

Abstract
Background
Survivin is proposed to play a central role in the progression and resistance to therapy of diverse tumour types. High levels of this molecule in tumour cells also correlate with loss of the TP53 tumour suppressor gene, suggesting a molecular connection between TP53 loss and transcriptional induction of Survivin. Patients with TP53 germline mutations, such as those with Li‐Fraumeni syndrome, are particularly susceptible to sarcomas, including rhabdomyosarcomas. Our study aimed to identify rhabdomyosarcoma tumours that express Survivin, in order to test novel Survivin‐targeted therapies in these tumours.
Methods
Tumour microarray slides composed of 63 primary rhabdomyosarcoma tumours were stained with a polyclonal antibody to Survivin to identify tumours expressing Survivin. Subcutaneous tumours were then established in NOD/SCID mice using RH30red cells, a red fluorescent clone of the RH30 human alveolar rhabdomyosarcoma cell line. Tumours were treated by hydrodynamic injection with a cocktail of Survivin‐shRNA‐encoding plasmids for a period of 2 weeks.
Results
Over 80% of primary rhabdomyosarcoma tumours expressed Survivin. Treatment of rhabdomyosarcoma xenografts showed greater than 70% reduction in growth when compared with control injected tumours at study completion (average tumour sizes: 1683 v 304 mm3, p<0.05).
Conclusions
Our findings support a role for Survivin in rhabdomyosarcoma biology and provide preliminary evidence for the therapeutic use of Survivin‐targeted RNA interference for human tumours that express high levels of this molecule.
Keywords: apoptosis, soft tissue sarcoma, Survivin, RNAi, xenografts
The Survivin gene has structural and functional similarities both to the inhibitor‐of‐apoptosis gene family, which specifically blocks the downstream effectors of cell death, and to the chromosomal passenger proteins which play an essential role in cytokinesis.1,2,3,4 The human Survivin gene locus encodes at least five alternatively spliced transcripts including Survivin, Survivin‐2B, Survivin‐ΔEx3, Survivin‐3B, and Survivin‐2α.5,6,7,8,9 Survivin is highly expressed during normal embryonic development and in a variety of transformed cell lines, but minimally expressed in normal, non‐transformed tissue. Adult tissues that do express Survivin include those composed of highly proliferating cells such as CD34+ bone marrow cells, vascular endothelium, endometrium, and neural stem cells.7,10,11 Human cancer cells that aberrantly express Survivin include epithelial tumours of lung, colon, pancreas, breast, stomach, CNS tumours, soft tissue sarcomas, and haematological malignancies (reviewed in Altieri12). Paediatric tumours that express Survivin include neuroblastoma, Wilms' tumours, and some paediatric CNS malignancies.11,13 High levels of expression within cancer cells have correlated with clinical outcome in many studies; most of these studies showed a direct correlation between higher levels of Survivin and a poor outcome.14
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma of childhood.15 It occurs at a disproportionately high rate among individuals with several heritable conditions including Gardner syndrome (autosomal dominant, APC mutation, 5q21, OMIM 175100), Werner syndrome (autosomal recessive, WRN mutation, 8p12, OMIM 277700), Nijmegen breakage syndrome (autosomal recessive, NBS1 mutation, 8q21, OMIM 602667), nevoid basal cell carcinoma syndrome (autosomal dominant, PTCH, 9q22.3‐q31, OMIM 601309), FAMMM (autosomal dominant, CDKN2A/p16Ink4a mutation, 9p21, OMIM 600160), and Li‐Fraumeni syndrome (LFS; autosomal dominant, TP53 mutation, 17p13, OMIM 191170).16 LFS is a rare autosomal dominant disorder characterised by a familial clustering of tumours with a predominance for sarcomas.17 The great majority of families with LFS carry germline mutations of the tumour suppressor gene TP53. The TP53 tumour suppressor gene has also been implicated as a critical gene in sporadic RMS.18 A substantial percentage of primary RMS tumours and cell lines have loss of function TP53 mutations, usually occurring in the DNA binding region.19 Mutations observed in germline and in sporadic cases of this tumour are very similar.18
Morphologically, RMS is composed of two main histological subtypes, alveolar and embryonal. Approximately 70% of tumours with alveolar histology are characterised molecularly by translocations that create aberrant fusion proteins between PAX7 or PAX3, two critical early muscle developmental regulators, and FKHR, a transcription factor.20 Development of these tumours likely represents a disruption in myogenic differentiation, as the tissue resembles primitive mesenchyme and expresses some myogenic markers including MyoD and myogenin.15,21 RMS can arise virtually anywhere within the body including the head/neck, chest, abdomen, pelvis, retroperitoneum, and extremities.15 Although morphologically similar, tumours presenting in different regions behave differently biologically and vary in their response to standard chemotherapeutic agents. New biological predictors of disease outcome and new therapeutic alternatives are essential as an average of 30–50% of patients succumb to their disease despite current modes of intensive chemotherapy, radiation, and surgery.
In this work, we demonstrate by tissue microarray that Survivin is expressed at high levels in most primary human alveolar (ARMS) and embryonal rhabdomyosarcoma (ERMS) tumours. We then use RNA interference to target Survivin and the Survivin splice variants Survivin‐2B and Survivin‐ΔEx3 in vitro and in vivo, using representative cell lines and xenograft models that harbour TP53 mutations commonly observed in germline mutations of families with LFS. Our results show that Survivin shRNAs significantly inhibit tumour growth in culture and in the animal models.
Methods
Tumour microarrays
Individual tumour microarray slides 4 μm in thickness, consisting of 32 alveolar and 31 embryonal RMS tumours along with adjacent normal muscle tissue were obtained from the Cooperative Human Tissue Network and prepared by the Center for Biopathology at Columbus Children's Research Institute. Each individual tumour was represented 3–5 times per slide. Permission from the Intergroup Rhabdomyosarcoma Study Group, Cooperative Human Tissue Network, and Columbus Children's Hospital Institutional Review Board was obtained for this study. Formalin fixed, paraffin embedded sections were immunostained with a polyclonal anti‐Survivin antibody (FL‐142, Santa Cruz Biotechnology, Santa Cruz, CA), as described previously.11 Survivin immunoreactivity was evaluated semi‐quantitatively based on the percentage of positive cells. Staining was assessed in 5–10 high powered fields at ×40 magnification. Slides were assigned scores of 1 if <1% cells were positive, 2 if 1–10% cells were positive, 3 if 10–50% cells were positive, and 4 if >50% cells were positive.
Tissue culture and chemotherapeutic treatment
CW9019, RH28, RD2, RH30, and RH30red cells were maintained under proliferating conditions in DMEM (CW9019, RH28, and RD2) or RPMI‐1640 (RH30 and RH30red) supplemented with 10% FBS and grown in 95% air, 5% CO2 at 37°C. Vincristine sulfate (Vincasar PFS, SICOR Pharmaceuticals, Irvine, CA) was used at a final concentration of 2 μM.
Generation of RH30red
RH30 RMS cells were transfected with DraIII digested pDsRed2‐N1 (Clontech, Palo Alto, CA ) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Stably transfected cells were selected in medium containing 200 μg/ml G418 sulfate (Mediatech, Herndon, VA) for 3 weeks. Cells stably expressing DsRed protein were then expanded in basic medium and further selected based on red fluorescence during sequential sorting rounds on a FACSVantage/DiVa cell sorter (Becton Dickson, Franklin Lakes, NJ). RH30red cells representing the brightest 20% red fluorescent cells were selected during each sterile sort. There were no apparent morphological or growth differences between the parental cell line and the red fluorescent cell population, and we verified that the signature fusion gene product (PAX3‐FKHR) was intact.
Total RNA isolation
Total RNA was isolated from 106 cells using the Trizol method (Invitrogen). cDNA was obtained in a random priming reaction using Omniscript reverse transcriptase (Qiagen, Valencia, CA).
Quantitative PCR
Primers to detect human Survivin and splice variants have been previously reported.22 TaqMan analysis was carried out according to the manufacturer's instructions by using an Applied Biosystems 7700 Sequence Detection System (Applied Biosystems, Foster City, CA). Results from each sample were compared with normal muscle as a calibrator using the relative standard curve method (Applied Biosystems). Genomic levels and cDNA expression levels were measured relative to 18S rRNA. Experiments were performed in triplicate and standard deviations were based on the average of three experiments.
Expression of Survivin shRNAs
Three human Survivin‐specific shRNAs were designed as 64‐mers containing a hairpin loop and cloned into pSUPER vector with the H1 RNA polymerase promoter as previously described.23 The complete sequences of the shRNAs are available upon request. For transient transfections, plasmid DNA was transfected into proliferating CW9019 or RH30 cells using Effectene transfection reagent (Qiagen) at a ratio of 1:30 (DNA:Effectene). The reporter plasmids pHcRed or pEGFP (Clontech) were co‐transfected with pSUPER or shRNA plasmids and used to sort transfected cells by FACS. A transfection efficiency of approximately 50% was achieved using this method, and the population of cells used for further experiments consisted of an enriched population of greater than 97% transfected cells.
Annexin V assays
Annexin V/propidium iodide staining was carried out using the Roche Annexin‐V‐Fluos Staining Kit (Roche, Indianapolis, IN) following the manufacturer's instructions, and analysed by FACS in a Coulter EPICS XL flow cytometer (Beckman Coulter, Fullerton, CA). Experiments were performed in triplicate.
Caspase assays
Two thousand cells from each experimental condition were assayed for caspase‐3 and caspase‐9 activity using the Caspase‐Glo 3/7 Assay and Caspase‐Glo 9 Assay (Promega, Madison, WI) according to the manufacturer's instructions. Caspase activity was measured in a Victor‐3 plate reader (Applied Biosystems) and expressed as relative luciferase units after background subtraction. Experiments were performed in triplicate.
Microscopy
Confocal microscopy was performed on RMS cells in Leibovitz L15 medium in glass‐bottomed dishes (MaTek, Ashland, MA). The cells were pulsed with Hoechst dye for labelling nucleic acids. Image acquisition was performed on a Zeiss LSM510 META confocal microscope equipped with a heated stage, using a blue diode laser. Measurement of nuclear diameter was performed using the microscope standard operating software.
Animal studies
For ex vivo studies, 6 week old NOD/SCID female mice (kindly provided by Dr Michael Boyer, Columbus Children's Research Institute) were injected with 1.5×106 viable RH30red cells (previously transfected for 24 h with Survivin shRNA cocktail or pSUPER control) subcutaneously into the right flank under isoflurane anaesthesia. Ten mice per study arm were used. For in vivo treatment studies, 6 week old NOD/SCID female mice were injected with 1.5×106 proliferating RH30red cells subcutaneously into the right flank under isoflurane anaesthesia. Approximately 21 days after inoculation, 85% of the mice developed palpable red fluorescent tumours. The tumours were imaged using a fluorescent Lightools Macroimager (Lightools, Encinitas, CA), as well as measured two‐dimensionally with electronic callipers. Tumour volume was calculated using the following formula:
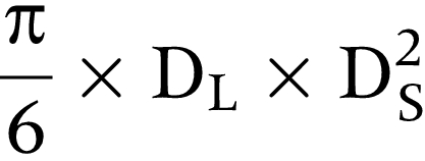 |
where DL is the largest diameter and DS is the smallest diameter. Injections were performed hydrodynamically directly into the tumour mass, in a final volume of 100 μl, containing a total of 15 μg of DNA. All experiments received prior approval from the Columbus Children's Research Institute Institutional Animal Use and Care Committee.
Histological analysis
Resected tumours were fixed in 10% neutral buffered formalin, and processed through an increasing ethanol series for paraffin embedding. Serial sections were taken at 5 μm on a Leica microtome and stained with H&E for histological analysis or immunostained with antibodies against Ki‐67, myoD1 (1:25, DakoCytomation, Carpinteria, CA), Survivin (1:400, Santa Cruz), Desmin (1:2000, Biomeda, Foster City, CA), or phospho (Ser10) histone H3 (1:250, Upstate Cell Signaling, Charlottesville, VA), or used in TUNEL assays (Calbiochem, EMD Biosciences, San Diego, CA) as described by the supplier. Quantification of staining (Ki‐67, Survivin, and TUNEL) was performed in a minimum of three high powered, well preserved areas of the slide measuring 0.066 mm2 per tumour sample. Each field considered contained at least 500 cells. Quantification of phospho‐histone H3 and mitotic figures was performed by counting the number of positive cells in a 0.26 mm2 field in at least 10 representative fields per experimental condition.24 Images were acquired with a Leica microscope.
Biostatistical analysis
The distribution of failure‐free survival (FFS) of patients and animals was estimated using the method of Kaplan and Meier, and significance assessed by log rank test. Statistical significances of tumour growth were assessed using a paired Student's t test.
Results
Survivin expression in paediatric rhabdomyosarcoma
Survivin is a top transcriptome expressed in many different human malignancies, including soft tissue sarcomas.25 A comprehensive analysis of its expression pattern in RMS tumours has not previously been demonstrated. To this end, we evaluated Survivin protein expression in 63 primary human RMS tumours that included 31 ERMS and 32 alveolar rhabdomyosarcoma (ARMS) tumours, by immunohistochemical staining with a polyclonal antibody against Survivin. Clinical data were available for the 63 patients. Most of the patients harbouring these tumours were treated on similar therapy protocols within the Intergroup Rhabdomyosarcoma Study Group or the Children's Oncology Group. Two patients were <1 year of age, 39 were 1–9 years old, and 22 were ⩾10 years old. There were 38 males and 25 females. There were five group I patients, 15 group II patients, 23 group III patients, and 14 group IV patients. The 5 year FFS for the entire analysable group of 63 patients was 50% (data not shown). Over 80% of the tumours analysed for Survivin expression (54/63 tumours) had a mean Survivin staining score greater than or equal to 3, indicating that at least 10–50% of tumour cells expressed Survivin (fig 1A). Of the 54 tumours, 25 were alveolar and 29 were embryonal. Only nine tumours (14%) had a Survivin score under 3; seven of these were of alveolar histology and two were of embryonal histology.

Figure 1 Survivin expression in rhabdomyosarcoma. (A) Representative immunohistochemical analysis of Survivin expression in a rhabdomyosarcoma tumour from a tissue microarray. Nuclear Survivin reactivity is strong in both alveolar and ERMS tumours, whereas no nuclear Survivin staining is observed in normal muscle tissue. (B) Transcriptional analysis of Survivin and the Survivin splice variants in representative cell lines of embryonal (ERMS; RD2) and alveolar (ARMS; CW9019, RH28, RH30) rhabdomyosarcoma compared to normal muscle RNA. Survivin and splice variants are expressed at high levels in all rhabdomyosarcoma cell lines compared to normal muscle controls.
As a prelude to the in vivo studies, we also examined Survivin expression in four representative RMS cell lines by quantitative PCR. Three cell lines were alveolar in origin (CW9019, RH28, and RH30) and one was embryonal (RD2). CW9019 cells are characterised by a PAX7;FKHR translocation,26 whereas RH28 and RH30 cells are characterised by a PAX3;FKHR translocation.27 All four lines also harbour non‐functional TP53 proteins. The ERMS cell line RD2 carries a missense mutation in codon 248 resulting in a Arg→Trp substitution. This mutation has also been observed in several families with LFS.28 The ARMS cell line RH30 carries a missense mutation in codon 273 resulting in a Arg→Cys substitution.19 This mutation is also commonly observed in LFS families.28,29 Mutations occurring at codons 248 and 273 (within the DNA binding domain) account for 22.7% of all TP53 mutations in LFS.30 Both mutations yield full‐length TP53 proteins that are functionally compromised.
Survivin, Survivin‐2B, and Survivin‐ΔEx3 were expressed at higher levels in all four cell lines than in their normal muscle tissue controls: Survivin expression was increased 10–100 fold, Survivin‐2B was increased 100–150 000 fold, and Survivin‐ΔEx3 was increased 50–300 fold above normal muscle (fig 1B). Increased expression of Survivin and Survivin‐ΔEx3 in these cell lines was further substantiated at the protein level by Western blot analysis (not shown). Taken together, these findings identify Survivin and its splice variants as potential biological targets for therapeutic intervention in RMS.
Cellular consequences of Survivin inhibition in vitro
Transient and stable disruption of Survivin expression within cancer and normal cells has been previously achieved by genetic deletion, anti‐sense oligonucleotides, dominant negative inhibitors, and ds siRNAs.3,31,32,33 Disruption of Survivin within several types of cancer cells has been shown to enhance programmed cell death, while overexpression of this gene has been shown to behave as an apoptotic inhibitor.14 For our study, we designed a cocktail of three DNA plasmids encoding Survivin‐specific shRNAs that targeted different regions of the human Survivin gene (fig 2). We validated the specificity of the shRNAs by demonstrating that each sequence specifically targeted Survivin. Our laboratory has previously shown that this combination of shRNAs effectively decreases the transcriptional expression of all three Survivin isoforms in a paediatric CNS tumour cell line by at least 50%.34 In ARMS cells we observed a decrease of approximately 60% in the levels of all Survivin transcripts, and at least a 50% reduction of Survivin and Survivin‐ΔEx3 at the protein level (fig 3A,B). Transient expression of Survivin shRNAs in CW9019 cells was achieved by transfection with an expression vector (pSUPER) containing either the Survivin‐shRNA sequences or control, in combination with the reporter vector pHcRed. The population of cells was sorted and enriched for transfected cells by FACS based on the red fluorescence of the HcRed protein. The resulting cell population consisted of greater than 97% of red expressing cells. The growth of these cells was followed for 48 h (fig 3C). Control transfected cells grew with a doubling time of approximately 31 h, whereas shRNA treated cells did not double over the course of 72 h. In the shRNA treated cells, approximately 45% of the original seeded cells were viable at 24 and 48 h post sorting (corresponding to 48 and 72 h post transfection). The same pattern of cell growth was observed in RH30 cells (not shown). These results suggest that Survivin shRNAs can both inhibit cell growth and increase cell death in alveolar RMS cell lines.

Figure 2 Schematic representation of Survivin gene structure and targeting with shRNAs. Grey boxes represent exons. The Survivin gene contains exons 1–4; Survivin‐2B contains exons 1–4 and an additional exon 2B; Survivin‐ΔEx3 contains exons 1, 2, and 4. The black lines and arrows above the figure show regions targeted by the different Survivin shRNAs.

Figure 3 Inhibition of Survivin by shRNA cocktail induces tumour cell death. RMS cells were transfected with Survivin shRNAs for a minimum of 24 h. The cells were sorted for an enriched cell population consisting of transfected cells. (A) Levels of Survivin gene family transcripts and (B) the levels of Survivin and Survivin‐ΔEx3 proteins were decreased after 48 h of treatment with Survivin shRNA cocktail. Growth of transfected cells was assessed by trypan blue exclusion at 24 h intervals. (C) shRNA treated cells had decreased viability compared to control treated cells, (D) had higher levels of apoptosis as assayed by annexin V staining, and (E) showed activation of caspase‐3 (RLU, relative luciferase units). (F) Nuclear alterations were observed in the form of enlarged nuclei and small nuclear clusters in shRNA treated cells stained with Hoechst (arrowheads). Images were acquired at the same magnification of ×400.
To further evaluate the cell death effects implicated by the growth curves in the shRNA treated tumour cells, we performed Annexin‐V/PI staining as an assay of early apoptosis, and caspase activity assays to demonstrate involvement of the caspase pathway. An increase in the percentage of apoptotic cells in shRNA treated cells was observed at all time points analysed (fig 3D). At 24 h, 14.3% of cells were Annexin V positive, at 48 h 16.9%, and at 72 h 39.2%. This is in contrast to the 5% positivity observed in control treated cells at the 72 h time point (fig 3D). Caspase‐3 activity was also markedly increased in shRNA treated cells compared with control treated cells at 72 h post transfection. The level of caspase‐3 activity in shRNA treated cells was comparable to that seen following 72 h of vincristine treatment (fig 3E). Caspase‐9 activity was also increased in shRNA treated cells when compared to control cells (not shown).
Recently, Bishop and co‐workers demonstrated that Survivin depletion by shRNAs in human retinal pigment epithelial (RPE) cells results in a number of nuclear abnormalities including multinucleation, bilobed nuclei, mininuclei, and deformed nuclei.35 Therefore, we analysed the nuclei of our shRNA treated tumour cells and compared them to control cells by Hoechst staining. Nuclei of shRNA treated cells appeared larger than those of control cells (fig 3F). A systematic analysis of nuclear diameter further substantiated this observation. The average diameter of shRNA treated cells was 118 μm compared to 87 μm for control treated cells, representing a 36.6% increase in nuclear size. Additionally, mitotic defects were evident. The formation of multiple small circular nuclear clusters was indicative of defects in cytokinesis. Similar findings were reported following Survivin shRNA treatment in TP53 null RPE cells.35 This suggests that loss of p53 is a contributing factor to the cytokinesis defects observed in Survivin‐depleted RMS cells.
Tumour engraftment potential of Survivin shRNA cocktail treated cells
To evaluate the effects of Survivin shRNA treatment on the potential for RMS cells to establish de novo tumours subcutaneously, we transfected RH30red cells, which stably express the red fluorescent protein DsRed, ex vivo with either the shRNA cocktail or control, in combination with pEGFP. After 24 h the cells were dislodged by trypsin digestion, enriched for transfected cells by FACS based on GFP fluorescence, and 1.5×106 viable transfected cells injected subcutaneously into anaesthetised NOD/SCID mice (n = 10 per study arm). An advantage of this cell line is that it allows for tracking and imaging of tumour establishment and growth in a non‐invasive fashion in vivo. Palpable tumours were established in 9/10 control injected animals by day 15 post inoculation. At the experimental end point (30 days), 9/10 control animals had very large tumours, with a mean volume of 1852 mm3. To allow for a possible slowed establishment of tumours from shRNA treated cells, we followed the animals for a period of 90 days. None of the animals injected with shRNA treated cells developed a tumour within the 90 day experimental period, either grossly or microscopically (fig 4).

Figure 4 Survivin shRNA cocktail prevents de novo RMS tumour establishment. RH30red cells were transfected ex vivo with Survivin shRNAs or control for 24 h before injection subcutaneously into the flanks of NOD/SCID mice. Control treated cells resulted in the formation and development of large tumours, whereas none of the animals injected with shRNA treated cells developed tumours.
In vivo modulation of Survivin activity
To evaluate the effects of Survivin targeted therapy on RMS tumour growth in vivo, we used our Survivin shRNA cocktail in the treatment of a human RMS xenograft model. We established ARMS tumours in NOD/SCID mice using the RH30red cell line. Subcutaneous palpable tumours were established between 12 and 15 days post inoculation. Treatment was initiated once the tumour volume was at least 100 mm3 (typically around 21 days post inoculation). We administered the plasmid cocktail hydrodynamically, using 5 μg of each shRNA construct (total 15 μg) or 15 μg of pSUPER control in PBS, by intratumoural injection at days 1, 3, 5, 6, 11, and 14 for a total experimental period of 15 days. Tumours from mice injected with the shRNA cocktail grew significantly slower than control injected mice (fig 5A). Differences in tumour volume were significant beginning at day 3 (48 h after the first treatment course, p<0.05). By day 4, 75% of shRNA treated animals showed a reduction in tumour size that persisted throughout the treatment period. Continuous tumour growth was observed for all control animals. At the completion of the study the mean tumour volume in the control treated mice was 5.5 times greater than that in the shRNA treated mice (1683 v 304 mm3, p<0.05), suggesting that the Survivin shRNA cocktail has significant antitumour activity in ARMS in vivo. Animal survival, defined by the time for any animal to reach an experimental endpoint, was evaluated for both treatment arms. The survival was represented by a Kaplan‐Meier curve. At the end of the experiment there was 25% survival in control animals compared to 100% in shRNA treated animals (fig 5B). The difference in survival between groups was significant as demonstrated by log rank test (p = 0.0042).

Figure 5 Survivin shRNA cocktail inhibits tumour growth in vivo. NOD/SCID mice subcutaneously injected with RH30red cells formed palpable tumours within 21 days. Mice were randomly assigned to treatment groups (control or treated) and injected with shRNA cocktail or control, as described in the Methods section. (A) Tumour growth during the experimental period. Tumour volume was measured as described in the Methods section, and the mean tumour volume used to generate the graph. A circle on the x axis represents treatment administration. Growth rate was calculated during treatment rest period. Overall growth is represented by the slope of the curve. (B) Survival analysis is represented by a Kaplan‐Meier curve. shRNA treated animals (dotted line) had improved survival as when compared with animals (solid line) as demonstrated by the significant log rank test.
At the end of the experimental period, tumours from the animals in both groups were visualised by fluorescence microscopy in vivo, and then resected. Tumour cells can be distinguished in these tumours from stroma and vasculature based on their red fluorescence. To validate a specific decrease of tumour cells, we evaluated the percentage of red fluorescent tumour cells in the resected tumours at study completion. By fluorescence microscopy it was estimated that 30% of cells in control tumours and 15% in shRNA treated tumours were red fluorescent (fig 6A). This estimate was further corroborated by FACS analysis of disaggregated tumour material. Cells isolated from control tumours consisted of 28.5% red fluorescent cells compared to 13.5% in shRNA treated tumours. These data indicate a 50% loss of tumour cells in shRNA treated tumours. Stained sections of resected tumours morphologically resembled human alveolar RMS on H&E staining and expressed the common human myogenic markers desmin and myoD (fig 6B–D).

Figure 6 Histology of RMS xenografts. Mice from control and shRNA treated groups were euthanised at the completion of the study. (A) Tumours were visualised for their red fluorescence using a Leica stereoscope equipped with a fluorescent light source at a magnification of ×7.5. (B, C, D) Resected tumours were fixed and paraffin embedded for histological analysis. (B) H&E staining of tumour sections demonstrating the increased incidence tumour giant cells as a result of cytokinesis defects in shRNA treated tumours (arrowheads). (C, D) Myogenic markers desmin (C) and myoD (D) expressed in human RMS xenografts.
Tumours treated with Survivin shRNAs had lower levels of Survivin staining than control treated tumours (16% v 48%; p<0.0005; fig 7A), supporting the hypothesis that the Survivin shRNA cocktail decreased Survivin protein levels in vivo. Furthermore, the levels of staining of Survivin‐ΔEx3 were also decreased in shRNA treated tumours (not shown). A high incidence of mitotic figures consistent with the highly proliferative nature of this type of tumour was observed in control treated mice (fig 7B). Tumours isolated from shRNA treated mice had a much lower incidence of mitotic figures (4.7% v 22.9%, p<0.00001) (fig 7B).

Figure 7 Immunohistochemical analysis of shRNA treated tumours. (A) Tissue sections from shRNA treated and control treated tumours were stained with a polyclonal anti‐Survivin antibody and quantified, as described. (B) Mitotic figures were quantified in multiple fields of similar area. The number of mitotic cells per area is represented on the graph by the circles. The lines represent the mean number of mitotic figures per field and the red bars represent the standard deviations. (C) Proliferation marker Ki‐67. (D) Apoptosis assay TUNEL. (E) Phospho‐histone H3. The percentage of positive cells is shown in the adjacent graphs for each marker. For phospho‐histone H3, the number of positive cells per predetermined area is shown (circles), with the line representing the mean number of cells and the red lines the standard deviation. Arrowheads indicate multinucleated clusters.
To investigate the aetiology of the diminished tumour growth, we assessed proliferation by Ki‐67 immunostaining and programmed cell death by TUNEL staining. We observed a decrease in proliferation (9% v 31%; p<0.005; fig 7C) and an increase in TUNEL staining (7.5% v 1.9%, p = 0.02; fig 7D) in shRNA treated tumours, consistent with the observed growth inhibition of treated tumours. We also assayed for phosphorylated histone H3, a protein phosphorylated by Aurora B kinase, as it is known that Aurora B activity is impaired by Survivin depletion. We observed a statistically significant (41.6% v 23.0%, p<0.0001) reduction in phosphorylated histone H3 in shRNA treated mice relative to control mice (fig 7E). These data further correlate with the observed decrease in mitotic figures in shRNA treated mice, as demonstrated in fig 7B. Multinuclear clusters were detected in all siRNA treated mice, but not in control treated mice, suggesting defective cytokinesis also occurs in vivo. Considering the above results together, the anti‐neoplastic effects of Survivin shRNAs in vivo are primarily due to a marked inhibition of tumour cell proliferation as a consequence of mitotic arrest and defective cytokinesis. These observations are consistent with the known role of Survivin in regulating a mitotic checkpoint.
Discussion
Paediatric RMS is the most common soft tissue sarcoma of childhood with an overall 5 year survival rate of approximately 70% using current multi‐modal therapy.36 The TP53 tumour suppressor gene has been implicated in the pathogenesis of RMS tumours as a number of primary tumours have mutations and/or deletions in this gene.37 Germline mutations in TP53 also occur in up to 10% of young patients with sporadic RMS and in patients with LFS who develop this tumour.37,38 Patients with LFS have a considerably higher incidence of rhabdomyosarcomas and soft tissue sarcomas than the general population,18,28 with population studies placing soft tissue sarcomas as the second most common neoplasm representing between 12 and 21% of all cancers in these patients.39,40 We show here that Survivin is highly expressed in paediatric RMS cell lines harbouring mutations in TP53 and in primary tumour tissue of varying histologies. Among a patient cohort of 63 primary tumours of either ERMS or ARMS histology, at least 80% of the tumours express Survivin at high levels. This identifies a large group of patients with RMS that might benefit from Survivin directed therapies. Disruption of Survivin expression in RMS cell lines through use of a Survivin‐specific shRNA cocktail resulted in a significant increase in tumour cell death. In vivo, disruption of Survivin expression by the same shRNA cocktail resulted in a significant reduction in tumour growth occurring as a consequence of decreased proliferation and increased programmed cell death.
Survivin's role in cell proliferation is primarily as a chromosomal passenger protein, ensuring proper nuclear and cytoplasmic division both directly and indirectly by interacting with partner proteins. Treatment with Survivin inhibitors results in an aberrant localisation of Aurora B kinase during assembly of the anaphase‐promoting complex.41,42 As a consequence, the cell loses its ability to phosphorylate critical mitotic targets, such as histone H3B, vimentin fibres at the cleavage furrow, kinesin‐like motor protein, spindle apparatus proteins, kinetochore protein, and the tumour suppressor protein p53.43,44,45 Aurora kinase inhibitors are currently being explored as therapeutic targets for a number of malignancies and recent reports show good therapeutic responses in pre‐clinical studies using this avenue.24 Our data, in combination with the previously reported findings described above, suggest that treatment of tumours with Survivin shRNAs could also be effective in the clinical treatment of tumours expressing this molecule.
Originally described as an inhibitor of apoptosis protein, Survivin inhibits PCD by interacting directly or indirectly with downstream proteins, thereby inhibiting activation of apoptotic signalling cascades. It would therefore be expected that the function of these proteins would be adversely affected by the elimination of Survivin through shRNA treatment. In addition to its inhibition of Smac/DIABLO,46 a pro‐apoptotic mitochondrial protein, mitochondrial Survivin inhibits the processing of caspase‐3 and caspase‐9 proteins to their active forms.35 Our data show that treatment with a Survivin‐shRNA cocktail activates both caspase‐3 and caspase‐9, suggesting that the shRNA cocktail is able to target the anti‐apoptotic mitochondrial pool of Survivin. Our laboratory also previously demonstrated that Survivin and Survivin‐ΔEx3 interact within the mitochondria to inhibit apoptosis.34 As our Survivin‐shRNA cocktail genetically targets all Survivin isoforms, our strategy should be highly effective at eliminating all tumour promoting activities of the Survivin family. Our results support a role for Survivin antagonists, like the Survivin shRNA cocktail, in therapeutic approaches to Survivin expressing tumours.
Due to its unique properties, Survivin has become a promising molecule for use in targeted biological cancer therapy. Several approaches have recently been utilised to disrupt Survivin activity in vivo, most of which rely on viral delivery of an interfering molecule, such as a dominant negative protein, antisense oligonucleotide, or siRNA. Blanc‐Brude et al demonstrated that a dominant negative form of Survivin delivered by an adenovirus both reduced tumour growth and decreased angiogenesis in a breast cancer xenograft.47 Tu et al exploited the use of both a dominant negative and an antisense molecule against Survivin in the treatment of a xenograft model of gastric cancer.48 In this study, however, the conclusions were based on the ex vivo treatment of the tumour cells, prior to tumour establishment in vivo. Most recently, Uchida et al reported the use of Survivin siRNAs transcribed as an RNA duplex from an adenoviral double promoter construct.49 Treatment of established glioma tumours in vivo resulted in a statistically significant decrease in tumour growth. However, molecular mechanisms for the observed decrease in tumour growth were not delineated.
In exploring the complexities of the delivery of DNA sequences in vivo, we chose a hydrodynamic approach with a promising potential to translate into clinical practice.50,51 Plasmid DNA itself offers an advantage in the ease of manufacturing large quantities for human treatment, and in its long shelf life, as it can be stored for prolonged periods in lyophilised form. Complications associated with the use of viral vectors such as retroviruses, lentiviruses, and human herpes viruses for DNA delivery, including insertional mutagenesis, generation of replication‐competent viruses, and pre‐existing or induced immune responses to viral proteins, are avoided by a hydrodynamic DNA based delivery method. Recently, efficient uptake and expression in skeletal muscle following intramuscular or intravenous hydrodynamic injection of DNA plasmids into rodents and dogs as well as non‐human primates has been demonstrated.50 This procedure has also been shown to effectively deliver DNA to other organs including liver, lungs, and brain.52 Our data support the use of this approach in future pre‐clinical therapeutic experiments and opens the possibility of the use of this modality of Survivin antagonists in malignancies expressing Survivin.
Acknowledgements
The authors thank Michael Boyer for providing the mice used in the study, Florinda Jaynes and Cindy McAllister for assistance with histology and flow cytometry, respectively, and Soledad Fernandez for statistical analysis.
Abbreviations
ARMS - alveolar rhabdomyosarcoma
ERMS - embryonal rhabdomyosarcoma
FFS - failure‐free survival
LFS - Li‐Fraumeni syndrome
RMS - rhabdomyosarcoma
RPE cells - retinal pigment epithelial cells
Footnotes
This work was supported in part by the Columbus Children's Research Institute, and by grants from the Elsa U. Pardee and the Hope Street Kids Foundations
Competing interests: none declared
References
- 1.Li F, Ambrosini G, Chu E Y, Plescia J, Tognin S, Marchisio P C, Altieri D C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998396(6711)580–584. [DOI] [PubMed] [Google Scholar]
- 2.Li F, Ackermann E J, Bennett C F, Rothermel A L, Plescia J, Tognin S, Villa A, Marchisio P C, Altieri D C. Pleiotropic cell‐division defects and apoptosis induced by interference with survivin function. Nat Cell Biol 19991(8)461–466. [DOI] [PubMed] [Google Scholar]
- 3.Uren A G, Wong L, Pakusch M, Fowler K J, Burrows F J, Vaux D L, Choo K H. Survivin and the inner centromere protein INCENP show similar cell‐cycle localization and gene knockout phenotype. Curr Biol 200010(21)1319–1328. [DOI] [PubMed] [Google Scholar]
- 4.Skoufias D A, Mollinari C, Lacroix F B, Margolis R L. Human survivin is a kinetochore‐associated passenger protein. J Cell Biol 2000151(7)1575–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conway E M, Pollefeyt S, Cornelissen J, DeBaere I, Steiner‐Mosonyi M, Ong K, Baens M, Collen D, Schuh A C. Three differentially expressed survivin cDNA variants encode proteins with distinct antiapoptotic functions. Blood 200095(4)1435–1442. [PubMed] [Google Scholar]
- 6.Mahotka C, Wenzel M, Springer E, Gabbert H E, Gerharz C D. Survivin‐deltaEx3 and survivin‐2B: two novel splice variants of the apoptosis inhibitor survivin with different antiapoptotic properties. Cancer Res 199959(24)6097–6102. [PubMed] [Google Scholar]
- 7.Konno R, Yamakawa H, Utsunomiya H, Ito K, Sato S, Yajima A. Expression of survivin and Bcl‐2 in the normal human endometrium. Mol Hum Reprod 20006(6)529–534. [DOI] [PubMed] [Google Scholar]
- 8.Caldas H, Honsey L E, Altura R A. Survivin 2alpha: a novel Survivin splice variant expressed in human malignancies. Mol Cancer 20054(1)11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Badran A, Yoshida A, Ishikawa K, Goi T, Yamaguchi A, Ueda T, Inuzuka M. Identification of a novel splice variant of the human anti‐apoptopsis gene survivin. Biochem Biophys Res Commun 2004314(3)902–907. [DOI] [PubMed] [Google Scholar]
- 10.Fukuda S, Pelus L M. Regulation of the inhibitor‐of‐apoptosis family member survivin in normal cord blood and bone marrow CD34(+) cells by hematopoietic growth factors: implication of survivin expression in normal hematopoiesis. Blood 200198(7)2091–2100. [DOI] [PubMed] [Google Scholar]
- 11.Altura R A, Olshefski R S, Jiang Y, Boue D R. Nuclear expression of Survivin in paediatric ependymomas and choroid plexus tumours correlates with morphologic tumour grade. Br J Cancer 200389(9)1743–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altieri D C. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 200322(53)8581–8589. [DOI] [PubMed] [Google Scholar]
- 13.Fangusaro J, Jiang Y, Holloway M P, Caldas H, Singh V, Boue D R, Hayes J, Altura R A. Survivin, survivin‐2B, and survivin‐deltaEx3 expression in medulloblastoma: biologic markers of tumor morphology and clinical outcome. Br J Cancer 200592(2)359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altieri D C. Validating survivin as a cancer therapeutic target. Nat Rev Cancer 20033(1)46–54. [DOI] [PubMed] [Google Scholar]
- 15.Wexler L H, Crist W M, Helman L J. Rhabdomyosarcoma and the undifferentiated sarcomas. In: Pizzo P, Poplack D, eds. Principles and practice of pediatric oncology. Philadelphia, PA: Lippincott Williams & Wilkins, 2002939–971.
- 16.Lynch H T, Deters C A, Hogg D, Lynch J F, Kinarsky Y, Gatalica Z. Familial sarcoma: challenging pedigrees. Cancer 200398(9)1947–1957. [DOI] [PubMed] [Google Scholar]
- 17.Chompret A. The Li‐Fraumeni syndrome. Biochimie 200284(1)75–82. [DOI] [PubMed] [Google Scholar]
- 18.Diller L, Sexsmith E, Gottlieb A, Li F P, Malkin D. Germline p53 mutations are frequently detected in young children with rhabdomyosarcoma. J Clin Invest 199595(4)1606–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson A A, Harwood F G, Tillman D M, Houghton J A. Selective sensitization to DNA‐damaging agents in a human rhabdomyosarcoma cell line with inducible wild‐type p53 overexpression. Clin Cancer Res 19984(1)145–152. [PubMed] [Google Scholar]
- 20.Barr F G. Molecular genetics and pathogenesis of rhabdomyosarcoma. J Pediatr Hematol Oncol 199719(6)483–491. [DOI] [PubMed] [Google Scholar]
- 21.Dias P, Chen B, Dilday B, Palmer H, Hosoi H, Singh S, Wu C, Li X, Thompson J, Parham D, Qualman S, Houghton P. Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol 2000156(2)399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fangusaro J R, Jiang Y, Holloway M P, Caldas H, Singh V, Boue D R, Hayes J, Altura R A. Survivin, Survivin‐2B, and Survivin‐deItaEx3 expression in medulloblastoma: biologic markers of tumour morphology and clinical outcome. Br J Cancer 200592(2)359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brummelkamp T R, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002296(5567)550–553. [DOI] [PubMed] [Google Scholar]
- 24.Harrington E A, Bebbington D, Moore J, Rasmussen R K, Ajose‐Adeogun A O, Nakayama T, Graham J A, Demur C, Hercend T, Diu‐Hercend A, Su M, Golec J M, Miller K M. VX‐680, a potent and selective small‐molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med 200410(3)262–267. [DOI] [PubMed] [Google Scholar]
- 25.Kappler M, Kotzsch M, Bartel F, Fussel S, Lautenschlager C, Schmidt U, Wurl P, Bache M, Schmidt H, Taubert H, Meye A. Elevated expression level of survivin protein in soft‐tissue sarcomas is a strong independent predictor of survival. Clin Cancer Res 20039(3)1098–1104. [PubMed] [Google Scholar]
- 26.Barr F G, Nauta L E, Davis R J, Schafer B W, Nycum L M, Biegel J A. In vivo amplification of the PAX3‐FKHR and PAX7‐FKHR fusion genes in alveolar rhabdomyosarcoma. Hum Mol Genet 19965(1)15–21. [DOI] [PubMed] [Google Scholar]
- 27.Hazelton B J, Houghton J A, Parham D M, Douglass E C, Torrance P M, Holt H, Houghton P J. Characterization of cell lines derived from xenografts of childhood rhabdomyosarcoma. Cancer Res 198747(16)4501–4507. [PubMed] [Google Scholar]
- 28.Birch J M, Blair V, Kelsey A M, Evans D G, Harris M, Tricker K J, Varley J M. Cancer phenotype correlates with constitutional TP53 genotype in families with the Li‐Fraumeni syndrome. Oncogene 199817(9)1061–1068. [DOI] [PubMed] [Google Scholar]
- 29.Bougeard G, Limacher J M, Martin C, Charbonnier F, Killian A, Delattre O, Longy M, Jonveaux P, Fricker J P, Stoppa‐Lyonnet D, Flaman J M, Frebourg T. Detection of 11 germline inactivating TP53 mutations and absence of TP63 and HCHK2 mutations in 17 French families with Li‐Fraumeni or Li‐Fraumeni‐like syndrome. J Med Genet 200138(4)253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olivier M, Goldgar D E, Sodha N, Ohgaki H, Kleihues P, Hainaut P, Eeles R A. Li‐Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res 200363(20)6643–6650. [PubMed] [Google Scholar]
- 31.Grossman D, McNiff J M, Li F, Altieri D C. Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J Invest Dermatol 1999113(6)1076–1081. [DOI] [PubMed] [Google Scholar]
- 32.O'Connor D S, Wall N R, Porter A C, Altieri D C. A p34(cdc2) survival checkpoint in cancer. Cancer Cell 20022(1)43–54. [DOI] [PubMed] [Google Scholar]
- 33.Carvalho A, Carmena M, Sambade C, Earnshaw W C, Wheatley S P. Survivin is required for stable checkpoint activation in taxol‐treated HeLa cells. J Cell Sci 2003116(Pt 14)2987–2998. [DOI] [PubMed] [Google Scholar]
- 34.Caldas H, Jiang Y, Holloway M P, Fangusaro J, Mahotka C, Conway E M, Altura R A. Survivin splice variants regulate the balance between proliferation and cell death. Oncogene 200524(12)1994–2007. [DOI] [PubMed] [Google Scholar]
- 35.Dohi T, Beltrami E, Wall N R, Plescia J, Altieri D C. Mitochondrial survivin inhibits apoptosis and promotes tumorigenesis. J Clin Invest 2004114(8)1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gurney J G, Young J L, Jr, Roffers S D, Smith M A, Bunin G R. Soft tissue sarcomas. In: Reis LA, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR, eds. Cancer incidence and survival among children and adolescents: United States SEER Program 1975–1995. Vol No. 99‐4649. Bethesda, MD: National Cancer Institute, SEER Program, 1999111–123.
- 37.Felix C A, Kappel C C, Mitsudomi T, Nau M M, Tsokos M, Crouch G D, Nisen P D, Winick N J, Helman L J. Frequency and diversity of p53 mutations in childhood rhabdomyosarcoma. Cancer Res 199252(8)2243–2247. [PubMed] [Google Scholar]
- 38.Malkin D, Li F P, Strong L C, Fraumeni J F, Jr, Nelson C E, Kim D H, Kassel J, Gryka M A, Bischoff F Z, Tainsky M A.et al Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990250(4985)1233–1238. [DOI] [PubMed] [Google Scholar]
- 39.Nichols K E, Malkin D, Garber J E, Fraumeni J F, Jr, Li F P. Germ‐line p53 mutations predispose to a wide spectrum of early‐onset cancers. Cancer Epidemiol Biomarkers Prev 200110(2)83–87. [PubMed] [Google Scholar]
- 40.Chompret A, Brugieres L, Ronsin M, Gardes M, Dessarps‐Freichey F, Abel A, Hua D, Ligot L, Dondon M G, Bressac‐de Paillerets B, Frebourg T, Lemerle J, Bonaiti‐Pellie C, Feunteun J. p53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 200082(12)1932–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J, Jin S, Tahir S K, Zhang H, Liu X, Sarthy A V, McGonigal T P, Liu Z, Rosenberg S H, Ng S C. Survivin enhances Aurora‐B kinase activity and localizes Aurora‐B in human cells. J Biol Chem 2003278(1)486–490. [DOI] [PubMed] [Google Scholar]
- 42.Honda R, Korner R, Nigg E A. Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol Biol Cell 200314(8)3325–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wheatley S P, Henzing A J, Dodson H, Khaled W, Earnshaw W C. Aurora‐B phosphorylation in vitro identifies a residue of survivin that is essential for its localization and binding to inner centromere protein (INCENP) in vivo. J Biol Chem 2004279(7)5655–5660. [DOI] [PubMed] [Google Scholar]
- 44.Ota T, Suto S, Katayama H, Han Z B, Suzuki F, Maeda M, Tanino M, Terada Y, Tatsuka M. Increased mitotic phosphorylation of histone H3 attributable to AIM‐1/Aurora‐B overexpression contributes to chromosome number instability. Cancer Res 200262(18)5168–5177. [PubMed] [Google Scholar]
- 45.Katayama H, Brinkley W R, Sen S. The Aurora kinases: role in cell transformation and tumorigenesis. Cancer Metastasis Rev 200322(4)451–464. [DOI] [PubMed] [Google Scholar]
- 46.Song Z, Yao X, Wu M. Direct interaction between survivin and Smac/DIABLO is essential for the anti‐apoptotic activity of survivin during taxol‐induced apoptosis. J Biol Chem 2003278(25)23130–23140. [DOI] [PubMed] [Google Scholar]
- 47.Blanc‐Brude O P, Mesri M, Wall N R, Plescia J, Dohi T, Altieri D C. Therapeutic targeting of the survivin pathway in cancer: initiation of mitochondrial apoptosis and suppression of tumor‐associated angiogenesis. Clin Cancer Res 20039(7)2683–2692. [PubMed] [Google Scholar]
- 48.Tu S P, Jiang X H, Lin M C, Cui J T, Yang Y, Lum C T, Zou B, Zhu Y B, Jiang S H, Wong W M, Chan A O, Yuen M F, Lam S K, Kung H F, Wong B C. Suppression of survivin expression inhibits in vivo tumorigenicity and angiogenesis in gastric cancer. Cancer Res 200363(22)7724–7732. [PubMed] [Google Scholar]
- 49.Uchida H, Tanaka T, Sasaki K, Kato K, Dehari H, Ito Y, Kobune M, Miyagishi M, Taira K, Tahara H, Hamada H. Adenovirus‐mediated transfer of siRNA against survivin induced apoptosis and attenuated tumor cell growth in vitro and in vivo. Mol Ther 200410(1)162–171. [DOI] [PubMed] [Google Scholar]
- 50.Hagstrom J E, Hegge J, Zhang G, Noble M, Budker V, Lewis D L, Herweijer H, Wolff J A. A facile nonviral method for delivering genes and siRNAs to skeletal muscle of mammalian limbs. Mol Ther 200410(2)386–398. [DOI] [PubMed] [Google Scholar]
- 51.Wells D J. Opening the floodgates: clinically applicable hydrodynamic delivery of plasmid DNA to skeletal muscle. Mol Ther 200410(2)207–208. [DOI] [PubMed] [Google Scholar]
- 52.Lewis D L, Hagstrom J E, Loomis A G, Wolff J A, Herweijer H. Efficient delivery of siRNA for inhibition of gene expression in postnatal mice. Nat Genet 200232(1)107–108. [DOI] [PubMed] [Google Scholar]