

Abstract
Progressive osseous heteroplasia (POH) is a rare, disabling disease of heterotopic ossification (HO) that progresses from skin and subcutaneous tissues into deep skeletal muscle. POH occurs in the absence of multiple developmental features of Albright hereditary osteodystrophy (AHO) or hormone resistance, clinical manifestations that are also associated with GNAS inactivation. However, occasional patients with AHO and pseudohypoparathyroidism 1a/c (PHP1a/c; AHO features plus hormone resistance) have also been described who have progressive HO. This study was undertaken to define the diagnostic and mutational spectrum of POH and progressive disorders of HO, and to distinguish them from related disorders in which HO remains confined to the skin and subcutaneous tissues. We reviewed the charts of 111 individuals who had cutaneous and subcutaneous ossification. All patients were assessed for eight characteristics: age of onset of HO, presence and location of HO, depth of HO, type of HO, progression of HO, features of AHO, PTH resistance, and GNAS mutation analysis. We found, based on clinical criteria that POH and progressive HO syndromes are at the severe end of a phenotypic spectrum of GNAS-inactivating conditions associated with extra-skeletal ossification. While most individuals with superficial or progressive ossification had mutations in GNAS, there were no specific genotype-phenotype correlations that distinguished the more progressive forms of HO (e.g., POH) from the non-progressive forms (osteoma cutis, AHO, and PHP1a/c).
Keywords: progressive osseous heteroplasia (POH), GNAS, Albright hereditary osteodystrophy, osteoma cutis, pseudohypoparathyroidism, heterotopic ossification
INTRODUCTION
Progressive osseous heteroplasia (POH) is a rare genetic condition of progressive ectopic ossification that was first described as a distinct disorder by Kaplan et al. [Kaplan et al., 1994]. Clinically, POH is defined by cutaneous ossification, characteristically presenting during childhood, that progresses to involve subcutaneous and deep connective tissues, including muscle and fascia, in the absence of multiple features of Albright hereditary osteodystrophy (AHO) or hormone resistance. POH is distinguished from fibrodysplasia ossificans progressiva (FOP), another rare autosomal dominant genetic condition of heterotopic ossification (HO), by the occurrence of cutaneous ossification, lack of congenital malformation of the great toes, and the absence of pre-osseous tumor-like inflammation or “flare-ups” [Kaplan et al., 2005; Kaplan et al., 2000]. Unlike FOP, which is caused by a recurrent activating missense mutation of the gene encoding the bone morphogenetic protein (BMP) type I receptor ACVR1 [Shore et al., 2006], most cases of POH are caused by heterozygous inactivating mutations of GNAS, the gene encoding the alpha subunit of the G-stimulatory protein of adenylyl cyclase [Shore et al., 2002].
POH is among a number of related genetic disorders, including Albright hereditary osteodystrophy (AHO), pseudohypoparathyroidism (PHP), and osteoma cutis (OC), that share the common features of sperficial ossification and association with inactivating mutations of GNAS [Bastepe et al., 2005b; Weinstein et al., 2004]. AHO describes a variable constellation of features, in addition to superficial ossification, including short adult stature, obesity, round facies, brachydactyly, neurobehavioral problems (including mental retardation), and superficial dermal ossification. PHP, or end-organ resistance to PTH, is subclassified into types 1a, 1b, and 1c [Bastepe et al., 2005b]. Clinically, PHP1a and 1c are identical and can include presentation with AHO features, defective responses to PTH, and multiple hormone resistance. However, PHP1a is distinguished from PHP1c by the presence of inactivating GNAS mutations and/or reduced activity of Gsα, the major protein product encoded by the GNAS locus. Patients with PHP1b also have hormone resistance, mostly limited to PTH target tissues, but show no AHO features or reduced Gsα activity. PHP1b is commonly associated with a GNAS imprinting defect, which in familial forms is caused by heterozygous deletions of a suspected imprinting control element [Bastepe et al., 2005a; Bastepe et al., 2005b; Bastepe et al., 2001; Jan De Beur et al., 2003; Juppner et al., 1998; Weinstein, 2001a; Weinstein et al., 2001]. Pseudopseudohypoparathyroidism (PPHP) refers to the condition in patients with AHO who have normal target-organ responses to PTH. Osteoma cutis (OC) describes HO that is limited to superficial tissues without any hormone resistance or AHO features.
Most cases of POH, PHP1a, and AHO result from heterozygous inactivating mutations of the GNAS gene, which is regulated by genomic imprinting [Ahmed et al., 1998; Ahrens et al., 2001; Aldred et al., 2000; Farfel et al., 1996; Fischer et al., 1998; Jan De Beur et al., 2003; Linglart et al., 2002; Luttikhuis et al., 1994; Miric et al., 1993; Nakamoto et al., 1998; Nakamoto et al., 1996; Patten et al., 1990; Schwindinger et al., 1994; Shapira et al., 1996; Shore et al., 2002; Walden et al., 1999; Warner et al., 1997; Warner et al., 1998; Weinstein et al., 1992; Weinstein et al., 1990; Wilson et al., 1994; Yokoyama et al., 1996; Yu et al., 1999; Yu et al., 1995]. Maternally-inherited mutations in GNAS lead to PHP1a, whereas paternally-inherited mutations are associated with POH. AHO is more frequently associated with maternally-inherited mutations; AHO caused by a paternally-inherited mutation has been referred to as PPHP.
A few case reports document that POH occasionally presents with additional features previously thought to occur exclusively in other GNAS-associated disorders of HO. Eddy et al. [2000] reported two cases in which patients exhibited progressive HO along with characteristics of AHO (short stature, round face, and brachydactyly) and reduced levels of Gsα protein with or without a heterozygous GNAS mutation. Another patient with progressive HO was described with severe plate-like osteoma cutis and also possessed a mutation in the GNAS gene [Tresserra et al., 1998; Yeh et al., 2000]. These cases support that POH is part of a clinical spectrum of HO disorders that are caused by inactivating GNAS mutations.
The current study was undertaken to examine a large cohort of patients with cutaneous and subcutaneous HO in order to define the clinical and molecular characteristics of POH and other conditions of progressive HO. We have identified criteria that will distinguish these conditions from related disorders in which the heterotopic ossification remains confined to superficial tissues only.
MATERIALS AND METHODS
Patients
We reviewed the charts of 111 individuals who presented to the University of Pennsylvania Orthopaedic Surgery Outpatient Clinic for evaluation of non-traumatic heterotopic ossification of the skin and subcutaneous tissues. Patients with a clear history of trauma-induced HO, or fibrodysplasia ossificans progressiva (FOP), were excluded. All other patients were assessed for eight characteristics associated with GNAS-based disorders of HO: (1) age of onset of HO, (2) presence and location of HO, (3) depth of HO, (4) progression of HO, (5) type of HO (endochondral or intramembraneous), (6) features of AHO, (7) PTH resistance, and (8) GNAS mutation analysis. The study was approved by the Institutional Review Board of the University of Pennsylvania.
Patients were subsequently categorized as having either progressive or superficial (non-progressive) HO. Those with progressive HO in the absence of multiple AHO features were defined as having POH [Kaplan et al, 1994; Kaplan and Shore, 2000). Those with progressive HO and multiple AHO features, in the absence or presence of hormone resistance, were newly defined here as having POH/AHO or POH/PHP 1a/1c, respectively. Patients with non-progressive forms of HO, including AHO, PHP 1a/1c, and osteoma cutis were defined as previously described [Bastepe and Juppner, 2005; Weinstein et al, 2004; Cottoni et al, 1993; Davis et al, 2002].
Evaluation of clinical features
Age of onset of HO was based on the history provided by patients and/or reliable informants at initial presentation. The presence and location of HO was confirmed by CT scan. The depth of HO was confirmed by CT scan or biopsy. Progression of HO was noted clinically, and bony lesions were designated as “progressive” if there was documentation on biopsy or CT scan of the presence of superficial (cutaneous and/or subcutaneous) ossification with extension of bone to within deep connective tissues, (i.e. muscle, tendon, fascia). If the presence of HO was confirmed by punch biopsy, the type of heterotopic ossification (intramembranous, endochondral, or both) was also noted. The presence or absence of AHO features was based on clinical observation of the following characteristics: short stature, obesity, round faces, brachydactyly, neurobehavioral abnormalities including mental retardation, and superficial heterotopic ossification. An endocrine evaluation included a survey of serum calcium, albumin, phosphorus, thyroid stimulating hormone (TSH), and intact parathyroid hormone (PTH) levels.
GNAS Mutation Analysis
Genomic DNA was isolated from blood or lymphoblastoid cell lines (LCLs) using DNA blood-isolation reagents (QIAamp, Qiagen, Valencia, Calif.). If sample size was small, total genomic DNA was amplified with the Qiagen Repli-g Kit. GNAS mutation analysis was conducted by polymerase chain reaction (PCR) amplification of genomic DNA using oligonucleotide primers (Sigma-Genosys; Woodlands, Texas) flanking each of the 13 exons of the human GNAS gene as previously described [Miric et al., 1993; Shore et al., 2002]. PCR reactions used genomic DNA (0.25μg) amplified in a 50 μL volume containing 5mM Tris-HCL (pH 8.3), 25mM KCl, 0.75mM MgCl2, 0.5 μM of each primer, 50μM each of dATP, dCTP, dGTP, and dTTP, and 1.25 U of Taq Polymerase (Invitrogen). For exon 1, PCR reactions were modified by using the Phusion High Fidelity Taq Polymerase (New England BioLabs, Inc., Ipswich, MA) according to the manufacturer's protocol. After an initial denaturation for 1 min at 95° C, 40 amplification cycles consisted of denaturation for 1 min at 94° C, annealing for 1 min at predetermined temperatures, and extension for 2 min at 72° C.
Amplified samples were electrophoresed through 1% agarose gels, stained with ethidium bromide (1μg/mL), and purified using the Qiagen Gene Clean Kit. Eluted products were sequenced by the DNA Sequencing core facilities of the University of Pennsylvania.
Statistical Analysis
Differences in continuous variables were analyzed by Graph Pad Prism 4 using unpaired, two-tailed, Student's t-test analysis with Bonferroni's adjustment. P values less than 0.01 were considered significant. For discontinuous (categorical) variables, the Chi-squared test was performed using double classification.
RESULTS
Clinical characteristics of POH and GNAS-based conditions of heterotopic ossification
We reviewed the charts of 111 patients who presented with heterotopic ossification of superficial tissues. Based on clinical characteristics, this group of 111 patients segregated into six diagnostic categories (Table I): (1) POH; (2) POH/AHO; (3) POH/PHP1a/1c; (4) osteoma cutis (OC); (5) AHO (PPHP); and (6) PHP1a/1c. Superficial (cutaneous/subcutaneous) HO was observed in all patients within each category. Patients with PHPIb were not observed, nor were they expected, since this condition is not associated with HO.
Table I.
Clinical characteristics of POH and other GNAS-based disorders of superficial heterotopic ossification (HO).
| Diagnosis | n | Superficial HO |
Deep HOa |
> 2 AHO Featuresb |
PTH Resistancec |
|---|---|---|---|---|---|
| POH | 52 | + | + | − | − |
| POH/AHO | 6 | + | + | +d | − |
| POH/PHP1a/1c | 5 | + | + | +d | +d |
| Osteoma cutis | 26 | + | − d | − | − |
| AHO | 10 | + | − d | +d | − |
| PHP 1a/1c | 12 | + | − d | +e | +d |
All (+) or no (−) patients within the diagnostic category displayed the indicated characteristic.
Deep HO refers to the extension of superficial (dermal) HO to deep tissues as described in the text.
AHO features included the following characteristics: short stature, obesity, round face, brachydactaly, and neurobehavioral abnormalities. The presence of heterotopic ossification was common to all presentations and excluded here.
An endocrine evaluation included a survey of calcium, phosphorus, TSH, and intact PTH blood levels. Two POH patients had endocrine abnormalities: one had a high TSH, and another had high calcium and phosphate levels. One POH/AHO patient had a high phosphate level. All POH/PHP 1a/1c and PHP 1a/1c patients had PTH resistance or PTH and thyroid hormone resistance. Differences found to be statistically significant when compared to POH are
p value < 0.0001
p value = 0.0002.
Abbreviations are as defined in the text.
POH is characterized by superficial HO that progresses into deeper tissues (Table I). Although some patients exhibit one AHO feature (in addition to HO), in this study all individuals with POH lacked multiple AHO features or hormone resistance (Table I and Table II). Interestingly, none (0/52; 0 percent) of the POH patients were obese, whereas 7/12 patients (58 percent) with PHP1a/1c were obese (Table II). Most POH patients had an average age-of-onset earlier than 1 year (41/52); however, 11 of the 52 POH patients had a much later age-of-onset of 8.8 years (range, 4 to 30 years).
Table II.
AHO features among patients with POH and other GNAS-based disorders of superficial heterotopic ossification.
| Diagnosis | Average no. of AHO features per patient (+/− SD) |
Short Stature (%) |
Obesity (%) |
Round face (%) |
Brachydactyly 1 (%) |
Mental retardation (%) |
|---|---|---|---|---|---|---|
| POH | 0.31 (0.61) | 7.7 | 0.0 | 3.8 | 15.4 | 3.8 |
| POH/AHO | 2.7 (0.5) | 66.7 | 33.3 | 66.7 | 83.3 | 16.6 |
| POH/ PHP1a/1c |
2.2 (1.5) | 80.0 | 40.0 | 20.0 | 40.0 | 40.0 |
| Osteoma cutis |
0.0 (0.0) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| AHO | 2.7 (1.9) | 50.0 | 40.0 | 50.0 | 80.0 | 20.0 |
| PHP1a/1c | 2.6 (1.4) | 50.0 | 58.3 | 66.7 | 66.7 | 8.3 |
We found that a small subset of patients with progressive heterotopic ossification (6/63; 9.5%) shared multiple features with AHO patients (POH/AHO), while another small subset (5/63; 7.9%) also had PTH resistance (POH/PHP1a/1c). We refer to POH/AHO and POH/PHP1a/1c as “progressive HO” syndromes, since the ectopic ossification in these patients is clinically progressive and indistinguishable from that seen in classic POH patients (Table I). Patients in the POH/AHO group presented with progressive HO and more than two AHO features (other than HO). Those in the POH/PHP1a/1c group displayed progressive HO and AHO features, but additionally presented with hormone resistance (Table I).
Patients with AHO/PPHP, osteoma cutis (OC), and PHP1a/1c who presented with extensive superficial HO either had dermal lesions that arose in multiple locations or had single or few lesions that expanded cutaneously, but never progressed to deeper tissue. Among these three groups (OC; AHO; or PHP1a/1c), patients were distinguished on the basis of AHO features and endocrine evaluation (Table I). Patients with OC were distinguished by the absence of AHO features (other than HO) and the absence of hormone resistance. While AHO patients (PPHP) also lack endocrine abnormalities, they exhibit characteristic features of an abnormal body habitus, with or without neurobehavioral abnormalities. PHP1a/1c patients have AHO features along with PTH resistance (Table I).
We found that neither the type of ossification (endochondral or intramembranous) nor the number of HO sites was helpful in distinguishing among these disorders of ectopic bone formation. In about 15% of all presenting patients with superficial HO, lesional biopsies were performed, with approximately 70% of all biopsied lesions (12/17) showing exclusive intramembraneous ossification and the remainder demonstrating either endochondral ossification (12%) or both intramembraneous and endochondral ossification (18%). Among POH patients studied, almost 20% (10/52) had lesional biopsies, with half demonstrating intramembraneous ossification, 20% showing endochondral ossification, and 30% both types. The number of discrete HO lesional sites varied from an average of four in patients with osteoma cutis to ten in patients with POH/PHP1a/1c, but no statistically significant differences were found among groups of GNAS-based disorders of superficial ossification. With respect to location of ossification, we were not able to identify any anatomical site(s) that were consistently associated with any specific disorder.
GNAS mutational analysis and phenotypic variability (variable expressivity) of POH
GNAS mutation analysis was conducted for 48/52 (92%) of POH patients in this study, and mutations were identified in 31/48 (64%) of patients. Those individuals without detectable mutations were clinically indistinguishable from those with mutations. Patients with POH showed mutations in several GNAS exons (Fig. 1). Our studies also detected a previously reported single nucleotide polymorphism located in exon 5, c. 393 T > C (I131I) [Miric et al., 1993].
Fig. 1.
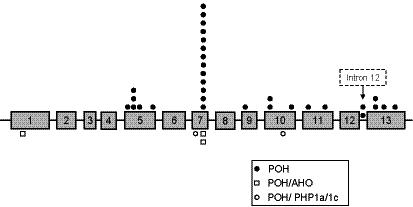
Distribution of GNAS mutations in POH and other conditions of progressive HO. Exons for the GNAS gene (Gsα mRNA) are identified by numbers and are approximately drawn to scale. Intronic sequences are represented by straight solid lines between exons. Information presented is from this study; recent reports of GNAS mutations in AHO and PHP1a/1c also show a wide distribution throughout the GNAS exons (e.g., Jan De Beur et al. [2003]; Linglart et al. [2002]; Aldred et al. [2000]). Symbols represent patients within each diagnostic group whose GNAS sequence analysis revealed specific mutations at the indicated approximate locations. A “hot spot” (4 base pair deletion) causing a frameshift mutation at c.565-568 occurs in exon 7.
The same GNAS mutation can present with variable severity and pleiotropy (phenotypic diversity), both for unrelated sporadic cases and within affected families who have more similar genetic backgrounds than unrelated individuals. For example, two families with a frameshift mutation in exon 7, a c.565-8 four base pair deletion (Fig. 2), show phenotypic variation of POH characteristics (Fig. 2A, B). In one family (Fig. 2A), each person with the mutation had POH with brachydactyly, suggesting contributions of similar genetic modifier loci within this family. However, in the second family (Fig. 2B), the same mutation produced mild POH in the father and delayed onset of more extensive ossification in an affected son. Neither the father nor affected son had brachydactyly. A second son was a non-penetrant carrier of the mutation at the time of presentation.
Fig. 2.
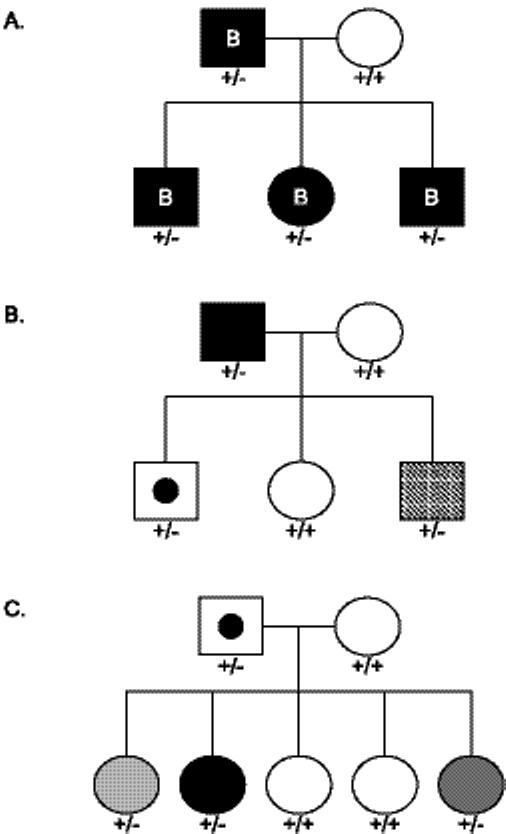
Family pedigrees showing phenotypic variability of POH. A and B, Phenotypic variability of POH in affected individuals with the same exon 7 4 bp deletion. All affected individuals in families A and B have POH and the same 4 bp deletion in exon 7 (as does the non-penetrant carrier in family B). Despite having the same GNAS mutation, the affected individuals express different clinical/phenotypic characteristics. In family A, affected individuals have brachdactyly (denoted by the letter B inside the filled symbol), a typical feature of AHO, whereas affected members of family B do not posses any AHO features. Furthermore, the affected offspring in family B is a late-onset POH variant (+/−; diagonally-shaded square) and his male sibling was a non-penetrant carrier (+/−; square with filled circle) at the time of presentation. C, Family pedigree showing non-penetrance and variability of severity of the POH phenotype. The father is a non-penetrant carrier of a mutant allele (c.725 del C in codon 242) which was inherited by three of his daughters. Each affected offspring clinically exhibits different degrees of severity, as represented by intensity of shading (dark to light represent more to less severe). These families were originally reported in Shore et al. [2002] and are shown here with respect to phenotypic variation. Unaffected (+/+; unfilled symbol); Affected heterozygote (+/−; filled symbol).
A further example of variability within a family is represented in Figure 2C. The father is a non-penetrant carrier of a GNAS exon 10 deletion (c. 725 del C) that was inherited by three of his five children. Although all three children have the same mutation, they exhibit varying degrees of severity based on the extent of progressive HO lesions and resultant functional impairment.
In addition to unique (‘private’) GNAS mutations in POH patients, we found a frameshift mutation located in exon 7, a four base pair deletion of c.565-8, in 13 POH cases (10 familial cases among 3 different families, and 3 individual spontaneous cases). This relatively common deletion “hotspot” has been previously reported in patients with AHO, PHP1a, and OC [Ahmed et al., 1998; Ahrens et al., 2001; Linglart et al., 2002; Nakamoto et al., 1996; Walden et al., 1999; Weinstein et al., 1992; Yokoyama et al., 1996].
Our findings do not identify genotype-phenotype correlations that differentiate POH or other progressive HO syndromes, including POH/AHO and POH/PHP 1a/1c (Fig. 1), from any of the GNAS-based conditions associated with more superficial HO. We performed GNAS mutational analysis on 27/48 individuals with non-progressive HO, and found mutations in 2/6 patients with AHO, in 5/7 with PHP 1a/1c, and in 3/14 with osteoma cutis (data not shown). Except for the non-penetrant carriers depicted in Fig. 2, unaffected family members did not have detectable GNAS mutations (data not shown).
DISCUSSION
In this study, we extensively analyzed clinical features and GNAS mutations in a large group of patients with heterotopic ossification of the skin and subcutaneous tissues. This non-random group of patients were self-selected for referral to an orthopaedic clinic, so it is no surprise that most individuals in our study group had POH or progressive HO syndromes (63/111;58%) rather than more superficial and non-progressive forms of heterotopic ossification (as in AHO, PHP1a, or osteoma cutis). Nevertheless, our data, together with previously published reports, support that the range of these disorders of superficial ossification is commonly associated with heterozygous inactivating GNAS mutations. Our data further show that these related disorders can be distinguished solely by clinical criteria.
Based on our evaluation, we broadly divide GNAS-based disorders of HO into those presenting with stable superficial bony lesions and those whose superficial lesions progress into deep connective tissue (Table I and Fig. 3). Among the non-progressive forms are osteoma cutis, AHO/PPHP, and PHP1a/c. The progressive types of GNAS-based HO are POH and the POH-related syndromes (Table I and Fig. 3). POH presents with superficial HO that progresses to deeper tissues in the absence of multiple other AHO features and without hormone resistance (Table I and Fig. 3). A small proportion of patients with progressive HO present with more extensive AHO features (POH/AHO) or with both AHO features and hormone resistance (POH/PHP1a/1c). It is possible that individuals without progressive HO could be too young at the time of initial diagnosis to have yet developed progressive HO. Similarly, individuals with POH could be too young at the time of diagnosis to have yet developed other features of AHO. Despite these formal possibilities, our study demonstrates that POH resides within a spectrum of diseases caused by inactivating mutations of GNAS (Fig. 4) with POH and progressive HO syndromes at the far end of the phenotypic spectrum of GNAS-based disorders of extra-skeletal ossification (Fig. 4).
Fig. 3.
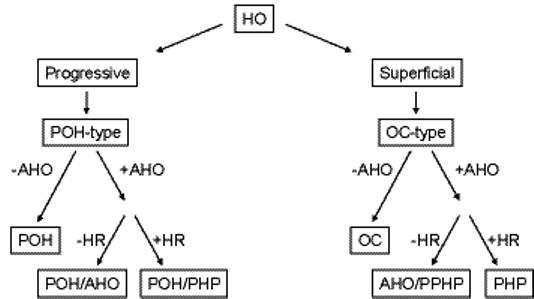
Algorithmic approach to the differential diagnosis of GNAS-based disorders of superficial heterotopic ossification. HR, hormone (PTH) resistance; other abbreviations are as defined in the text.
Fig. 4.
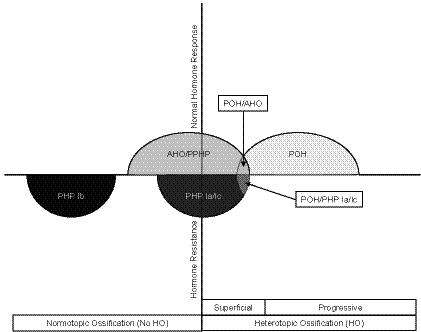
Spectrum of POH and other GNAS-based conditions of superficial HO. Conditions that are associated with heterozygous inactivating GNAS mutations are depicted along axes of heterotopic ossification and hormone resistance. PHP1b and PHP1a/1c are associated with hormone resistance and no or variable/occasional superficial heterotopic ossification (HO), respectively. AHO (PPHP) and POH display no hormone resistance. Superficial HO is a variable feature of AHO (PPHP) while progressive HO is characteristic of POH. Overlapping syndromes (POH/AHO and POH/PHP1a/1c) are also indicated.
Although some POH patients possess limited AHO features, they are never obese. Weinstein et al. [2001] reported mouse models with heterozygous disruption of GNAS exon 2 that are paternally (+/p−) or maternally inherited (m−/+) which have contrasting metabolic phenotypes. The (+/p−) mice are very lean, hypermetabolic, and hyperactive; conversely, (m−/+) mice are obese, hypometabolic, and hypoactive. Since familial POH cases previously reported were all paternally inherited [Shore et al., 2002], and our current study failed to demonstrate any obese POH patients, these findings suggest that fat stores and metabolic activity are related to the parental allele expression of GNAS in both mouse and human. Our data on maternal versus paternal transmission of POH are limited, but thus far suggests exclusive paternal transmission (data not shown). These observations are consistent with recent findings which indicate that obesity is associated with maternal transmission, and may be considered a feature of PHP1a and associated with hormone resistance rather than a general feature of AHO [Long et al., 2007]. Our data showing that a majority of PHP1a/1c patients are obese supports this idea.
The presence of inactivating GNAS mutations cannot distinguish POH or progressive HO syndromes from the GNAS-based conditions that present with more superficial forms of HO, nor can it predict whether patients with superficial HO in infancy will progress to develop POH. Additionally, there is no specific mutation, domain-localizing cluster, or set of mutations that can predict or establish a clinical diagnosis in any patient with a GNAS-mediated disorder of heterotopic ossification.
It is well-established that phenotypic expression of autosomal dominant Mendelian disorders can be variable [Chiba-Falek et al., 2001; Nicholls et al., 1998; Shore et al., 2002; Stoll et al., 2000]. Patients with heterozygous inactivating mutations in GNAS, including the spectrum of disorders characterized above as POH, POH-related disorders, AHO, PHP1a/1c, and OC, are consistent with such observations.
One possible explanation for this phenotypic variation is epigenetic regulation. For example, differential DNA methylation appears to be an important epigenetic factor that influences expression from the GNAS locus and governs parental imprinting [Bastepe et al., 2003; Coombes et al., 2003; El-Maarri et al., 2003; Hayward et al., 1998a; Hayward et al., 1998b; Liu et al., 2005; Liu et al., 2000; Weinstein, 2001b]. An alternative possibility is that complex regulatory mechanisms may contribute to phenotypic variation. Direct transcriptional regulation of GNAS has recently been described by Bertaux et al. [2006], indicating that Gsα expression may be affected by Runx2 via a protein-DNA complex formed in the promoter region of Gsα, and suggesting that mutations in the promoter region of GNAS would disrupt important regulatory pathways.
Other influences on variable gene expression and phenotypic variation may arise from effects of genes outside of the GNAS locus or may be due to environmental (non-genetic) factors, although evidence of the former is not established despite reports of AHO-like and PHP1a-like cases that are associated with chromosome 2 or chromosome 22 deletions, respectively [Bertaux et al., 2006; Craigen et al., 1997; Phelan et al., 1995]. In this context, it is important to note that the GNAS promoter and regulatory regions, as well as other genes known to regulate Gsα expression, are not well-defined and were not investigated for mutations in affected individuals within this study nor in other previous reports, to our knowledge.
In summary, this report clearly establishes that POH and progressive HO syndromes can be distinguished from other GNAS-based disorders by one clinical parameter alone: the extension of HO from superficial to deep tissue. We also demonstrate that the presence of GNAS inactivating mutations is important in establishing a distinct genetic basis for this spectrum of disorders of HO. However, our data further indicate that GNAS inactivating mutations, either by presence alone or by mutation pattern within GNAS, do not predict a specific disorder, variability of phenotype, or severity of progression within this spectrum.
ACKNOWLEDGMENTS
The authors wish to acknowledge Krystyna Knight for technical assistance in the isolation of DNA from peripheral lymphocytes, as well as the services of the DNA Sequencing core facilities of the University of Pennsylvania, in the School of Veterinary Medicine and School of Medicine. This work was supported by the National Institutes of Health (NIH R01-AR46831 to EMS), the Progressive Osseous Heteroplasia Association (POHA), the International Fibrodysplasia Ossificans Progressiva Association (IFOPA), the Ian Cali Fund, the University of Pennsylvania Center for Research in FOP and Related Disorders, and the Isaac and Rose Nassau Professorship of Orthopaedic Molecular Medicine (to FSK).
REFERENCES
- Ahmed SF, Dixon PH, Bonthron DT, Stirling HF, Barr DG, Kelnar CJ, Thakker RV. GNAS1 mutational analysis in pseudohypoparathyroidism. Clin Endocrinol (Oxf) 1998;49:525–531. [PubMed] [Google Scholar]
- Ahrens W, Hiort O, Staedt P, Kirschner T, Marschke C, Kruse K. Analysis of the GNAS1 gene in Albright's hereditary osteodystrophy. J Clin Endocrinol Metab. 2001;86:4630–4634. doi: 10.1210/jcem.86.10.7946. [DOI] [PubMed] [Google Scholar]
- Aldred MA, Trembath RC. Activating and inactivating mutations in the human GNAS1 gene. Hum Mutat. 2000;16:183–189. doi: 10.1002/1098-1004(200009)16:3<183::AID-HUMU1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Bastepe M, Frohlich LF, Hendy GN, Indridason OS, Josse RG, Koshiyama H, Korkko J, Nakamoto JM, Rosenbloom AL, Slyper AH, Sugimoto T, Tsatsoulis A, Crawford JD, Juppner H. Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J Clin Invest. 2003;112:1255–1263. doi: 10.1172/JCI19159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastepe M, Frohlich LF, Linglart A, Abu-Zahra HS, Tojo K, Ward LM, Juppner H. Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypoparathyroidism type Ib. Nat Genet. 2005a;37:25–27. doi: 10.1038/ng1487. [DOI] [PubMed] [Google Scholar]
- Bastepe M, Juppner H. GNAS locus and pseudohypoparathyroidism. Horm Res. 2005b;63:65–74. doi: 10.1159/000083895. [DOI] [PubMed] [Google Scholar]
- Bastepe M, Lane AH, Juppner H. Paternal uniparental isodisomy of chromosome 20q--and the resulting changes in GNAS1 methylation--as a plausible cause of pseudohypoparathyroidism. Am J Hum Genet. 2001;68:1283–1289. doi: 10.1086/320117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertaux K, Broux O, Chauveau C, Hardouin P, Jeanfils J, Devedjian JC. Runx2 regulates the expression of GNAS on SaOs-2 cells. Bone. 2006;38:943–50. doi: 10.1016/j.bone.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Chiba-Falek O, Nussbaum RL. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum Mol Genet. 2001;10:3101–3109. doi: 10.1093/hmg/10.26.3101. [DOI] [PubMed] [Google Scholar]
- Coombes C, Arnaud P, Gordon E, Dean W, Coar EA, Williamson CM, Feil R, Peters J, Kelsey G. Epigenetic properties and identification of an imprint mark in the Nesp-Gnasxl domain of the mouse Gnas imprinted locus. Mol Cell Biol. 2003;23:5475–5488. doi: 10.1128/MCB.23.16.5475-5488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craigen WJ, Lindsay EA, Bricker JT, Hawkins EP, Baldini A. Deletion of chromosome 22q11 and pseudohypoparathyroidism. Am J Med Genet. 1997;72:63–65. [PubMed] [Google Scholar]
- Eddy MC, Jan De Beur SM, Yandow SM, McAlister WH, Shore EM, Kaplan FS, Whyte MP, Levine MA. Deficiency of the alpha-subunit of the stimulatory G protein and severe extraskeletal ossification. J Bone Miner Res. 2000;15:2074–2083. doi: 10.1359/jbmr.2000.15.11.2074. [DOI] [PubMed] [Google Scholar]
- El-Maarri O, Seoud M, Coullin P, Herbiniaux U, Oldenburg J, Rouleau G, Slim R. Maternal alleles acquiring paternal methylation patterns in biparental complete hydatidiform moles. Hum Mol Genet. 2003;12:1405–1413. doi: 10.1093/hmg/ddg152. [DOI] [PubMed] [Google Scholar]
- Farfel Z, Iiri T, Shapira H, Roitman A, Mouallem M, Bourne HR. Pseudohypoparathyroidism, a novel mutation in the betagamma-contact region of Gsalpha impairs receptor stimulation. J Biol Chem. 1996;271:19653–19655. doi: 10.1074/jbc.271.33.19653. [DOI] [PubMed] [Google Scholar]
- Fischer JA, Egert F, Werder E, Born W. An inherited mutation associated with functional deficiency of the alpha-subunit of the guanine nucleotide-binding protein Gs in pseudo- and pseudopseudohypoparathyroidism. J Clin Endocrinol Metab. 1998;83:935–938. doi: 10.1210/jcem.83.3.4656. [DOI] [PubMed] [Google Scholar]
- Hayward BE, Kamiya M, Strain L, Moran V, Campbell R, Hayashizaki Y, Bonthron DT. The human GNAS1 gene is imprinted and encodes distinct paternally and biallelically expressed G proteins. Proc Natl Acad Sci U S A. 1998a;95:10038–10043. doi: 10.1073/pnas.95.17.10038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward BE, Moran V, Strain L, Bonthron DT. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc Natl Acad Sci U S A. 1998b;95:15475–15480. doi: 10.1073/pnas.95.26.15475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan De Beur SM, O'Connell JR, Peila R, Cho J, Deng Z, Kam S, Levine MA. The pseudohypoparathyroidism type lb locus is linked to a region including GNAS1 at 20q13.3. J Bone Miner Res. 2003;18:424–433. doi: 10.1359/jbmr.2003.18.3.424. [DOI] [PubMed] [Google Scholar]
- Juppner H, Schipani E, Bastepe M, Cole DE, Lawson ML, Mannstadt M, Hendy GN, Plotkin H, Koshiyama H, Koh T, Crawford JD, Olsen BR, Vikkula M. The gene responsible for pseudohypoparathyroidism type Ib is paternally imprinted and maps in four unrelated kindreds to chromosome 20q13.3. Proc Natl Acad Sci U S A. 1998;95:11798–11803. doi: 10.1073/pnas.95.20.11798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan FS, Craver R, MacEwen GD, Gannon FH, Finkel G, Hahn G, Tabas J, Gardner RJ, Zasloff MA. Progressive osseous heteroplasia: a distinct developmental disorder of heterotopic ossification. Two new case reports and follow-up of three previously reported cases. J Bone Joint Surg Am. 1994;76:425–436. [PubMed] [Google Scholar]
- Kaplan FS, Glaser DL, Shore EM, Deirmengian GK, Gupta R, Delai P, Morhart R, Smith R, Le Merrer M, Rogers JG, Connor JM, Kitterman JA. The phenotype of fibrodyplasia ossificans progressiva. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:183–188. [Google Scholar]
- Kaplan FS, Shore EM. Progressive osseous heteroplasia. J Bone Miner Res. 2000;15:2084–2094. doi: 10.1359/jbmr.2000.15.11.2084. [DOI] [PubMed] [Google Scholar]
- Linglart A, Carel JC, Garabedian M, Le T, Mallet E, Kottler ML. GNAS1 lesions in pseudohypoparathyroidism Ia and Ic: genotype phenotype relationship and evidence of the maternal transmission of the hormonal resistance. J Clin Endocrinol Metab. 2002;87:189–197. doi: 10.1210/jcem.87.1.8133. [DOI] [PubMed] [Google Scholar]
- Liu J, Nealon JG, Weinstein LS. Distinct patterns of abnormal GNAS imprinting in familial and sporadic pseudohypoparathyroidism type IB. Hum Mol Genet. 2005;14:95–102. doi: 10.1093/hmg/ddi009. [DOI] [PubMed] [Google Scholar]
- Liu J, Yu S, Litman D, Chen W, Weinstein LS. Identification of a methylation imprint mark within the mouse Gnas locus. Mol Cell Biol. 2000;20:5808–5817. doi: 10.1128/mcb.20.16.5808-5817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DN, McGuire S, Levine MA, Weinstein LS, Germain-Lee EL. Body Mass Index Differences in Pseudohypoparathyroidism Type 1a Versus Pseudopseudohypoparathyroidism May Implicate Paternal Imprinting of G{alpha}s in the Development of Human Obesity. J Clin Endocrinol Metab. 2007;92:1073–1079. doi: 10.1210/jc.2006-1497. [DOI] [PubMed] [Google Scholar]
- Luttikhuis ME, Wilson LC, Leonard JV, Trembath RC. Characterization of a de novo 43-bp deletion of the Gs alpha gene (GNAS1) in Albright hereditary osteodystrophy. Genomics. 1994;21:455–457. doi: 10.1006/geno.1994.1297. [DOI] [PubMed] [Google Scholar]
- Miric A, Vechio JD, Levine MA. Heterogeneous mutations in the gene encoding the alpha-subunit of the stimulatory G protein of adenylyl cyclase in Albright hereditary osteodystrophy. J Clin Endocrinol Metab. 1993;76:1560–1568. doi: 10.1210/jcem.76.6.8388883. [DOI] [PubMed] [Google Scholar]
- Nakamoto JM, Sandstrom AT, Brickman AS, Christenson RA, Van Dop C. Pseudohypoparathyroidism type Ia from maternal but not paternal transmission of a Gsalpha gene mutation. Am J Med Genet. 1998;77:261–267. [PubMed] [Google Scholar]
- Nakamoto JM, Zimmerman D, Jones EA, Loke KY, Siddiq K, Donlan MA, Brickman AS, Van Dop C. Concurrent hormone resistance (pseudohypoparathyroidism type Ia) and hormone independence (testotoxicosis) caused by a unique mutation in the G alpha s gene. Biochem Mol Med. 1996;58:18–24. doi: 10.1006/bmme.1996.0027. [DOI] [PubMed] [Google Scholar]
- Nicholls RD, Saitoh S, Horsthemke B. Imprinting in Prader-Willi and Angelman syndromes. Trends Genet. 1998;14:194–200. doi: 10.1016/s0168-9525(98)01432-2. [DOI] [PubMed] [Google Scholar]
- Patten JL, Johns DR, Valle D, Eil C, Gruppuso PA, Steele G, Smallwood PM, Levine MA. Mutation in the gene encoding the stimulatory G protein of adenylate cyclase in Albright's hereditary osteodystrophy. N Engl J Med. 1990;322:1412–1419. doi: 10.1056/NEJM199005173222002. [DOI] [PubMed] [Google Scholar]
- Phelan MC, Rogers RC, Clarkson KB, Bowyer FP, Levine MA, Estabrooks LL, Severson MC, Dobyns WB. Albright hereditary osteodystrophy and del(2) (q37.3) in four unrelated individuals. Am J Med Genet. 1995;58:1–7. doi: 10.1002/ajmg.1320580102. [DOI] [PubMed] [Google Scholar]
- Schwindinger WF, Miric A, Zimmerman D, Levine MA. A novel Gs alpha mutant in a patient with Albright hereditary osteodystrophy uncouples cell surface receptors from adenylyl cyclase. J Biol Chem. 1994;269:25387–25391. [PubMed] [Google Scholar]
- Shapira H, Mouallem M, Shapiro MS, Weisman Y, Farfel Z. Pseudohypoparathyroidism type Ia: two new heterozygous frameshift mutations in exons 5 and 10 of the Gs alpha gene. Hum Genet. 1996;97:73–75. doi: 10.1007/BF00218836. [DOI] [PubMed] [Google Scholar]
- Shore EM, Ahn J, Jan de Beur S, Li M, Xu M, Gardner RJ, Zasloff MA, Whyte MP, Levine MA, Kaplan FS. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med. 2002;346:99–106. doi: 10.1056/NEJMoa011262. [DOI] [PubMed] [Google Scholar]
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- Stoll C, Javier MR, Bellocq JP. Progressive osseous heteroplasia: an uncommon cause of ossification of soft tissues. Ann Genet. 2000;43:75–80. doi: 10.1016/s0003-3995(00)00020-4. [DOI] [PubMed] [Google Scholar]
- Tresserra L, Tresserra F, Grases PJ, Badosa J, Tresserra M. Congenital plate-like osteoma cutis of the forehead: an atypical presentation form. J Craniomaxillofac Surg. 1998;26:102–106. doi: 10.1016/s1010-5182(98)80048-6. [DOI] [PubMed] [Google Scholar]
- Walden U, Weissortel R, Corria Z, Yu D, Weinstein L, Kruse K, Dorr HG. Stimulatory guanine nucleotide binding protein subunit 1 mutation in two siblings with pseudohypoparathyroidism type 1a and mother with pseudopseudohypoparathyroidism. Eur J Pediatr. 1999;158:200–203. doi: 10.1007/s004310051048. [DOI] [PubMed] [Google Scholar]
- Warner DR, Gejman PV, Collins RM, Weinstein LS. A novel mutation adjacent to the switch III domain of G(S alpha) in a patient with pseudohypoparathyroidism. Mol Endocrinol. 1997;11:1718–1727. doi: 10.1210/mend.11.11.0013. [DOI] [PubMed] [Google Scholar]
- Warner DR, Weng G, Yu S, Matalon R, Weinstein LS. A novel mutation in the switch 3 region of Gsalpha in a patient with Albright hereditary osteodystrophy impairs GDP binding and receptor activation. J Biol Chem. 1998;273:23976–23983. doi: 10.1074/jbc.273.37.23976. [DOI] [PubMed] [Google Scholar]
- Weinstein LS. The stimulatory G protein alpha-subunit gene: mutations and imprinting lead to complex phenotypes. J Clin Endocrinol Metab. 2001a;86:4622–4626. doi: 10.1210/jcem.86.10.8007. [DOI] [PubMed] [Google Scholar]
- Weinstein LS. The role of tissue-specific imprinting as a source of phenotypic heterogeneity in human disease. Biol Psychiatry. 2001b;50:927–931. doi: 10.1016/s0006-3223(01)01295-1. [DOI] [PubMed] [Google Scholar]
- Weinstein LS, Gejman PV, de Mazancourt P, American N, Spiegel AM. A heterozygous 4-bp deletion mutation in the Gs alpha gene (GNAS1) in a patient with Albright hereditary osteodystrophy. Genomics. 1992;13:1319–1321. doi: 10.1016/0888-7543(92)90056-x. [DOI] [PubMed] [Google Scholar]
- Weinstein LS, Gejman PV, Friedman E, Kadowaki T, Collins RM, Gershon ES, Spiegel AM. Mutations of the Gs alpha-subunit gene in Albright hereditary osteodystrophy detected by denaturing gradient gel electrophoresis. Proc Natl Acad Sci U S A. 1990;87:8287–82890. doi: 10.1073/pnas.87.21.8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein LS, Liu J, Sakamoto A, Xie T, Chen M. Minireview: GNAS: normal and abnormal functions. Endocrinology. 2004;145:5459–5464. doi: 10.1210/en.2004-0865. [DOI] [PubMed] [Google Scholar]
- Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001;22:675–705. doi: 10.1210/edrv.22.5.0439. [DOI] [PubMed] [Google Scholar]
- Wilson LC, Oude Luttikhuis ME, Clayton PT, Fraser WD, Trembath RC. Parental origin of Gs alpha gene mutations in Albright's hereditary osteodystrophy. J Med Genet. 1994;31:835–839. doi: 10.1136/jmg.31.11.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh GL, Mathur S, Wivel A, Li M, Gannon FH, Ulied A, Audi L, Olmstead EA, Kaplan FS, Shore EM. GNAS1 mutation and Cbfa1 misexpression in a child with severe congenital platelike osteoma cutis. J Bone Miner Res. 2000;15:2063–2073. doi: 10.1359/jbmr.2000.15.11.2063. [DOI] [PubMed] [Google Scholar]
- Yokoyama M, Takeda K, Iyota K, Okabayashi T, Hashimoto K. A 4-base pair deletion mutation of Gs alpha gene in a Japanese patient with pseudohypoparathyroidism. J Endocrinol Invest. 1996;19:236–241. doi: 10.1007/BF03349874. [DOI] [PubMed] [Google Scholar]
- Yu D, Yu S, Schuster V, Kruse K, Clericuzio CL, Weinstein LS. Identification of two novel deletion mutations within the Gs alpha gene (GNAS1) in Albright hereditary osteodystrophy. J Clin Endocrinol Metab. 1999;84:3254–3259. doi: 10.1210/jcem.84.9.5970. [DOI] [PubMed] [Google Scholar]
- Yu S, Yu D, Hainline BE, Brener JL, Wilson KA, Wilson LC, Oude-Luttikhuis ME, Trembath RC, Weinstein LS. A deletion hot-spot in exon 7 of the Gs alpha gene (GNAS1) in patients with Albright hereditary osteodystrophy. Hum Mol Genet. 1995;4:2001–2002. doi: 10.1093/hmg/4.10.2001. [DOI] [PubMed] [Google Scholar]