

Abstract
A xerogel-based optical pH sensor capable of releasing low levels of nitric oxide (NO) and measuring changes in solution pH is reported. Through simple dip-coating procedures, aminoalkoxysilane-based xerogel films modified with N-diazeniumdiolate NO donor precursors and the fluorescent pH indicator seminaphthorhodamine-1 carboxylate (SNARF-1) were sequentially deposited onto optical fibers. The resulting sensors were characterized by fast and linear response to pH throughout the physiological range (pH 7.0-7.8). Real-time chemiluminescence measurements confirmed the presence of the overlying SNARF-1 containing TMOS layer did not have an inhibitory effect on N-diazeniumdiolate formation or NO release, and the NO-releasing coatings were capable of maintaining NO fluxes >0.4 pmol/cm2s up to 16 h. In vitro blood compatibility studies using porcine platelets confirmed the expected thromboresistivity of the NO-releasing xerogel coatings.
INTRODUCTION
Blood pH, when measured in conjunction with PCO2, provides a useful window into respiratory, cardiovascular, and renal function.1, 2 Strong shifts from arterial pH values of 7.4 due to alkalosis and acidosis conditions are often indicative of respiratory or metabolic distress. During thoracic surgeries and mechanical lung ventilation, blood gases (e.g., PO2, PCO2) and pH may fluctuate rapidly.3 Throughout these critical periods, the common clinical practice of periodic arterial blood sampling followed by bench-top laboratory blood gas analysis does not provide an adequately fast means of tracking respiratory function. As such, real-time analysis strategies must be employed. Techniques such as capnometry and transcutaneous blood gas measurement provide real-time detection, but have important limitations. Capnometry requires normal pulmonary function to correctly estimate CO2 concentration, which can lead to unreliable readings in critically-ill patients.3 Transcutaneous sensors are often hampered by skin and tissue variations in adult subjects.4 These issues may be circumvented by direct determination of blood gases through the implantation of miniaturized catheter sensors or sensor arrays into the arterial blood stream.
To date, most intravascular blood gas sensors have been based on amperometric (e.g., Clark-style dissolved oxygen electrodes) or fiber-optic-based photochemical platforms.5 Fiber-optic sensors may have greater appeal because of their high sensitivity, reduced electrical interference, and ease of miniaturization for incorporation into bundled arrays.6, 7 Analyte concentrations are measured via their interaction with transducer dye molecules immobilized at the fiber surface, typically resulting in changes of absorbance or fluorescence intensity, lifetime, or phase.8 For example, the Paratrend 7+® (Diametrics Medical Inc.), a multiparameter continuous intravascular blood gas monitoring probe commercially available from 1999 to 2004, employed a bundle of 3 optical fiber probes modified to measure PO2 via ruthenium dye fluorescence quenching, PCO2 and pH via phenol red absorbance, and temperature via a thermocouple. Clinical trials confirmed that the real-time measurements made by these optical sensors compared well with intermittent laboratory blood gas analysis, the current clinical standard.3, 5, 9
Despite favorable results in most cases, blood clot formation at the surface of fiber-optic intravascular sensors has been reported.10, 11 Platelet adhesion and aggregation at a sensor limits analyte diffusion to the transduction element, rendering the sensor largely inoperable.12 At worst, subsequent dislodgement of the thrombus may lead to potentially serious health effects such as vessel occlusion. Recent research has focused on the synthesis and characterization of nitric oxide (NO)-releasing polymers as antithrombogenic sensor membranes to combat blood clot formation. Nitric oxide is released by human endothelial cells at femto- to picomolar fluxes where it helps regulate vasodilation and platelet activation.13 Indeed, polymers modified to release NO at physiological concentrations have demonstrated much lower proclivities for thrombus formation at a blood/material interface.14-16 Unlike other systemically administered anticoagulants such as heparin, NO has a short half-life in blood (e.g., on the order of seconds), ensuring its antithrombogenic effects remain localized near the sensor/blood interface, precluding potentially detrimental side-effects.17
Nitric oxide release capability has been imparted to polymers through the inclusion of a number of NO donor functionalities, including nitrosothiols, nitrosamines, metal complexes, and diazeniumdiolates into the polymer via doping or covalent attachment.18 To date, nitrogen-bound diazeniumdiolates (N-diazeniumdiolates) represent the most widely employed NO donors for spontaneously generating NO from materials. Briefly, N-diazeniumdiolates are formed by exposing secondary amine precursors to high pressures of NO under basic conditions, resulting in the reversible binding of two NO molecules. Subsequent exposure to a proton source such as water initiates N-diazeniumdiolate decomposition and release of two equivalents of NO.19 Coatings utilizing this system have been employed to improve the biocompatibility of catheter and subcutaneous sensors for biologically relevant analytes. Previously described examples include amperometric PO220, 21 and glucose sensors,22 and potentiometric PCO2 sensors.23 Nitric oxide-releasing polymeric coatings suitable for fluorescence-based PO2 detection have also been described, although miniaturized sensors were neither fabricated nor evaluated.24
Herein, we describe a NO-releasing optical fiber-based pH sensor fabricated through sol-gel chemistry. During the sol-gel process, hydrolysis and condensation of silicon alkoxide precursors form a robust, glassy silica network referred to as a xerogel.25 Incorporation of aminoalkoxysilane precursors into the siloxane polymer backbone allows for the subsequent formation of covalently bound N-diazeniumdiolates, introducing NO-release capability to the xerogel.14, 26, 27 Nitric oxide-releasing xerogels with tunable NO-release kinetics have been reported previously, and their effectiveness in improving the biocompatibility of amperometric oxygen28 and glucose29, 30 sensors has been demonstrated. Unfortunately, exposure of aminoalkoxysilane-based xerogels to high pressures of NO during N-diazeniumdiolate formation was shown to accelerate sol-gel polycondensation, resulting in dense (i.e., non-permeable) xerogel coatings.29 As such, sensors fabricated with such coatings often suffer from limited sensitivity and lengthened response times. Strategies for overcoming the reduced analyte diffusion have included doping xerogel particles into conventional sensor membranes29 and utilizing soft-lithographic micropatterning techniques to selectively modify regions of the underlying electrode.30 While effective, these techniques make sensor fabrication more complicated and limit the overall flux of NO attainable at such surfaces.
The dense nature of NO-releasing xerogel coatings may make them better suited to optical sensing platforms, where analyte diffusion to an electrode surface is not required. Optically transparent and chemically inert xerogels have long been recognized as a useful host matrix for fluorescent indicator molecules.31-33 The siloxane network properties including membrane porosity and permeability are easily manipulated by varying the composition of the sol (e.g., water:silane ratio and/or acid concentration). Furthermore, physical entrapment of indicator dyes in the gel is straightforward. Herein, we report the development of a miniaturized, NO-releasing xerogel-derived optical pH sensor suitable for intravascular use that combines the simplicity of sol-gel-based optical sensor fabrication with the added biocompatibility benefits of NO release. Using a two-layer immobilization strategy, xerogels containing both NO-producing N-diazeniumdiolates and the pH-sensitive fluorophore seminaphthorhodamine-1 carboxylate (SNARF-1) are immobilized onto optical fiber supports. The fabrication, analytical performance, and enhanced blood biocompatibility of the NO-releasing optical pH sensors are discussed.
EXPERIMENTAL SECTION
Materials
Ethyltrimethoxysilane (ETMOS), isobutyltrimethoxysilane (BTMOS) methyltrimethoxysilane (MTMOS), (aminoethylaminomethyl)phenethyltrimethoxysilane (AEMP3), N-(6-aminohexyl)aminopropyltrimethoxysilane (AHAP3), and aminopropyltriethoxysilane (APTES), and were purchased from Gelest (Tullytown, PA). Tetramethyl orthosilicate (TMOS), glutaraldehyde (50% w:w in water), and hexamethyldisilazane (HMDS) were purchased from Aldrich (Milwaukee, WI). Seminaphthorhodamine-1 carboxylate (SNARF-1) pH indicator was purchased from Invitrogen (Carlsbad, CA). Nitric oxide (99.5%) was purchased from National Welders Supply (Durham, NC). Whole blood was obtained from healthy pigs at the Francis Owen Blood Research Laboratory (Chapel Hill, NC). Other solvents and chemicals were analytical-reagent grade and used as received. Distilled water was purified to 18.2 MΩ·cm with a Millipore Milli-Q Gradient A-10 water purification system (Bedford, MA). Optical fiber materials were purchased from Ocean Optics (Dunedin, FL).
NO-releasing Xerogel Preparation
NO-releasing xerogels were prepared through the inclusion of 20 or 40% AHAP3 in ETMOS gels (v:v total silane). Ethanol (300 µL) was mixed with ETMOS (160 or 120 µL) and AHAP3 (40 or 80 µL) in a vortex shaker for 5 min. Water (10 µL) was added and shaken for an additional 5 min. The sol was then immobilized on optical fibers via dip-coating or drop-cast onto glass slides.
pH-Sensing Xerogel Preparation
Xerogels containing the SNARF-1 pH indicator were fabricated in a manner similar to that described by Grant and Glass.34 Water (1000 µL) was combined with TMOS (500 µL) and 0.04 M HCl (10 µL) and sonicated in an ice bath. After 30 min, a 75 µL aliquot of the sol was combined with 20 µL of a 5 mM SNARF-1/water stock solution and diluted with 300 µL pH 7.4 phosphate buffered saline (PBS). The sol was sonicated on ice for an additional minute prior to dip-coating procedures.
Sensor Fabrication
Two-layer NO-releasing pH sensors were created by sequential dip-coating of optical fibers in the AHAP3/ETMOS and TMOS sols. Unjacketed silica core/silica clad optical fiber (400 µm o.d., NA = 0.22) was cut into 8 cm segments and the polyimide sheathing around the fiber was removed by immersion in hot concentrated sulfuric acid. The fibers were tapered to enhance the coupling of fluorescent modes into the fiber core.35 Approximately 1.0 cm of the bare silica fiber was immersed in a concentrated hydrofluoric acid bath with an overlying layer of toluene for 2.5 h,36 resulting in the self-terminated tapering of approximately 0.5 cm of the distal end of the fiber down to 80 µm o.d. After rinsing, 2 cm of the tapered optical fibers were manually dip coated twice into the AHAP/ETMOS sol, drying 1 min in a 75 °C oven between coats. Sensors were allowed to age for a minimum of 3 d under ambient conditions before 1 coat of the pH-sensing sol was applied in a similar manner. Again, sensors were aged for a minimum of 3 d under ambient conditions before diazeniumdiolate formation. Sensors were placed in an in-house reaction vessel and thoroughly flushed with Ar prior to exposure to 5 atm NO for 3 d. After removing unreacted NO from by vessel by purging with Ar, the sensors were stored at -20 °C until use.
Apparatus
Sensors were evaluated with a simple optical fiber-based spectrophotometer system. The xerogel-coated probes were coupled directly to the common leg bundle of a solarization resistant 200 µm o.d. bifurcated optical fiber assembly via an SMA905 connector and Ocean Optics SMA905 bare fiber adaptor. Excitation light was supplied through one leg of the bifurcated assembly by a Power Technology Inc. LDCU3/3663 10 mW, 532 nm laser (Alexander, AR) after passing through a 90% attenuation neutral density filter. Fluorescent emission from the xerogel coating was coupled back into the probe and partially transmitted through the second leg of the bifurcated assembly for detection at an Ocean Optics USB2000 Spectrometer. Sensor response characteristics were determined by sequential immersion of the probes in PBS solutions buffered across the common physiological pH range of blood (pH 7.0-7.8). Complete spectra in the 400-800 nm range were generated and saved with Ocean Optics OOIBase32 software. Fluorescence intensities at the 580 and 640 nm peak emission wavelengths of the SNARF-1 pH indicator, were later extracted and ratios generated using LabVIEWTM (National Instruments).
Characterization of NO Release
Real-time NO flux from AHAP/ETMOS xerogels prepared with and without TMOS overlayers was monitored using a Sievers NOATM 280 Chemiluminescence Nitric Oxide Analyzer (Boulder, CO). Xerogel films were cast onto glass slides via glutaraldehyde fixation procedures. The slides were cleaned in 10% nitric acid (v:v, water) at 70 °C for 30 min, rinsed with purified water, immersed in 10% APTES (v:v, PBS, pH 7.0) at 70 °C for 90 min, rinsed again, and then immersed in 10% glutaraldehyde (v:v, water) at 25 °C for 60 min. After rinsing the slides thoroughly with water, AHAP3/ETMOS gel solution (30 µL) was drop-cast onto the slides and allowed to dry under ambient conditions. After drying for 24 h, a TMOS overlayer was dip-coated over select gels by complete immersion of the xerogel-coated glass slides in the TMOS sol. Dip-coated gels were allowed to dry for an additional 3 days under ambient conditions. The resulting xerogel films were then exposed to 5 atm NO for 3 d to form N-diazeniumdiolates as described above and stored at -20 °C until use. The Sievers NOA 280 instrument was calibrated before each experiment using air passed through a Sievers zero NO filter and 24.1 ppm NO gas (balance N2). Individual slides were immersed in 20 mL PBS buffer (pH 7.4) at 37 °C and sparged with a 200 mL/min N2 stream. The detected chemiluminescence due to NO was monitored continuously in 1 sec intervals for 16 h.
Platelet Adhesion Studies
Healthy porcine blood was drawn into acid citrate dextrose (ACD)-anticoagulated tubes (1 part ACD to 9 parts whole blood) and maintained at 37 °C for 1 h prior to use. Platelet enriched plasma (PRP) was obtained by centrifugation at 150g for 20 min at room temperature.37 Normal platelet activity was reestablished by the addition of CaCl2 to achieve a 0.5mM Ca2+ concentration. Individual NO-releasing xerogel films (prepared as described above) and controls were immersed in 2 mL PRP at 37 °C for 1 h. Loose platelets and cells were removed by rinsing with 37 °C Tyrodes buffer (137 mM NaCl, 2.7 mM KCl, 3.3 mM KH2PO4, 5.6 mM glucose, pH 7.35). The remaining adhered platelets were fixed by immersing the slides in a 1% glutaraldehyde solution (v:v, Tyrodes buffer) at 37 °C for 30 min. The slides were rinsed with Tyrodes buffer and water, and chemically dried as follows to preserve cell morphology. Slides were immersed in 50, 75, and 95% ethanol (v:v, water) for 5 min, 100% ethanol for 10 min, and HMDS overnight. Representative scanning electron microscope (SEM) images of all samples were obtained. Semiquantitative platelet surface coverages on NO-releasing samples were calculated via digital image processing and black/white thresholding from phase contrast images obtained with a Zeiss Axiovert 200 inverted microscope (Chester, VA). Average percent surface coverages were calculated from the overall average of five slides, with 5 images taken per slide.
RESULTS AND DISCUSSION
The near-neutral dynamic range (pKa = 7.5) and useful dual emission properties of the SNARF-1 fluorescent pH indicator make it an attractive choice for intravascular pH sensing.38 Both acid and base isomers fluoresce strongly at 580 and 640 nm, respectively, and may be excited at a single wavelength. By monitoring the ratio of the fluorescent intensity at two emission maxima instead of intensity changes at only one wavelength, the dye provides a self-referencing means to compensate for common sources of signal drift such as photobleaching and light source inconsistencies. Fabrication of NO-releasing optical pH sensors was first attempted by direct encapsulation of SNARF-1 indicator within several aminoalkoxysilane/alkoxysilane systems, including AEMP3/MTMOS, AHAP3/ETMOS, and AHAP3/BTMOS xerogels. Unfortunately, sensors fabricated by direct SNARF-1 encapsulation in the aminoalkoxysilanes-modified xerogels proved unresponsive.. No changes in fluorescent emmision were observed when such sensors were immersed in PBS of pH 7.0 - 7.8 (data not shown). The lack of response can be explained by the nature of the aminoalkoxysilane xerogels used to host the SNARF-1 indicator. Stable, NO-releasing xerogels require aminosilane precursors in conjunction with more inert ethyl- or methyl-substituted alkoxysilanes.14 Such xerogel networks are inherently more hydrophobic than those formed strictly from inorganic silanes like tetramethyl and tetraethyl orthosilicates.39 Additionally, when exposed to high pressures of NO to form N-diazeniumdiolates, aminoalkoxysilane xerogels suffer an extreme loss of permeability.29 As such, it is unlikely that buffer was penetrating into the bulk of the xerogel and thus, the majority of the fluorescent emission measured did not reflect the pH conditions of the surrounding buffer but rather the static interior pH of the bulk xerogel matrix.
Two-layer Sensors
To improve the responsiveness of the NO-releasing optical pH sensors, a benefit of optical sensing platforms was utilized. Unlike amperometric sensors, fiber-optic sensors do not require that the analyte permeate through a polymeric membrane to an electrode. As long as fluorescent emission from the pH indicator is coupled back into the optical fiber, the indicator may be separated physically from the fiber surface. While xerogels modified with N-diazeniumdiolates suffer from low analyte permeability, they remain optically transparent. Thus, the application of a thin, NO-releasing xerogel directly to a fiber-optic surface will present minimal interference to the coupling of excitation and emission modes to and from an overlying xerogel layer encapsulating the pH indicator. Two-layer NO-releasing optodes comprised of a 40:60% (v:v) AHAP3/ETMOS NO-releasing xerogel as a base layer and a TMOS xerogel overlayer doped with the SNARF-1 indicator showed a significantly improved response (i.e., change in fluorescent emission at 580 and 640 nm) to buffer pH compared to single-layer sensors (Figure 1).
Figure 1.
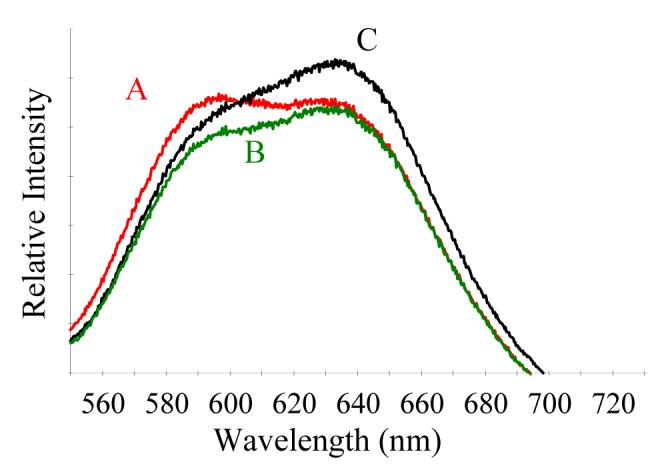
Fluorescence emission spectra from a 40:60% (v:v) AHAP3/ETMOS-TMOS optical pH sensor immersed in phosphate buffered saline at pH (A) 7.0; (B) 7.4; and, (C) 7.8.
Sensitivity and Reproducibility
The response of the two-layer NO-releasing sensors was measured by sequential immersion of the optical fiber in PBS solutions buffered between pH 7.0 and 7.8 (Figure 2). The response of the sensors was linear (R2 ≥ 0.986) throughout the tested pH range. From the calibration curves generated, a minimum resolvable pH shift of approximately 0.04 pH units was determined, as estimated by doubling the average standard deviation of emission ratios at individual pH values. The sensitivity and signal reproducibility of five individual sensors, as well as the average emission ratio of each sensor at pH 7.4, are provided in Table 1. As expected, variation between sensors was apparent. Such variability is attributed to inconsistencies in fiber tapering and coating. The TMOS gel employed for SNARF-1 encapsulation reacts and solidifies rapidly. Thus, a natural evolution of the gelation can be observed during the dip coating procedure. The sol coated onto fibers later in succession is generally more viscous, resulting in the deposition of a thicker xerogel layer.40 While more stringent timing control and automated dip-coating procedures would likely reduce sensor-to-sensor variations, calibration of individual sensors prior to use is still necessary.
Figure 2.

Time-resolved response from 40:60% (v:v) AHAP3/ETMOS-TMOS optical pH sensor immersed in pH 7.0-7.8 PBS solutions.
Table 1.
Reference emission ratios, sensitivity, linearity, and signal reproducibility of 40:60% (v:v) AHAP3/ETMOS-TMOS NO-releasing sensors
| Sensor | 580 / 640 nm Emission Ratioa | Δ Emission Ratio / Δ pH | R2 | Average standard deviation | Resolvable pH Shift |
|---|---|---|---|---|---|
| 1 | 0.850 | 0.196 | 0.986 | 0.00392 | 0.040 |
| 2 | 0.589 | 0.113 | 0.999 | 0.00203 | 0.036 |
| 3 | 0.707 | 0.116 | 0.999 | 0.00173 | 0.030 |
| 4 | 0.502 | 0.075 | 0.999 | 0.00138 | 0.037 |
| 5 | 0.581 | 0.079 | 0.998 | 0.00173 | 0.044 |
pH=7.4
Pretreatment and Response Time
A common concern reported for xerogel-based sensors employed in aqueous solution is lengthy response times, which can range from seconds up to hours.41-46 While porosity and gel hydrophobicity play an important role in determining diffusion rates through the material, Ismail et al. have demonstrated that hydrogen bonding at the gel/liquid interface has an equal, if not greater effect in many cases.47 Xerogels may contain a large number of unreacted free silanol groups capable of forming hydrogen bonds with other nearby free silanols either directly or through the mediation of bridging water molecules. These hydrogen bond networks can serve as a barrier to rapid equilibrium of the exterior aqueous environment with the interior of the xerogel. Proton-exchange indicator molecules trapped in the xerogel can associate with residual protons and water molecules retained by hydrogen bonding until complete equilibrium of the material with its surrounding environment is achieved. The result is long response times and lengthy soaking requirements of xerogel-based sensors prior to use. Obviously, long response times are less than optimal for most sensing applications. Long presoak requirements are especially detrimental for sensors modified with NO-releasing coatings as NO release is initiated from diazeniumdiolate NO donors upon solution immersion. For NO-releasing xerogel based amperometric oxygen sensors, Marxer et al. was able to reduce the minimum presoaking time to 30 min (to obtain adequate sensitivity) by increasing the permeability of the xerogel via the inclusion of 1% (v:v) hydrophilic polyurethane in the gel.28 Doping xerogel coatings to be used for optical sensors with such polymers, however, would lead to optically opaque films.
To ensure rapid response with minimal presoak requirements, the xerogel optical sensors were immersed in 0.5 M sodium hydroxide solution for 30 s prior to use, as described previously by Ismail et al.47 Brief exposure to strongly basic solution allowed for rapid disruption of the silanol hydrogen bond networks present and substitution by sodium cations while having minimal effect on the proton-induced decomposition of N-diazeniumdiolates. The encapsulated SNARF-1 indicator was thus free to equilibrate with the surrounding buffer. The brief immersion in base may also serve to break siloxane bonds, further opening up the xerogel for more rapid analyte diffusion throughout the material. Such polymer breakdown was limited, however, as successive base immersions did not lead to changes in response or further reductions in fluorescent signal intensity. Following base immersion, the sensors were immediately used to monitor pH. Sensor response times were determined by tracking the shift of fluorescent emission ratios in 0.9 s increments as the two-layer sensor was moved from pH 7.8 to pH 7.0 buffer solutions. The time required to achieve 90% of the final response (τ90) was determined by fitting the resulting data to a first order exponential decay model (Figure 3). The 40:60% (v:v) AHAP3/ETMOS-TMOS modified sensors exhibited extremely rapid response characteristics, with a calculated average τ90 of 14 ± 2 s (n = 3). Indicator leaching after base immersion was evaluated by continuously monitoring the average fluorescent intensity of the 640 nm SNARF-1 emission peak at hourly intervals from a sensor immersed in pH 7.4 PBS. Over the course of 20 h, the fluorescence intensity measured at 640 nm did not change (1070.1 ± 5.3 and 1068.0 ± 9.5 a.u. at 0 and 20 h, respectively). Thus, the AHAP3/ETMOS-TMOS coatings allowed immediate sensor use and rapid response with only a 30 s base pretreatment step.
Figure 3.
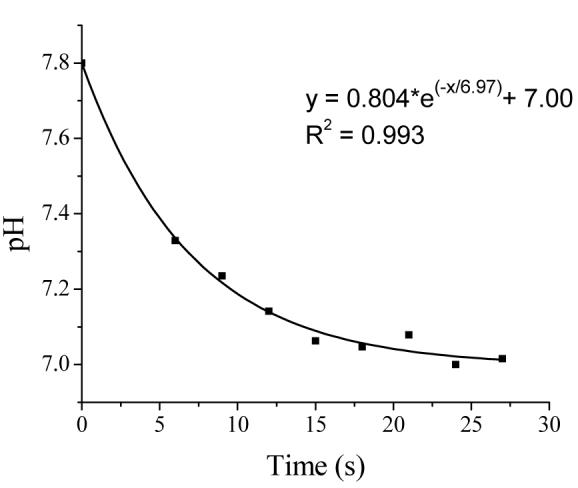
Response of 40:60% (v:v) AHAP3/ETMOS-TMOS optical pH sensor as a function of pH (7.0-7.8).
Nitric Oxide Release
Manipulation of the identity and percentage of aminosilane employed in the original sol mixture allows control over the magnitude and duration of NO released by a N-diazeniumdiolate-modified xerogel film.27 To evaluate the NO-release potential of the two-layer AHAP3/ETMOS-TMOS coatings employed in this study, real-time NO flux measurements were performed via chemiluminescence. Example NO release profiles demonstrating the NO flux generated by coatings comprised of 20 and 40% AHAP3 (v:v; balance ETMOS) are shown in Figure 4. The optimal NO flux for reducing thrombus formation at a blood/material interface is generally believed to fluctuate depending on the localized abundance of NO-scavengers (e.g., proteins, thiols, etc.) in the surrounding environment. Ramamurthi and Lewis have reported that surface NO fluxes as low as 0.6 fmol/cm2s were effective at reducing in vitro platelet adhesion.48 Likewise, Robbins et al. demonstrated maintained thromboresistivity for up to 24 h at micropatterned xerogel arrays with NO surface fluxes of 0.4 pmol/cm2s.49 Considering 0.4 pmol/cm2s as a minimum threshold value for inhibiting platelet adhesion, sensors fabricated with 20% AHAP3 would be thromboresistant for approximately 6 h. Likewise, sensors prepared with 40% AHAP3 xerogels would maintain fluxes > 0.4 pmol/cm2s for at least 16 h. Notably, the addition of a TMOS overlayer to the NO-releasing AHAP3/ETMOS films prior to diazeniumdiolate formation did not alter the NO-release profile significantly (Figure 4), suggesting that the presence of the porous pH-sensing TMOS layer does not serve as an impediment to NO or proton transport through the gel.
Figure 4.

Nitric oxide release from (A) 40:60% and (B) 20:80% (v:v) AHAP3/ETMOS xerogels; and, (C) 20:80% (v:v) AHAP3/ETMOS xerogel with a TMOS overlayer film, immersed in 37 °C PBS (pH 7.4).
In vitro Platelet Adhesion Studies
Platelet adhesion to slides coated with NO-releasing AHAP3/ETMOS (40:60%, v:v) xerogels with and without a TMOS overlayer was examined to evaluate the potential blood compatibility of the xerogel coatings. Representative scanning electron micrographs of platelet adhesion to the films are shown in Figure 5. After 1 h exposure to platelet enriched porcine plasma, platelet adhesion at both the single and two-layer NO-releasing films was significantly decreased compared to non-NO-releasing controls. As expected, NO contributes a strongly inhibitory effect on platelet adhesion, resulting in drastic reductions in overall coverage of platelets at xerogel coatings. As suggested by NO-flux measurements, the presence of the TMOS overcoat layer did not inhibit the effectiveness of the underlying NO-releasing film. In fact, TMOS-coated films showed slightly less platelet adhesion than AHAP3/ETMOS films (5.7 ± 1.5 versus 2.0 ± 0.9 percent coverage, respectively). Intravascular fiber optic pH sensors may be expected to exhibit similar biocompatibility benefits in vivo throughout the window of NO-release.
Figure 5.

Representative scanning electron micrographs illustrating platelet adhesion to (A) NO-releasing and (C) control 40:60% (v:v) AHAP3/ETMOS films, and equivalent (B) NO-releasing and (D) control films with TMOS overcoats.
CONCLUSIONS
Introducing NO-release capability to a device implanted in the bloodstream may allow for significant reductions in platelet adhesion and thrombus formation at the material's surface and a concomitant improvement in the device's performance. For sensors, xerogel coatings modified with N-diazeniumdiolates provide a useful means of generating such NO release. To avoid the issue of low permeability observed for NO-releasing xerogel amperometric sensors, an optical sensing strategy was employed. Through straightforward dip-coating procedures, two-layer NO-releasing pH optical sensors were fabricated by depositing a porous pH-sensing xerogel layer over AHAP3/ETMOS NO-releasing xerogel-coated optical fibers. Sufficient resolution and response times were obtained from the resulting pH sensors after only a 30 s pretreatment step in sodium hydroxide solution. In vitro platelet adhesion studies indicated that the flux of NO (>0.4 pmol/cm2s for 16 h) from the xerogel coatings was effective at reducing platelet adhesion. It is anticipated that the pH sensors and techniques described herein may be utilized in the fabrication of more biocompatible elements for intravascular blood gas sensor arrays and as the basic transduction mechanism for physiologically relevant chemical (e.g., CO2, NH4+) and enzyme-based (e.g., glucose, urea) biosensors.50
ACKNOWLEDGEMENTS
This research was supported by the National Institutes of Health (NIH EB000708). The authors thank the University of North Carolina at Chapel Hill Francis Owen Blood Research Laboratory for providing porcine blood samples. K.P.D. gratefully acknowledges a National Science Foundation Graduate Research Fellowship. G.W.C. acknowledges the support of the Frances C. and William P. Smallwood Foundation.
REFERENCES
- (1).Zhang HB, Vincent JL. Am. Rev. Resp. Dis. 1993;148:867–871. doi: 10.1164/ajrccm/148.4_Pt_1.867. [DOI] [PubMed] [Google Scholar]
- (2).Grundler W, Weil MH, Rackow EC. Circulation. 1986;74:1071–1074. doi: 10.1161/01.cir.74.5.1071. [DOI] [PubMed] [Google Scholar]
- (3).Ganter MT, Hofer CK, Zollinger A, Spahr T, Pasch T, Zalunardo MP. J. of Cardiothorac. Vasc. Anesth. 2004;18:587–591. doi: 10.1053/j.jvca.2004.07.017. [DOI] [PubMed] [Google Scholar]
- (4).Rithalia SVS. J. Med. Eng. Technol. 1991;15:143–153. doi: 10.3109/03091909109023701. [DOI] [PubMed] [Google Scholar]
- (5).Ganter M, Zollinger A. Br. J. Anaesth. 2003;91:397–407. doi: 10.1093/bja/aeg176. [DOI] [PubMed] [Google Scholar]
- (6).Frost MC, Meyerhoff ME. Curr. Opin. Chem. Biol. 2002;6:633–641. doi: 10.1016/s1367-5931(02)00371-x. [DOI] [PubMed] [Google Scholar]
- (7).Udd E. Rev. Sci. Instrum. 1995;66:4015–4030. [Google Scholar]
- (8).Wolfbeis OS. Anal. Chem. 2004;76:3269–3283. doi: 10.1021/ac040049d. [DOI] [PubMed] [Google Scholar]
- (9).Oropello JM, Manasia A, Hannon E, Leibowitz A, Benjamin E. Chest. 1996;109:1049–1055. doi: 10.1378/chest.109.4.1049. [DOI] [PubMed] [Google Scholar]
- (10).Ishikawa S, Makita K, Nakazawa K, Amaha K. Can. J. Anaesth. 1998;45:273–276. doi: 10.1007/BF03012916. [DOI] [PubMed] [Google Scholar]
- (11).Weiss IK, Fink S, Harrison R, Feldman JD, Brill JE. Pediatrics. 1999;103:440–445. doi: 10.1542/peds.103.2.440. [DOI] [PubMed] [Google Scholar]
- (12).Wisniewski N, Reichert M. Colloid Surf. B-Biointerfaces. 2000;18:197–219. doi: 10.1016/s0927-7765(99)00148-4. [DOI] [PubMed] [Google Scholar]
- (13).Radomski MW, Rees DD, Dutra A, Moncada S. Brit. J. Pharmacol. 1992;107:745–749. doi: 10.1111/j.1476-5381.1992.tb14517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Marxer SM, Rothrock AR, Nablo BJ, Robbins ME, Schoenfisch MH. Chem. Mat. 2003;15:4193–4199. [Google Scholar]
- (15).Zhou ZR, Meyerhoff ME. Biomaterials. 2005;26:6506–6517. doi: 10.1016/j.biomaterials.2005.04.046. [DOI] [PubMed] [Google Scholar]
- (16).Mowery KA, Schoenfisch MH, Saavedra JE, Keefer LK, Meyerhoff ME. Biomaterials. 2000;21:9–21. doi: 10.1016/s0142-9612(99)00127-1. [DOI] [PubMed] [Google Scholar]
- (17).Kelm M, Yoshida K. In: Methods in Nitric Oxide Research. Feelisch M, Stamler JS, editors. John Wiley and Sons Inc.; New York: 1996. pp. 47–58. [Google Scholar]
- (18).Shin JH, Schoenfisch MH. Analyst. 2006;131:609–615. doi: 10.1039/b600129g. [DOI] [PubMed] [Google Scholar]
- (19).Hrabie JA, Klose JR, Wink DA, Keefer LK. J. Org. Chem. 1993;58:1472–1476. [Google Scholar]
- (20).Frost MC, Rudich SM, Zhang HP, Maraschio MA, Meyerhoff ME. Anal. Chem. 2002;74:5942–5947. doi: 10.1021/ac025944g. [DOI] [PubMed] [Google Scholar]
- (21).Schoenfisch MH, Mowery KA, Rader MV, Baliga N, Wahr JA, Meyerhoff ME. Anal. Chem. 2000;72:1119–1126. doi: 10.1021/ac991370c. [DOI] [PubMed] [Google Scholar]
- (22).Gifford R, Batchelor MM, Lee Y, Gokulrangan G, Meyerhoff ME, Wilson GS. J. Biomed. Mat. Res. Part A. 2005;75A:755–766. doi: 10.1002/jbm.a.30359. [DOI] [PubMed] [Google Scholar]
- (23).Frost MC, Batchelor MM, Lee YM, Zhang HP, Kang YJ, Oh BK, Wilson GS, Gifford R, Rudich SM, Meyerhoff ME. Microchem. J. 2003;74:277–288. [Google Scholar]
- (24).Schoenfisch MH, Zhang HP, Frost MC, Meyerhoff ME. Anal. Chem. 2002;74:5937–5941. doi: 10.1021/ac026075y. [DOI] [PubMed] [Google Scholar]
- (25).Brinker J, Scherer G. Sol-Gel Science. Academic Press; New York: 1989. [Google Scholar]
- (26).Nablo BJ, Schoenfisch MH. J. Biomed. Mat. Res. Part A. 2003;67A:1276–1283. doi: 10.1002/jbm.a.20030. [DOI] [PubMed] [Google Scholar]
- (27).Nablo BJ, Chen TY, Schoenfisch MH. J. Amer. Chem. Soc. 2001;123:9712–9713. doi: 10.1021/ja0165077. [DOI] [PubMed] [Google Scholar]
- (28).Marxer SM, Robbins ME, Schoenfisch MH. Analyst. 2005;130:206–212. doi: 10.1039/b412079e. [DOI] [PubMed] [Google Scholar]
- (29).Shin JH, Marxer SM, Schoenfisch MH. Anal. Chem. 2004;76:4543–4549. doi: 10.1021/ac049776z. [DOI] [PubMed] [Google Scholar]
- (30).Oh BK, Robbins ME, Nablo BJ, Schoenfisch MH. Biosens. Bioelectron. 2005;21:749–757. doi: 10.1016/j.bios.2005.01.010. [DOI] [PubMed] [Google Scholar]
- (31).Grattan KTV, Badini GE, Palmer AW, Tseung ACC. Sens. Actuator A-Phys. 1991;26:483–487. [Google Scholar]
- (32).MacCraith BD, McDonagh C. J. Fluoresc. 2002;12:333–342. [Google Scholar]
- (33).Maccraith BD, McDonagh CM, Okeeffe G, McEvoy AK, Butler T, Sheridan FR. Sens. Actuator B-Chem.l. 1995;29:51–57. [Google Scholar]
- (34).Grant SA, Glass RS. Sensors and Actuators B-Chemical. 1997;45:35–42. [Google Scholar]
- (35).Golden JP, Anderson GP, Rabbany SY, Ligler FS. IEEE Trans Biomed. Eng. 1994;41:585–591. doi: 10.1109/10.293246. [DOI] [PubMed] [Google Scholar]
- (36).Hoffmann P, Dutoit B, Salathe RP. Ultramicroscopy. 1995;61:165–170. [Google Scholar]
- (37).Cazenave J, Mulvihill J. In: The role of platelets in blood-biomaterial interactions. Missirlis Y, Wautier J-L, editors. Kluwer; Dordrecht: 1993. pp. 69–80. [Google Scholar]
- (38).Parker JW, Laksin O, Yu C, Lau ML, Klima S, Fisher R, Scott I, Atwater BW. Anal. Chem. 1993;65:2329–2334. [Google Scholar]
- (39).Schubert U, Husing N, Lorenz A. Chem. Mat. 1995;7:2010–2027. [Google Scholar]
- (40).McDonagh C, Sheridan F, Butler T, MacCraith BD. J. Non-Cryst. Solids. 1996;194:72–77. [Google Scholar]
- (41).Sanchez-Barragan I, Costa-Fernandez JM, Sanz-Medel A. Sens. Actuator B-Chem. 2005;107:69–76. [Google Scholar]
- (42).Nivens DA, Schiza MV, Angel SM. Talanta. 2002;58:543–550. doi: 10.1016/s0039-9140(02)00323-5. [DOI] [PubMed] [Google Scholar]
- (43).Cajlakovic M, Lobnik A, Werner T. Anal. Chim. Acta. 2002;455:207–213. [Google Scholar]
- (44).Wallace PA, Elliott N, Uttamlal M, Holmes-Smith AS, Campbell M. Meas. Sci. Technol. 2001;12:882–886. [Google Scholar]
- (45).Makote R, Collinson MM. Anal. Chim. Acta. 1999;394:195–200. [Google Scholar]
- (46).Lobnik A, Oehme I, Murkovic I, Wolfbeis OS. Anal. Chim. Acta. 1998;367:159–165. [Google Scholar]
- (47).Ismail F, Malins C, Goddard NJ. Analyst. 2002;127:253–257. [Google Scholar]
- (48).Kanungo M, Collinson MM. Anal. Chem. 2003;75:6555–6559. doi: 10.1021/ac034658h. [DOI] [PubMed] [Google Scholar]
- (49).Ramamurthi A, Lewis RS. Biomed. Sci. Instr. 1999;35:333–338. [PubMed] [Google Scholar]
- (50).Robbins ME, Hopper ED, Schoenfisch MH. Langmuir. 2004;20:10296–10302. doi: 10.1021/la048368n. [DOI] [PubMed] [Google Scholar]
- (51).Muller C, Hitzmann B, Schubert F, Scheper T. Sens. Actuator B-Chem. 1997;40:71–77. [Google Scholar]