

Abstract
Background
Heterozygous mutations in the COL1A1 or COL1A2 gene encoding the α1 and α2 chain of type I collagen generally cause either osteogenesis imperfecta or the arthrochalasis form of Ehlers‐Danlos syndrome (EDS). Homozygous or compound heterozygous COL1A2 mutations resulting in complete deficiency of the proα2(I) collagen chains are extremely rare and have been reported in only a few patients, albeit with variable phenotypic outcome.
Methods
The clinical features of the proband, a 6 year old boy, were recorded. Analysis of proα and α‐collagen chains was performed by SDS‐polyacrylamide gel electrophoresis using the Laemmli buffer system. Single stranded conformation polymorphism analysis of the proband's DNA was also carried out.
Results
In this report we show that complete lack of proα2(I) collagen chains can present as a phenotype reminiscent of mild hypermobility EDS during childhood.
Conclusions
Biochemical analysis of collagens extracted from skin fibroblasts is a powerful tool to detect the subset of patients with complete absence of proα2(I) collagen chains, and in these patients, careful cardiac follow up with ultrasonography is highly recommended because of the risk for cardiac valvular problems in adulthood.
Keywords: collagen, Ehlers‐Danlos syndrome, joint hypermobility, homozygous mutation
Type I collagen is a heterotrimer consisting of two α1(I) chains and one α2(I) chain, encoded, respectively, by the COL1A1 and COL1A2 genes. It is the most abundant extracellular matrix protein in humans and is the major structural protein of skin, bone, tendon, ligaments, and cornea. Mutations in the COL1A1 and COL1A2 genes are commonly found in all forms of osteogenesis imperfecta (OI).1 OI comprises a heterogeneous group of conditions characterised by a variable degree of bone fragility with diverse manifestations such as blue sclerae, hearing loss, thin skin, dentinogenesis imperfecta, and sometimes mild joint laxity. Patients with type I OI are usually heterozygous for a non‐functional COL1A1 allele, whereas more severe forms of OI result from structural changes in the proα1(I) or proα2(I) chain of type I procollagen. At the protein level, either decreased synthesis of type I procollagen or production of structurally abnormal type I procollagen is observed.
Occasionally, mutations in COL1A1 or COL1A2 result in a rare form of Ehlers‐Danlos syndrome (EDS). EDS comprises a heterogeneous group of connective tissue diseases, the major clinical features of which are hyperextensibility of the skin, hypermobility of the joints, and generalised connective tissue fragility.2 The latest classification of EDS (Villefranche, 1997) recognises six genetic subtypes, which differ as regards clinical features, underlying biochemical and molecular defects, and mode of inheritance.3 Whereas heterozygous mutations in the structural genes for type III and type V collagen are responsible for the vascular and classic subtypes of EDS, respectively, mutations in the genes for type I collagen or in genes involved in the processing of type I collagen are causal in several other, rare EDS subtypes. A specific subset of mutations in COL1A1 or COL1A2 that prohibit cleavage of the amino‐propeptide of procollagen type I results in the arthrochalasis type of EDS. This disorder is characterised by severe generalised joint hypermobility, congenital hip dislocation, multiple dislocations of joints other than the hip, and hyperelasticity and fragility of the skin.2 Homozygous or compound heterozygous mutations in the gene encoding the enzyme that cleaves off the amino‐terminal propeptide of type I procollagen (ADAMTS2), are responsible for EDS type VIIC or dermatosparaxis type, which is characterised by extreme skin fragility and excessive bruising.4
Here we report a total deficiency of collagen α2(I) chains in a boy with a phenotype of hypermobile EDS, an EDS subtype for which the underlying genetic defect is at present largely unknown. Complete deficiency of α2(I) chains due to homozygosity or compound heterozygosity for a non‐functional COL1A2 gene was reported earlier in eight patients.5,6,7,8,9,10,11,12 The clinical features of these patients, however, are strikingly variable, ranging from severe OI to a mild EDS/OI‐like phenotype, and associated in some adult patients with severe cardiac valvular abnormalities, necessitating cardiac surgery.
We provide a review of the literature and speculate on the mechanisms determining the varying phenotypic outcomes.
Methods
Clinical features
The proband, a 6 year old Portuguese boy, is the only child of a first cousin marriage. His father is also the only child of a first cousin marriage. The proband was born at term after a normal pregnancy. At the age of 2 months he underwent surgery for hypertrophic pyloric stenosis. At the age of 5 months he underwent surgery for inguinal hernia. He has been receiving physiotherapy since his first year because of equilibrium problems due to muscle hypotonia and joint hypermobility with deformities of the feet. Up to the age of 6 years he had not suffered from any joint dislocations or bone fractures. Wound healing was not delayed, scar formation was normal, and he did not bruise easily. Clinical examination at the age of 6 years revealed stature between the 50th and 75th percentile (121 cm). He presented generalised joint hypermobility. Passive apposition of the thumbs to the flexor aspect of the forearm was positive (fig 1), as well as passive dorsiflexion of the little fingers beyond 90° and hyperextension of the knees beyond 10°. This yielded a positive Beighton score of 6/9. He presented genua vara recurvata and severe pes planus with valgus heels, hallux valgus, and subluxations of the toes (fig 2). The skin was moderately hyperextensible. He had white sclerae. His fingers were long and thin. There were no signs of dentinogenesis imperfecta. Ophthalmological examination and a hearing test were normal. Echocardiography was performed and revealed no gross mitral or aortic valve abnormalities, although there were signs of mitral valve bulging. Bone mineral density was estimated by measuring the cortical width of the left metacarpal phalanges, and this was slightly diminished, reflecting some decrease in bone mineral density. Based on the clinical findings, the diagnosis of EDS, hypermobility type was made.

Figure 1 Passive apposition of the thumb to the flexor aspect of the forearm. (Written informed consent was obtained from the patient and his parents for the publication of this image.)
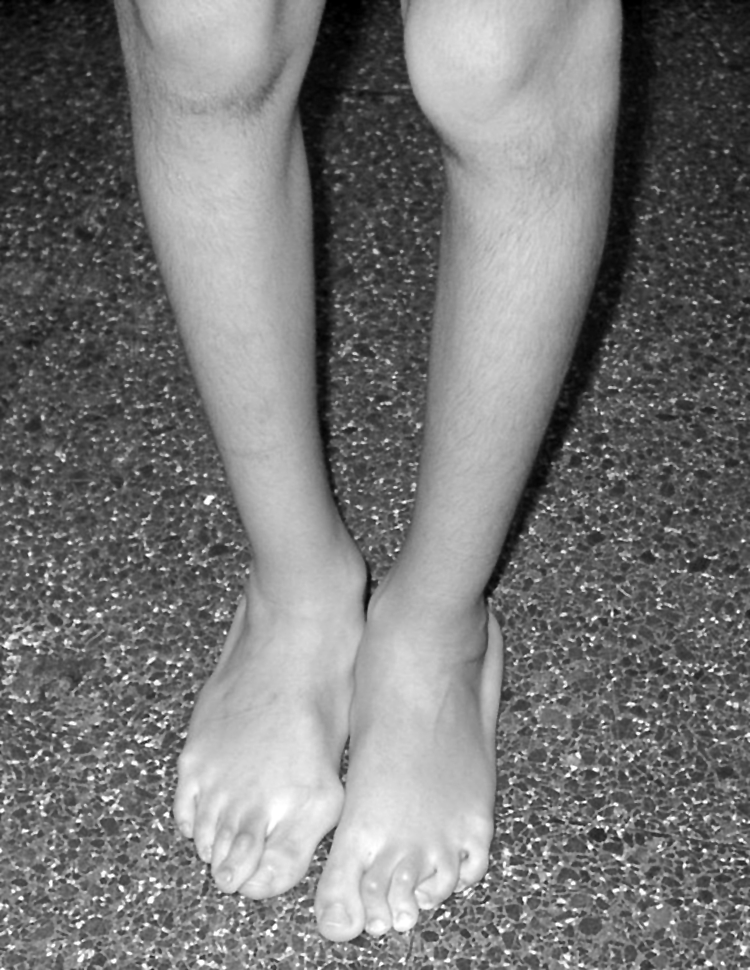
Figure 2 Genua vara and pedes plano valgi with hallux valgus and subluxations of the toes. (Written informed consent was obtained from the patient and his parents for the publication of this image.)
Both parents were clinically normal. There was no history of fractures or dislocations, and they presented no joint hypermobility. Metacarpal cortical width was normal for the father but slightly decreased in the mother. Echocardiography in the parents revealed no cardiac abnormalities.
Biochemical studies
Skin fibroblast cultures were established from the proband and grown under standard conditions. The cells were incubated with [14C]proline at 37° for 20 h, medium and cell layer were harvested separately, supplemented with protein inhibitors, and stored at −80°C prior to analysis. Medium and cell layer procollagens were converted to the respective collagen molecules by pepsin digestion, lyophilised, and re‐dissolved in sample buffers. Analysis of proα and α‐collagen chains was performed by SDS‐polyacrylamide gel electrophoresis (SDS‐PAGE) using the Laemmli buffer system.13
Molecular analysis
Genomic DNA was isolated from the fibroblast cultures using the Easy‐DNA Kit (Invitrogen, Carlsbad, CA). PCR was performed using primers for the COL1A2 coding region and the amplimers were analysed by single stranded conformation polymorphism (SSCP) analysis as described.14 Fragments containing abnormal shifts were then cloned and sequenced using the ABI 3100 sequencer.
In order to confirm nonsense mediated mRNA decay (NMD) of the patient's COL1A2 mRNA, total RNA was isolated from cultured skin fibroblasts from the proband and from two controls samples by TRIZOL (Life Technologies, Gaithersburg, MD). For conversion to cDNA, Moloney murine leukaemia virus transcriptase was used in combination with random hexanucleotide primers. A COL1A2 cDNA fragment downstream of the mutation was PCR amplified using the primers 5′GCCCTGGGGAGCCTGGTCTC 3′ (COL1A2 exon 23) and 5′ACCTCGGCTTCCAATAGGAC 3′ (COL1A2 exon 31). The length of this fragment was 509 bp. In the same PCR reaction, a COL1A1 cDNA fragment was amplified using the primers 5′CCCCCTGAAAGAATGGAGA 3′ (COL1A1 exon 9) and 5′GCACCAGTAGCACCATCATT 3′ (COL1A1 exon 15). The length of this fragment was 316 bp. PCR cycling conditions were as follows: 95°C for 5 min; then 20 cycles, 25 cycles, and 30 cycles of 1 min at 94°C, 1 min at 58°C, and 1.5 min at 72°C, respectively. The amplified products were separated on 2% agarose.
Results
Biochemical analysis
SDS‐PAGE of the procollagens secreted by the proband's cells showed the presence of both proα1(I) and α1(I) collagen chains, but there was no evidence of proα2(I), pNα2(I), or α2(I) collagen chains. After pepsin digestion, no α2(I) collagen chains could be detected either in the medium or in the cell layer. The aspect of the α1(I) collagen chains was slightly broadened compared with that in control cell lines, compatible with mild post‐translational overmodification (fig 3).

Figure 3 Biochemical analysis of the patient's fibroblasts showing the total absence of α2(I) collagen chains.
Molecular analysis
SSCP analysis of the proband's DNA showed migration shifts in the genomic DNA fragment encoding a part of IVS 6, exon 7, and a part of IVS 7. Sequencing of the corresponding genomic DNA revealed a homozygous insertion of a C (c.292_293insC) at codon 8 of COL1A2 exon 7 (fig 4). The resulting frameshift introduces a premature termination codon (PTC) in the next amino acid position. Sequencing of the same fragment was performed in both parents revealing the presence of the same C insertion in exon 7 in the heterozygous state. This observation was in agreement with the consanguinity that is present in the family.

Figure 4 Part of the normal and mutant gDNA sequence of exon 7 of the COL1A2 gene.
PCR amplification of a COL1A2 cDNA fragment downstream of the homozygous mutation showed absence of the proband's COL1A2 cDNA, as compared with his COL1A1 cDNA and as compared with normal controls, after 20, 25, and 30 cycles of PCR amplification (fig 5).

Figure 5 Agarose gel showing absence of the proband's COL1A2 cDNA after 20, 25, and 30 cycles of PCR amplification of a COL1A2 cDNA fragment downstream of the mutation, and of a COL1A1 cDNA fragment of the patient (P) and two normal controls (C).
Discussion
Total deficiency of collagen proα chains is rare. Complete deficiency of proα1(I) chains appears to be incompatible with life.15 In this report we describe a boy with clinical features reminiscent of mild hypermobility EDS due to a defect in the production of the proα2(I) collagen chains. To date there are only few reports of patients with total α2(I) collagen deficiency, who show markedly different phenotypes (table 1).
Table 1 Clinical and molecular findings in reported index patients with total absence of proα2(I) collagen chains.
| Reference | Nicholls et al6 | Sasaki et al9 | Hata et al10 | Nicholls et al11 | Schwarze et al12 (P1) | Schwarze et al12 (P2) | Schwarze et al12 (P3) | This report |
|---|---|---|---|---|---|---|---|---|
| Age (years) | 5 | 30 | 35 | 9 | 45 | 38 | 30 | 6 |
| Clinical | OI type III | Classic | Classic | OI/EDS | Classic | Classic | Classic | Hypermobile |
| phenotype | EDS‐like | EDS‐like | EDS‐like | EDS‐like | EDS‐like | EDS | ||
| Cardiac valve | No | AI | MI | No | AI and MI | MI | AI and MI | Mitral valve |
| involvement | bulging | |||||||
| mRNA | No | Not | Yes | Yes | Yes | Yes | Yes | Yes |
| instability | reported | |||||||
| Mutation in | Homozygous | Not | Not | Homozygous | IVS11+5G>A | IVS1+717A>G | Homozygous | Homozygous |
| COL1A2 | C‐terminal 4 | reported | reported | IVS46+2t>C | and | and | c.3601G>T | c.292_293insC |
| bp deletion | IVS24+1G>C | IVS24+1G>A | → E1201X |
AI, aortic valve insufficiency; MI, mitral valve insufficiency.
The original patient reported by Nicholls et al was a 5 year old boy with severe, progressively deforming OI (OI type III) with multiple fractures, skeletal deformities, and popcorn deformity of the epiphyses.6 A homozygous four‐nucleotide frameshift deletion within the carboxy‐terminal propeptide of proα2(I) collagen was identified.8 This mutation prevented incorporation of proα2(I) chains into the normal type I procollagen molecules and resulted in the production of proα1(I) collagen homotrimers.7 No cardiac abnormalities were reported in the index patient. The development of valvular disease later on can, however, not be excluded, since there is no follow‐up report of this patient when he was older.
Subsequently, two Japanese patients were reported with total α2(I) collagen deficiency, both presenting EDS‐like features, including joint hypermobility, hyperextensibility of the skin, and abnormal wound healing.9,10 The first of these patients also suffered from severe aortic regurgitation, necessitating aortic valve replacement in adulthood,9 while the second patient had mitral valve regurgitation.10
In 2001 Nicholls et al reported a total absence of α2(I) collagen chains in a girl with phenotypic manifestations of both OI, including blue sclerae and slightly increased bone fragility, and EDS, including generalised joint laxity, patellar dislocations, and pedes plano valgi with secondary shortening of the Achilles tendon. Her skin was normal. No cardiovascular anomalies were observed at the age of 9 years. A homozygous splice site mutation in IVS 46 led to the introduction of a PTC.11
Finally, three adult patients with complete α2(I) collagen chain deficiency were reported, all showing features of classic EDS, including hyperextensible skin, hypermobile joints, abnormal wound healing with atrophic scarring, and/or easy bruising. The brother of patient 3 presented more subtle EDS signs, with hypermobility of small joints and soft skin. All three patients developed valvular heart disease in adulthood, involving either the aortic or mitral valve, or both, and necessitating valve replacement surgery. In these patients, homozygous nonsense mutations or compound heterozygous splice site mutations that lead to COL1A2 mRNA instability were identified.12 Because of the severe cardiac valve problems in most adult patients with α2(I) chain deficiency, this phenotype was called the “cardiac valvular form of EDS” by Schwarze et al.12 Since the patient described here already shows some signs of mitral valve bulging at the age of 6 years, continued cardiac follow up with echocardiography will be necessary.
The difference in clinical phenotype in patients with total lack of α2(I) collagen chains is remarkable, ranging from a very severe OI phenotype with multiple fractures and deformities of the long bones, to a much milder, EDS‐like phenotype, albeit with potentially serious cardiac problems in adulthood. It is well established that, in eukaryotic cells, mRNAs containing a PTC are rapidly degraded by a process called nonsense mediated RNA decay (NMD). Thus, potentially deleterious truncated proteins are not expressed at high levels. Indeed, in the patients with total α2(I) collagen chain deficiency who presented an EDS phenotype, COL1A2 mRNA was shown to be markedly decreased. In vertebrates, it has been shown that PTCs located in the last coding exon do not lead to NMD.16 Hence the stable but mutant mRNA is translated into abnormal protein chains. This may explain the severe phenotypic consequences in the OI patient with the 4 bp deletion located in the last exon of the COL1A2 gene. Indeed, it was shown that the patient's fibroblasts produced structurally abnormal proα2(I) chains, which were not incorporated into procollagen molecules.7 Probably these mutant proteins, although not participating in collagen trimerisation, accumulate intracellularly and exert a deleterious effect on the normal fraction of collagen chains.
In conclusion, in the majority of cases, the underlying COL1A2 mutations result in NMD and a loss of function effect. The phenotypic consequences are those of a form of EDS, with hypermobility in childhood and complicated by cardiac valve disease in adulthood. On the other hand, COL1A2 mutations which do not result in NMD produce a gain of function effect with the production of abnormal collagen type I chains that disturb interaction with the normal collagen chains and lead to a severe OI phenotype.
We show that the autosomal recessive cardiac valvular form of EDS can overlap with a mild EDS hypermobility type during childhood, while the cardiac valvular phenotype may evolve during adulthood. EDS hypermobility type is usually an autosomal dominant disorder, mainly characterised by joint hypermobility and joint dislocations, but usually without major cardiac valvular problems, although mitral valve prolapse and aortic root dilation may be more common than in the general population.2,17 Apart from the association with tenascin‐X haploinsufficiency in a small subset of patients with autosomal dominant hypermobile EDS,18 the underlying defect of this subtype is largely unknown.
Biochemical SDS‐PAGE analysis of collagen extracted from skin fibroblasts can show the absence of proα2(I) collagen chains in patients with cardiac valvular EDS. In this subset of patients, careful clinical and echocardiographic follow up throughout adulthood is mandatory.
Acknowledgements
We would like to thank the patient and his family for their cooperation.
Abbreviations
EDS - Ehlers‐Danlos syndrome
NMD - nonsense‐mediated mRNA decay
OI - osteogenesis imperfecta
PTC - premature termination codon
SDS‐PAGE - SDS‐polyacrylamide gel electrophoresis
SSCP analysis - single stranded conformation polymorphism analysis
Footnotes
This work is supported by grant G.0171.05 from the Fund for Scientific Research‐Flanders and a GOA research grant from Ghent University to ADP. FS is a research fellow of the Fund for Scientific Research‐Flanders.
Competing interests: none declared
Written informed consent was obtained from the patient and his parents for the publication of his details
References
- 1.Byers P, Cole W G. Osteogenesis imperfecta. In: Royce P, Steinmann B, eds. Connective tissue and its heritable disorders. 2nd ed. New York: Wiley‐Liss, 2002385–443.
- 2.Steinmann B, Royce P, Superti‐Furga A. The Ehlers‐Danlos syndrome. In: Royce P, Steinmann B, eds. Connective tissue and its heritable disorders. 2nd ed. New York: Wiley‐Liss, 2002431–523.
- 3.Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup R J. Ehlers‐Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers‐Danlos National Foundation (USA) and Ehlers‐Danlos Support Group (UK). Am J Med Genet 199877(1)31–37. [DOI] [PubMed] [Google Scholar]
- 4.Malfait F, De Coster P, Hausser I, van Essen A J, Franck P, Colige A, Nusgens B, Martens L, De Paepe A. The natural history, including orofacial features of three patients with Ehlers‐Danlos syndrome, dermatosparaxis type (EDS type VIIC). Am J Med Genet 2004131(1)18–28. [DOI] [PubMed] [Google Scholar]
- 5.Nicholls A C, Pope F M, Schloon H G. Biochemical heterogeneity of osteogenesis imperfecta: new variant. Lancet 19791(8127)1193. [DOI] [PubMed] [Google Scholar]
- 6.Nicholls A C, Osse G, Schloon H G, Lenard H G, Deak S, Myers J C, Prockop D J, Weigel W R, Fryer P, Pope F M. The clinical features of homozygous alpha 2(I) collagen deficient osteogenesis imperfecta. J Med Genet 198421(4)257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deak S B, Nicholls A, Pope F M, Prockop D J. The molecular defect in a nonlethal variant of osteogenesis imperfecta. Synthesis of pro‐alpha 2(I) chains which are not incorporated into trimers of type I procollagen. J Biol Chem 1983258(24)15192–15197. [PubMed] [Google Scholar]
- 8.Pihlajaniemi T, Dickson L A, Pope F M, Korhonen V R, Nicholls A, Prockop D J, Myers J C. Osteogenesis imperfecta: cloning of a pro‐alpha 2(I) collagen gene with a frameshift mutation. J Biol Chem 1984259(21)12941–12944. [PubMed] [Google Scholar]
- 9.Sasaki T, Arai K, Ono M, Yamaguchi T, Furuta S, Nagai Y. Ehlers‐Danlos syndrome. A variant characterized by the deficiency of pro alpha 2 chain of type I procollagen. Arch Dermatol 1987123(1)76–79. [DOI] [PubMed] [Google Scholar]
- 10.Hata R, Kurata S, Shinkai H. Existence of malfunctioning pro alpha2(I) collagen genes in a patient with a pro alpha 2(I)‐chain‐defective variant of Ehlers‐Danlos syndrome. Eur J Biochem 1988174(2)231–237. [DOI] [PubMed] [Google Scholar]
- 11.Nicholls A C, Valler D, Wallis S, Pope F M. Homozygosity for a splice site mutation of the COL1A2 gene yields a non‐functional pro(alpha)2(I) chain and an EDS/OI clinical phenotype. J Med Genet 200138(2)132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwarze U, Hata R, McKusick V A, Shinkai H, Hoyme H E, Pyeritz R E, Byers P H. Rare autosomal recessive cardiac valvular form of Ehlers‐Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense‐mediated RNA decay pathway. Am J Hum Genet 200474(5)917–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970227(259)680–685. [DOI] [PubMed] [Google Scholar]
- 14.Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T. Detection of polymorphisms of human DNA by gel electrophoresis as single‐strand conformation polymorphisms. Proc Natl Acad Sci U S A 198986(8)2766–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schnieke A, Harbers K, Jaenisch R. Embryonic lethal mutation in mice induced by retrovirus insertion into the alpha 1(I) collagen gene. Nature 1983304(5924)315–320. [DOI] [PubMed] [Google Scholar]
- 16.Hentze M W, Kulozik A E. A perfect message: RNA surveillance and nonsense‐mediated decay. Cell 199996(3)307–310. [DOI] [PubMed] [Google Scholar]
- 17.Wenstrup R J, Meyer R A, Lyle J S, Hoechstetter L, Rose P S, Levy H P, Francomano C A. Prevalence of aortic root dilation in the Ehlers‐Danlos syndrome. Genet Med 20024(3)112–117. [DOI] [PubMed] [Google Scholar]
- 18.Zweers M C, Bristow J, Steijlen P M, Dean W B, Hamel B C, Otero M, Kucharekova M, Boezeman J B, Schalkwijk J. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers‐Danlos syndrome. Am J Hum Genet 200373(1)214–217. [DOI] [PMC free article] [PubMed] [Google Scholar]