Porteous syndromes) and in several small families with similar clinical findings.7,8,9,10 These disorders are characterised by microcephaly, a long narrow face, and a lean body build with short stature. Spastic diplegia is frequent.
Use of the Greenwood‐Miami XLMR database1 was instrumental in the search for the gene causing Golabi‐Ito‐Hall syndrome, as the syndrome had not been regionally mapped on the X chromosome. The XLMR database lists 55 entries with short stature and 26 with microcephaly. Twenty five have both microcephaly and short stature. Congenital heart lesions, however, are uncommon in XLMR disorders. Among the 200 listings in our XLMR database1 only eight include any type of congenital heart disease and five of these are not further described or defined. Three have atrial or ventricular septal defects (PQBP1 related disorders, BCOR (oculo‐facial cardio‐dental syndrome) related disorders, and MIDAS (microphthalmia with linear skin defect) syndrome). Since the latter two syndromes have ocular and other distinctive clinical manifestations, PQBP1 was given priority as a candidate gene for Golabi‐Ito‐Hall syndrome.
A review of the clinical and molecular findings illustrates the differences as well as the similarities in the PQBP1 related disorders.8,9,10,11 Two clinical differences between Golabi‐Ito‐Hall syndrome and other PQPB1 related disorders are evident. The growth of affected males with Golabi‐Ito‐Hall syndrome appears to be more severely restricted than that of other males with PQBP1 mutations; head circumference and length are most notably restricted. As radiological and autopsy studies did not document major CNS malformations, it is likely that the microcephaly was a manifestation of the overall growth deficiency.
The second difference of interest is the influence on organogenesis. Atrial septal defects were present in two of the three affected males in the Golabi‐Ito‐Hall syndrome family. Among subjects with PQBP1 mutations, 4/4 of patients with Hamel cerebro‐palato‐cardiac syndrome12 and 1/4 in a recently reported family with Renpenning syndrome11 had cardiac malformations. One carrier mother had an atrial septal defect.10 The mechanism by which mutations in PQBP1 predispose to congenital cardiac anomalies is not known at present. In addition, small testes, which have reported in the other PQBP1 related syndromes, were not observed in the present family, another difference that may have a molecular basis.8,9,10,11
Six of the seven known mutations in PQBP1 result in a truncated protein. These affect a cell's pre‐RNA splicing mechanism and disrupt the nuclear localisation signal.8,9,10 The missense mutation in Golabi‐Ito‐Hall syndrome is, therefore, of considerable interest. This mutation, p.Y65C, alters the highly conserved hydrophobic triplet from YYW of the WW domain to YCW (fig 3). It is known that mutations in this triplet destroy the transcription promoting activity of RNA polymerase II.13 The wide range of interactions of this polymerase likely explains the unusual cardiac findings and small testes, but the details of such possible interactions remain unknown at this time. A more detailed comparison of the different mutations with clinical findings in Renpenning syndrome is provided in the reports by Kleefstra et al10 and Stevenson et al.11 The combined information on the overall effects on growth and on the specific effects on organogenesis should provide an important clue for the process of determining the exact mechanism(s) by which this specific mutation acts.

Figure 3 Evolutionary conservation of the amino acids in the WW domain of the PQBP1 protein. A comparison of the WW domain from human, mouse, rat, and zebra fish showing the conserved amino acids in italics. The mutated amino acid (Y) at position 65 is shaded in grey.
Just as our XLMR database1 has been helpful in directing the molecular search for this PQPB1 mutation, it should also be of importance in guiding diagnostic molecular studies for mutations in XLMR families, sibships with two affected males, and even sporadic males with mental retardation.
Acknowledgements
We express appreciation to the family (K8275) for their cooperation in this research. Dedicated to the memory of Ethan Francis Schwartz 1996–1998.
Abbreviations
DHPLC - denaturing high performance liquid chromatography
IPS - incorporation PCR SSCP
MRX - non‐syndromic XLMR
PQBP1 - polyglutamine tract binding protein 1 gene
XLMR - X linked mental retardation
Footnotes
Grant support was provided by NICHD (HD 26202)
Competing interests: none declared
Patient details are published with consent
References
- 1.Greenwood Genetic Center – University of Miami XLMR Database July 2005. http://xlmrdb.ggc.org
- 2.Stevenson R E, Schwartz C E. Clinical and molecular contributions to the understanding of X‐linked mental retardation. Cytogenet Genome Res 200299265–275. [DOI] [PubMed] [Google Scholar]
- 3.Greenwood Genetic Center XLMR update. July 2005. http://www.ggc.org/xlmr.htm
- 4.Golabi M, Ito M, Hall B D. A new X‐linked multiple congenital anomalies/mental retardation syndrome. Am J Med Genet 198417367–374. [DOI] [PubMed] [Google Scholar]
- 5.Sossey‐Alaoui K, Lyon J A, Jones L, Abidi F E, Hartung A J, Hane B, Schwartz C E, Stevenson R E, Srivastava A K. Molecular cloning and characterization of TRPC5 (HTRP5), the human homologue of a mouse brain receptor‐activated capacitative Ca2+ entry channel. Genomics 199960330–340. [DOI] [PubMed] [Google Scholar]
- 6.Vervoort V S, Beachem M A, Edwards P S, Ladd S, Miller K E, de Mollerat X, Clarskon K, DuPont B, Schwartz C E, Stevenson R E, Boyd E, Srivastava A K. AGTR2 mutations in X‐linked mental retardation. Science 20022962401–2403. [DOI] [PubMed] [Google Scholar]
- 7.Cason A L, Ikeguchi Y, Skinner C, Wood T C, Holden K R, Lubs H A, Martinez F, Simensen R J, Stevenson R E, Pegg A E, Schwartz C E. X‐linked spermine synthase gene (SMS) defect: the first polyamine deficiency syndrome. Eur J Hum Genet 200311937–977. [DOI] [PubMed] [Google Scholar]
- 8.Kalscheuer V M, Freude K, Musante L, Jensen L R, Yntema H G, Gecz J, Sefiani A, Hoffmann K, Moser B, Haas S, Gurok U, Haesler S, Aranda B, Nshedjan A, Tzschach A, Hartmann N, Roloff T C, Shoichet S, Hagens O, Tao J, Van Bokhoven H, Turner G, Chelly J, Moraine C, Fryns J P, Nuber U, Hoeltzenbein M, Scharff C, Scherthan H, Lenzner S, Hamel B C, Schweiger S, Ropers H H. Mutations in the polyglutamine‐binding protein 1 gene cause X‐linked mental retardation. Nat Genet 200335313–315. [DOI] [PubMed] [Google Scholar]
- 9.Lenski C, Abidi F, Meindl A, Gibson A, Platzer M, Frank Kooy R, Lubs H A, Stevenson R E, Ramser J, Schwartz C E. Novel truncating mutations in the polyglutamine tract binding protein 1 gene (PQBP1) cause Renpenning syndrome and X‐linked mental retardation in another family with microcephaly. Am J Hum Genet 200474777–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleefstra T, Franken C E, Arens Y H, Ramakers G J, Yntema H G, Sistermans E A, Hulsmans C F, Nillesen W N, van Bokhoven H, de Vries B B, Hamel B C. Genotype‐phenotype studies in three families with mutations in the polyglutamine‐binding protein 1 gene (PQBP1). Clin Genet 200466318–326. [DOI] [PubMed] [Google Scholar]
- 11.Stevenson R E, Bennett C W, Abidi F, Kleefstra T, Porteous M, Simensen R J, Lubs H A, Hamel B C, Schwartz C E. Renpenning syndrome comes into focus. Am J Med Genet 2005134A415–421. [DOI] [PubMed] [Google Scholar]
- 12.Hamel B C, Mariman E C, van Beersum S E, Schoonbrood‐Lenssen A M, Ropers H H. Mental retardation, congenital heart defect, cleft palate, short stature, and facial anomalies: a new X‐linked multiple congenital anomalies/mental retardation syndrome: clinical description and molecular studies. Am J Med Genet 199451591–597. [DOI] [PubMed] [Google Scholar]
- 13.Komuro A, Saeki M, Kato S. Npw38, a novel nuclear protein possessing a WW domain capable of activating basal transcription. Nucleic Acids Res 1999271957–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]