

Abstract
Previously, we have shown that 4-hydroxynonenal (4-HNE) induces Fas-mediated apoptosis in HLE B-3 cells through a pathway which is independent of FasL, FADD, procaspase8-and DISC (Li, J. et al. Biochemistry, 45, 12253-12264). The involvement of Daxx has also been suggested in this pathway but its role is not clear. Here, we report that Daxx plays an important regulatory role during 4-HNE induced, Fas-mediated apoptosis in Jurkat cells. 4-HNE induces Fas-dependent apoptosis in procaspase8 deficient Jurkat cells via the activation of ASK1, JNK and caspase3 and the apoptosis can be inhibited by masking Fas with the antagonistic anti-Fas antibodies. We demonstrate that 4-HNE exposure to Jurkat cells leads to the induction of both Fas and Daxx. 4-HNE binds to both Fas and Daxx and promotes the export of Daxx from nucleus to cytosol where it binds to Fas and inhibits apoptosis. Depletion of Daxx results in increase in the activation of ASK1, JNK, and caspase3 along with exacerbation of 4-HNE-induced apoptosis suggesting that Daxx inhibits apoptosis by binding to Fas. 4-HNE-induced translocation of the Daxx is also accompanied with the activation of the transcription factor HSF1. Results of these studies are consistent with a model in which by interacting with Fas, 4-HNE promotes pro-apoptotic signaling via ASK1, JNK and caspase3. In parallel, 4-HNE induces Daxx and promotes its export from the nucleus to cytosol where it interacts with Fas to self-limit the extent of apoptosis by inhibiting the downstream pro-apoptotic signaling. Cytoplasmic translocation of Daxx also results in up-regulation of HSF1 associated stress responsive genes.
Keywords: 4-HNE, Fas, Daxx, apoptosis, Jurkat cells, HSF1
4-Hydroxynonenal (4-HNE), one of the major products of lipid peroxidation generated during oxidative stress is invariably present in aerobic organisms (1). At higher concentrations, 4-HNE has been shown to be an inducer of apoptosis in various cell types (2-7). Because apoptotic stressors such as UV radiation, heat shock and many xenobiotics markedly elevate the intracellular concentration of 4-HNE above its basic constitutive level in cells, it has been suggested that 4-HNE plays a crucial role in the mechanisms of programmed cell death (3-5). A number of studies have revealed that depending on the intracellular concentrations, 4-HNE is also involved in a wide variety of signaling processes including the activation of tyrosine kinase receptors on the cell membrane (8, 9). The role of 4-HNE in signal transduction is complex; at “sub-physiologic” concentrations it appears to promote cell survival signals while at high concentrations it induces cell death (10).
Cellular apoptotic signals have been suggested to be of two types, extrinsic and intrinsic. Extrinsic apoptotic signaling is mediated by the activation of cell surface death receptors that transmit the signals after ligation with specific ligands. Fas (Apo1/CD95) is one of the death receptors belonging to the tumor necrosis factor receptor (TNFR) gene superfamily (11,12). Fas and other members of the TNFR family consist of cysteine rich extracellular subdomains which recognize their ligands with specificity, resulting in the trimerization and activation of the respective death receptor. Subsequent signaling is mediated by the cytoplasmic part of the death receptor which contains a conserved sequence termed as the death domain (DD). Adapter molecules like FADD are recruited to the DD of the activated death receptor, thereby forming death inducing signaling complex (DISC). In addition to DD, the adaptor FADD also contains a death effector domain (DED) which sequesters procaspase8 in the DISC (12). Recruitment of procaspase8 to the DISC leads to its autocatalytic activation and release of active caspase8 which then processes downstream effector caspases leading to the cell death.
Recent studies in our laboratory have demonstrated that 4-HNE affects a number of signaling pathways (3-5, 13) including signaling for apoptosis mediated by Fas. Our studies revealed that the regulation of Fas-mediated apoptotic signaling by 4-HNE in human lens epithelial (HLE B-3) cells was independent of the classical extrinsic apoptotic pathway (13) summarized above and suggested an involvement of another death associated protein, Daxx (14). Daxx is primarily a nuclear protein which translocates to cytoplasm during stress and acts as a death receptor adaptor at the cell surface (15-18). Nuclear Daxx can associate directly with different DNA-binding transcription factors and has been shown to be a transcriptional repressor (15-18). Previous studies by other investigators have suggested that Daxx binds to Fas and activates Fas-mediated apoptosis via the apoptosis signal regulating kinase 1 (ASK1) which in turn activates the c-jun N-terminal kinase (JNK) pathway and that this pathway is independent of the Fas-FADD-caspase8 pathway operative in various cell types (14).
The critical role of caspase8 in Fas-mediated apoptosis has been demonstrated in a number of studies involving caspase8 mutant cells (19,20). In these cells, procaspase8 is inactivated by mutations at the active site that render it incapable of interacting with FADD. One such mutant cell line, CRL2571 (clone I 9.2), has been developed from the wild type Jurkat cell line A3 (19). CRL2571 cells have been shown to be resistant to apoptosis caused by toxic Fas antibodies and are also partially resistant to apoptosis caused by UV-radiations and doxorubicin (DOX) (19). Because 4-HNE induces apoptosis in HLE B-3 cells through a caspase8/DISC-independent pathway, we used CRL2571 cells to examine the role of the Fas-Daxx-ASK1-JNK pathway in 4-HNE-triggered apoptotic signaling. Results of these studies indicate that exposure of CRL2571 cells to 4-HNE causes a dose-and time-dependent apoptosis in these cells through activation of Fas via a direct interaction of 4-HNE with the Fas receptor. This is accompanied by activation of ASK1 and JNK, and results in the cleavage of the effector procaspase3 and PARP. 4-HNE also induces an activation and translocation of Daxx from the nucleus to the cytoplasm. Surprisingly, silencing of Daxx rendered the cells more susceptible to 4-HNE-triggered apoptosis suggesting that Daxx plays a regulatory role during 4-HNE- induced signaling for apoptosis.
Materials and Methods
Materials
4-Hydroxynonenal was purchased from Cayman Chemical (Ann Arbor, MI). Bradford reagent, bis-acrylamide and SDS for SDS-PAGE were obtained from BioRad (Hercules, CA). Apoptosis detection system (CaspACE™ FITC-VAD-FMK in situ marker) was purchased from Promega Inc. (Madison, WI). Cytoplasmic and nuclear protein extraction kit was acquired from Imgenex Co. (San Diego, CA), Protein A/G-Agarose from Santa Cruz Biotechnology (Santa Cruz, CA), JNK inhibitor SP6000125 from A-G Scientific (San Deigo, CA), and the Western blot stripping buffer were from Pierce Co. (Rockford, IL). All other reagents and chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Cell culture medium and Fetal Bovine serum were from GIBCO (Invitrogen, Carlsbad, CA)
Cells
CRL2571(ATCC), a caspase8 mutant cell line developed from Jurkat cells was purchased from ATCC (Manassas, VA) and maintained in RPMI medium supplemented with 10% FBS, L-glutamine 300mg/L, 1 mM sodium pyruvate, 10 mM HEPES and 1.5 g/L sodium bicarbonate, 4.5 g/L glucose and 1% penicillin and streptomycin.
Jurkat cells were purchased from the Tissue culture core facility of the University of Texas Medical Branch, Galveston and maintained in RPMI medium supplemented with L-glutamine 300mg/L, 10% FBS and 1% penicillin and streptomycin. Both cell lines were maintained at 37°C in 5% CO2 atmosphere.
Antibodies
Antibodies against Fas receptor (IgM, CH11) were purchased from MBL International Corporation (Woburn, MA) whereas Fas mouse monoclonal (B-10) and polyclonal antibodies against PARP, caspase3, Daxx, p53, HSF1 and Bax were purchased from SantaCruz Biotech. (Santa Cruz, CA). c-Jun fusion protein bound to agarose beads and phospho c-jun antibodies were procured from Cell Signaling Technologies (Boston, MA). Polyclonal antibodies against 4-HNE-protein adducts (4-HNE 11-S) used in this study were from Alpha Diagnostics (San Antonio, TX). Daxx siRNA (h), a pool of 3 target-specific 20-25 nucleotide siRNA designed to knock down Daxx gene expression was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and siRNA–A (sc-37007), a non-targeting 20-25-nucleotide siRNA was used as a negative control.
Preparation of cell extracts
Cells were pelleted at 357g washed twice with cold PBS, and resuspended in radioimmunoprecipitation assay (RIPA) buffer containing 1× phenylmethylsulfonyl fluoride (PMSF), and 2 μg/ml pepstatin. To prepare cytoplasmic protein extracts, cells were washed with ice-cold PBS and resuspended in hypotonic lysis buffer (Imgenex, San Deigo CA) for 15 min, mixed with 30 μl of 10% NP-40 and vortexed for 10 s. The cell lysate was centrifuged for 30 s at 10000g, and the supernatant was collected. The pellet was extracted in 100μl of nuclear extraction buffer, vortexed and incubated at 4°C for 30min on a shaker. Suspension was once again vortexed for 30s, centrifuged at 10000g rpm for 10min and the supernatant containing nuclear extract was collected. Cytoplasmic and nuclear extracts were then used for further analysis.
Western blot analysis
Cell extracts containing 50-60 μg of protein were separated on SDS polyacrylamide gels (4-20%), and transferred onto nitrocellulose (Bio-Rad) or PVDF membrane (Millipore). Membranes were blocked with 1% fat-free milk and 1% BSA at room temperature for 30 min, and incubated overnight at 4°C with the appropriate primary antibody in 1% milk, 1% BSA in Tris-buffered saline (TBS) containing 50mM NaF and 0.05% Tween 20 (T-TBS). After washing with T-TBS, the membrane was incubated with the appropriate secondary antibodies at room temperature for 1h. After washing again with T-TBS, the membrane was treated with Super signal ‘West Pico’ chemiluminescent reagent (Pierce, Rockford, IL) as per manufacturer's instructions, and exposed to Hyperfilm ECL film (Amersham) at room temperature.
Detection of PARP
For the detection of PARP, 1 × 107 cells were suspended in 100 μl of denaturing lysis buffer containing 62.5 mM Tris-HCl, pH 6.8, 6.0 M urea, 2% SDS, 10% glycerol, 1.4 mM β-mercaptoethanol, 0.00125% bromphenol blue, 0.5% Triton X-100, and 1 mM PMSF. Cells were sonicated (3×5 s) on ice to disrupt protein-DNA binding and incubated at 65°C for 15 min. Samples containing 30 μg protein were applied to 10% SDS-PAGE gels, and Western blot analysis was performed using PARP antibodies.
In situ caspase3 assay for Apoptosis
1 × 105 CRL2571 cells were treated with 0-20 μM 4-HNE or with Fas agonistic CH11 antibodies (250ng/ml) for 2 h at 37°C. Apoptotic cells were detected by staining with 5 or 10μM CaspACE FITC-VAD-FMK (Promega) in situ marker for 30min in the dark. The cells were cytospun on polylysine coated glass slides at 500 rpm (5min). The slides were fixed with 4% paraformaldehyde for 1h, rinsed with PBS twice, mounted in a medium containing DAPI (1.5μg/ml), and observed under fluorescence microscope (Nikon, Japan).
Immunoprecipitation Studies
Cells were washed twice with cold PBS and pellets were resuspended in RIPA buffer containing 1× PBS, pH 7.4, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, and 2 μg/ml pepstatin. Following sonication on ice, the homogenates were centrifuged at 10,000g for 15min at 4°C and aliquots of the supernatant containing 500μg protein were incubated with anti-Fas or anti-Daxx antibodies (1:100) at 4°C overnight. 50 μl protein A/G PLUS agarose beads were then added to the reaction mixture and incubated again overnight at 4°C. The agarose beads were collected by pulse centrifugation (5s at 14000 rpm in a microfuge), washed three times with ice-cold RIPA buffer, resuspended in 60 μl 2× sample buffer, and boiled for 5 min to dissociate the immunocomplexes from the beads. The supernatant collected after centrifugation (10000g) was subjected to Western blot analysis using specific antibodies.
Cytotoxicity assay
The sensitivity of the cells to 4-HNE was measured by the MTT assay as described by Mosmann (21) with slight modifications. Briefly, 2 × 10 4 cells in 190 μl of medium were plated to each well in 96-well flat bottomed microtiter plates. 10 μl of PBS containing various amounts of 4-HNE were added. Eight replicate wells were used for each concentration of 4-HNE used in these experiments. Following incubation of the plates at 37°C for 2h, 10 μl of MTT solution (5mg/ml in PBS) was added to each well, and the plates were incubated for additional 4h at 37°C. The plates were centrifuged at 2000g for 10 min. The medium within the wells was aspirated and 100μl of dimethylsulfoxide (DMSO) was added to each well. The intracellular formazan dye crystals were dissolved by shaking the plates at room temperature for 2h. The absorbance of formazan at 562nm was measured using a microtiter plate reader (Elx808 BioTek Instruments, Inc). The concentration of 4-HNE resulting in a 50% decrease in formazan formation (IC50) was obtained by plotting a dose-response curve.
Kinase assays
Solid phase kinase assays were performed by the method of Uchida et al (22). Briefly, cells were treated with 20μM 4-HNE for different time periods. Whole-cell lysates were prepared in lysis buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA and 1 mM EGTA, 1% Triton X100, 2.5 mM sodium pyrophosphate, 1mM β-glycerophosphate, 1mM of sodium vanadate, 1mM PMSF, and 1μg/ml leupeptin. JNK activity was precipitated from cell lysates by incubating overnight at 4°C with 2μg of GST c-jun (1-89) fusion protein linked to GSH-agarose beads. The beads were then washed twice with lysis buffer and then twice with kinase buffer (25mM Tris-HCl, pH7.5, 5 mM β-glycerophosphate, 2mM DTT, 0.1mM sodium-vanadate,10mM MgCl2) and resuspended in 50μl of kinase buffer containing 100μM ATP for 30min at 37°C. The solid phase kinase reaction was terminated by the addition of Laemmli sample buffer and phophorylation of GST c-jun on Ser-63 was examined after SDS-PAGE and immunoblotting with anti-phospho (Ser-63) c-jun antibodies.
Transfection of Daxx siRNA in CRL2571 cells
The Daxx siRNA transfection was essentially carried out according to the manufacturer's instructions. Briefly, CRL2571 cells (2×105 cells per well) were plated in a six-well tissue culture plate, in 2 ml normal growth medium supplemented with FBS. Cells were cultured at 37°C until 60%-80% confluency. For each transfection, 50 pmol of siRNA was diluted with 100μl siRNA transfection medium (Solution A) and 6μl of siRNA transfection reagent was diluted with 100μl siRNA transfection medium (Solution B). Solution A was directly added to Solution B, mixed gently and the mixture was incubated for 30min at room temperature. Cells were washed with 2ml of siRNA transfection medium and 0.8ml of siRNA transfection medium was added to the mixture of the solutions A and B, gently mixed and overlaid onto the washed cells and incubated for 24h at 37°C. After 24h, 2ml of fresh normal medium was added to each well and cells were further incubated for 48h. Control cells were treated in a similar manner with a mixture of scrambled siRNA. Cells were harvested at appropriate time points and the silencing of Daxx was examined.
Protein estimation
Bradford (23) assay was used to determine protein concentration in cell lysates.
Statistical analysis
Data are expressed as mean± SD for each group. Statistical significance was determined by Student's t test and was set at 0.05.
Results
4-HNE causes apoptosis in wild type as well as caspase8 mutant cells
CRL2571 cells have been used to establish the requirement of procaspase8 in Fas-mediated apoptosis through the extrinsic pathway involving DISC formation (19). In these cells, the active wild-type procaspase8 of Jurkat cells has been replaced by an inactive procaspase8 mutant. These cells are resistant to apoptosis induced by toxic Fas antibodies (CH11) which cause apoptosis in wild-type Jurkat cells. In the course of the present studies, the results of experiments comparing the effect of CH11 antibodies confirmed that these antibodies were more toxic to wild-type Jurkat cells than to CRL2571 cells (Fig. 1 Panel A). CH11antibodies induced apoptosis only in the wild type cells as indicated by characteristic morphological changes such as cell shrinkage, nuclear condensation (data not presented) and the activation of caspase3. In the caspase8 mutant CRL2571 cells these antibodies did not cause apoptosis as confirmed by the lack of caspase 3 activation (Fig. 2). These results confirmed the previous findings that the CH11 antibodies do not cause apoptosis in CRL2571 cells and established the validity of this cell model for use in present studies. In contrast to the differential cytotoxicity of CH11 antibodies to the wild type and caspase8 mutant (CRL2571) Jurkat cells, 4-HNE had similar cytotoxic effects on both cells as indicated by comparable IC50 values of 4-HNE (14.3±1.2 μM Vs 16.8± 2.3 μM) for these cell lines (Fig.1 Panel B). 4-HNE also induced apoptosis in both wild-type and mutant (CRL2571) cells. However, these results strongly indicated that the wild type Jurkat cells were noticeably more sensitive to 4-HNE-induced apoptosis as compared to the CRL2571 cells. In situ immunohistochemical analysis to assess the activation of caspase3 as the end-point of apoptosis showed that when exposed to 10 μM 4-HNE for 2h, the wild type cells had a noticeably higher number of apoptotic cells as compared to CRL2571 cells (Fig. 2 Panel A). This inference was further supported by the results of Western blot analyses which showed only a minimal activation of caspase3 in 10 μM 4-HNE treated CRL2571 cells while under the identical conditions the wild type cells showed a robust activation of caspase3. Furthermore, exposure to 5 μM 4-HNE caused a clearly noticeable activation of caspase3 only in the wild type cells and not in CRL2571 cells (Fig. 2 Panel B). The differential effect of 4-HNE showing that CRL2571 are relatively more resistant to 4-HNE induced apoptosis as compared to the wild type cells parallels the reported relatively higher resistance of the CRL2571 cells to UV and DOX-induced apoptosis as compared to the wild type cells (19). This is consistent with the earlier studies (3-9, 24) suggesting that 4-HNE is involved in the mechanisms of stress signaling.
Fig.1. Cytotoxicity of 4-HNE to wild type (WT) Jurkat and CRL2571 cells.
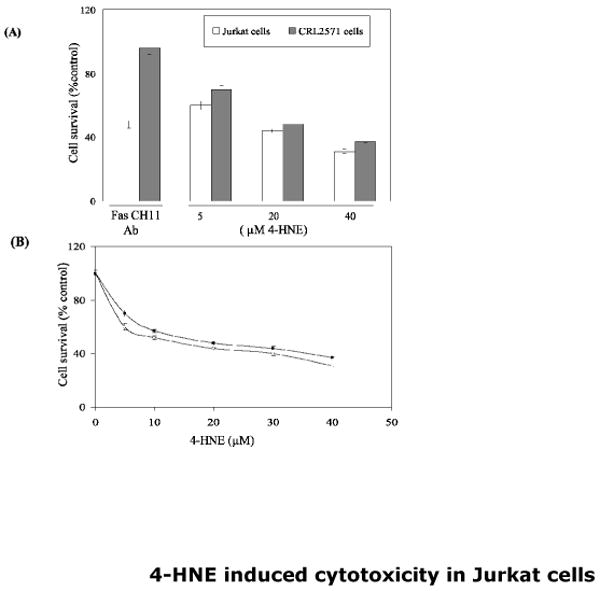
Cells (2× 104) were plated in 96 well plates in complete growth medium and treated with increasing concentration of 4-HNE (0-40μM) for 2h at 37°C. Same number of cells in duplicate columns of 96 well plates was separately treated for 2h with Fas-agonistic CH11 antibodies (50ng/well) and MTT cytotoxicity assay was performed as described in Materials and Methods section. (A): Bar graph showing the percentage of cell survival after exposure of Jurkat and CRL2571 cells with anti-Fas CH11 antibodies or different concentrations of 4-HNE. (B) Cell survival plots for 4-HNE treated WT Jurkat and CRL2571 cells (n=4).
Fig.2. 4-HNE and anti-Fas CH11 antibodies-induced apoptosis in WT and CRL2571 Jurkat cells.
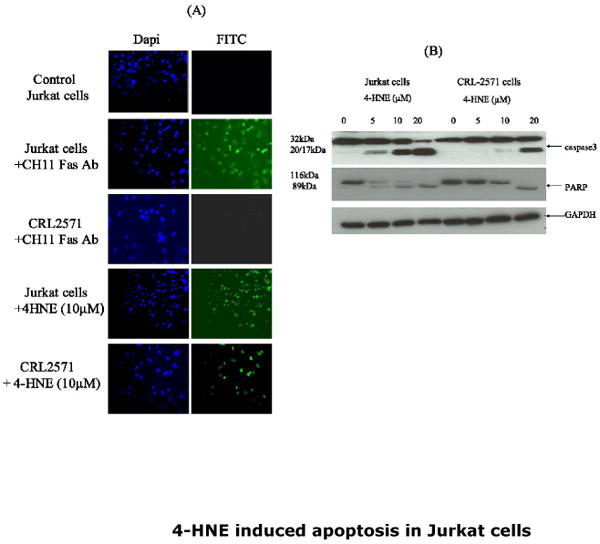
(A) In-situ activation of caspase3: Cells (2×105) were either treated with anti-Fas CH 11 antibodies (250ng/ml) or with 10μM 4-HNE for 2h at 37°C in 8ml complete growth medium. The activation of caspase was examined by staining with 10 μM CaspACE FITC-VAD-FMK in situ marker as per the manufacturer's instructions. The slides were mounted with Vectashield DAPI mounting medium and observed under fluorescence microscope (Nikon). The photographs were taken with 20× objective lens. DAPI and FITC stained cells are appropriately marked in panels (B) Western blot analysis showing the activation of caspase3 and PARP cleavage: WT and CRL2571 Jurkat cells (2×105) were separately treated with different concentrations of 4-HNE (0-20μM) for 2h at 37°C in complete growth medium. The cells were pelleted and washed 2× with PBS and the cell lysates were prepared as described in Materials and Methods section. Cell extracts (50μg protein) were then subjected to Western blot analysis using anti-caspase3 or anti-PARP antibodies. Anti-GAPDH antibodies were used as loading control.
4-HNE causes up-regulation of Fas and induces Fas-mediated apoptosis
To examine the possible role of Fas in 4-HNE-induced apoptosis in Jurkat cells, two series of experiments were carried out. First, we determined the effect of 4-HNE on the expression of Fas. As shown in Fig. 3 (Panels A and B), 4-HNE treatment caused a time- and concentration-dependent increase of Fas. These results are consistent with the reported induction of Fas by 4-HNE in HLE B-3 cells (13) and suggest that induction of Fas by 4-HNE is not limited to a particular cell type. These results, however, did not provide information about the involvement of Fas in 4-HNE induced apoptosis. In order to address this question, a second set of experiments were conducted in which cells were coated with the antagonistic monoclonal antibodies (B-10) to mask Fas. Unlike CH11, B-10 antibodies do not induce apoptosis even though they bind to Fas (25). The results of these experiments showed that cells pre-incubated with B-10 antibodies acquired resistance to apoptosis induced by 4-HNE (Fig.3 Panels C and D). The combined results of the above experiments indicated that Fas was induced by 4-HNE and that it was required for 4-HNE-induced –apoptosis in Jurkat cells through a pathway which was independent of procaspase 8.
Fig.3. Effect of 4-HNE on the expression of Fas in CRL2571 cells.
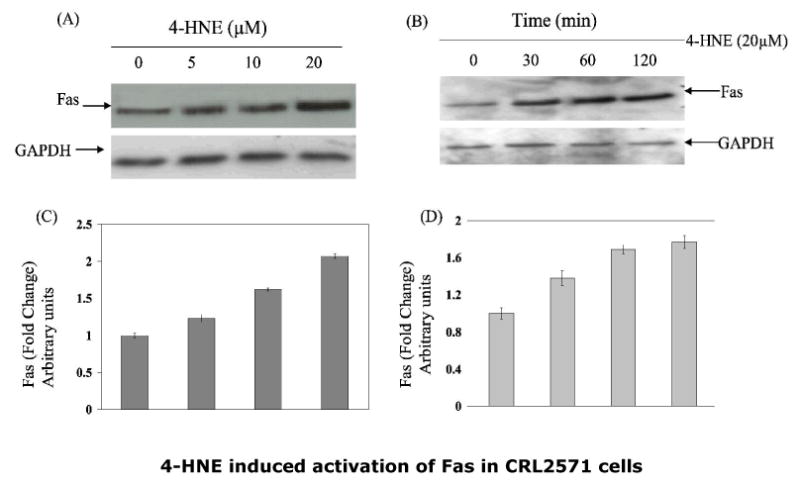
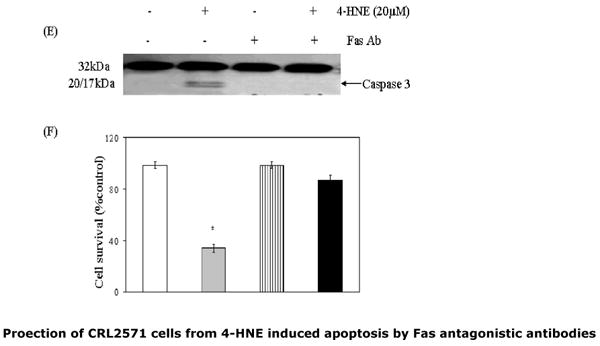
To examine the dose (panel A) or time-dependent (panel B) effect of 4-HNE, cells (2×105) were treated with 4-HNE (0-20μM) for 2h or with 20μM 4-HNE at different time points (0-120min) in complete growth medium at 37°C. After incubation the cells were pelleted, washed 2× with PBS and homogenized in radioimmunoprecipitation assay (RIPA) buffer and centrifuged at 10,000g. Supernatants containing 50μg protein were subjected to Western blot analysis using anti-Fas monoclonal antibodies (B-10) as described in the Methods section. Blots were developed by West Pico-chemiluminescence reagent (Pierce). (C and D) Bar graphs showing the fold change (mean ± SD, n=3) in densitometric analysis of the Fas bands relative to GAPDH bands using Kodak 1D 3.6 image analysis software. (E) Effect of antagonistic anti-Fas antibodies on 4-HNE- induced caspase3 activation: Cells (2×105) were plated in Petri-dishes and pretreated with anti-Fas (B-10) monoclonal antibodies (2μg IgG protein) for 2h followed by 20μM 4-HNE treatment for another 2h at 37°C. Cells were washed with PBS (2×) and counted in a hemocytometer using Trypan blue dye exclusion method. Rest of the cell pellets were extracted in RIPA buffer as described above and the extracts were analyzed for the activation of caspase3 on immunoblots using anti-caspase3 antibodies. (F) Attenuation of 4-HNE toxicity in anti-Fas antibodies (B-10) coated cells: Bar chart showing the percent cell survival in control and Fas antibodies treated cells after treatment with 4-HNE. Data presented are mean ± SD of two separate experiments done in triplicate. (* significantly different from the control cells p<0.01)
4-HNE activates ASK1 and JNK
Previous studies have shown that binding of Daxx to Fas is observed during stress-induced, Fas-mediated apoptosis in several cell types (14-18, 26), and is accompanied by activation of ASK1 and JNK, the kinases involved in the down stream signaling for apoptosis. We have therefore examined the effect of 4-HNE on these parameters in CRL2571 cells, and observed a dose dependent activation of ASK1 and JNK (Fig 4). The activation of both ASK1 and JNK by 4-HNE was rapid and sustained as indicated by our results showing that, for ASK1, activation was maximal at 30 min, and was sustained for both kinases for at least 120 min. These results are consistent with earlier studies (2-5) suggesting that a sustained activation of JNK is required for 4-HNE induced apoptosis. This contention was further supported by the results of experiments showing that pretreatment of CRL2571 cells with the JNK inhibitor, SP600125, made these cells resistant to apoptosis by 4-HNE. (Fig.4 Panel C)
Fig.4. Activation of ASK1 and JNK by 4-HNE and inhibition of 4-HNE induced apoptosis by JNK inhibitor in CRL2571 cells.
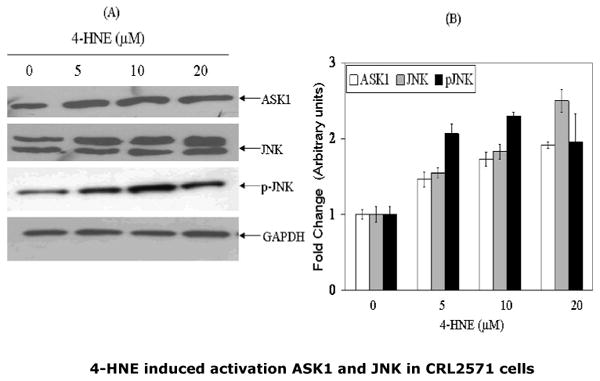
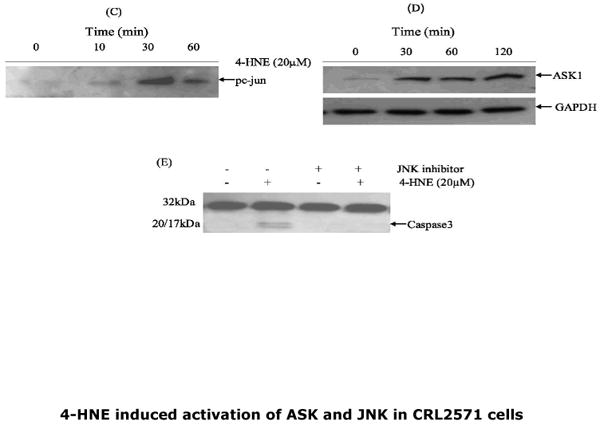
For concentration dependence, cells (2×105) were treated with 0-20μM 4-HNE for 2h. For time course studies, cells were incubated with 20μM 4-HNE for indicated time points at 37°C and processed as described in the Methods section. (A) Dose dependent activation of ASK1, JNK and pJNK by 4-HNE. (B) Histogram showing the fold change in the densitometric analysis of the bands of the Western blots obtained using Kodak ID 3.6 image analysis software. The values (mean ± SD, n=3) of different bands are normalized with those obtained from GAPDH bands used as loading control (C and D) Time course of phospho c-jun induction and ASK1 activation upon treatment with 20μM 4-HNE. (E) Inhibition of 4-HNE induced caspase3 activation by pretreatment of cells with JNK inhibitor SP600125 (40nM).
Effect of 4-HNE on Daxx
Daxx has been shown to bind to the cytosolic domain of Fas during FADD-DISC independent apoptosis caused by oxidative stress in Jurkat, HeLa, and HEK-293 cells (14-18). Therefore, we studied the effect of 4-HNE on Daxx and its interaction with Fas. The results of these experiments showed that 4-HNE treatment caused a rapid and sustained upregulation of Daxx expression in CRL2571 cells (Fig. 5). The induction of Daxx by 4-HNE was rapid and reached a maximum within 30 to 60 min exposure of 4-HNE (Fig. 5 Panel B). Daxx is known to be essentially localized in the nucleus associated with PML and nuclear 10 domains (14-18). When analysis of Daxx was performed in the nuclear and cytoplasmic compartments of 4-HNE treated cells, an increase of Daxx in the cytoplasmic compartment was observed (Fig. 5 Panel C), indicating that 4-HNE treatment facilitated the export of Daxx from nucleus to cytoplasm. Since 4-HNE induces Fas-mediated apoptosis and causes a concomitant induction and translocation of Daxx to cytoplasm, we examined the interaction of 4-HNE with Fas and Daxx and the relevance, if any, of these interactions to the mechanisms of 4-HNE-induced apoptosis in Jurkat cells. Whole-cell lysates of 4-HNE-treated cells were immunoprecipitated with Fas antibodies and these immunoprecipitates were probed with anti-4-HNE-11-S antibodies (Fig. 6). The appearance of bands corresponding to the molecular weights of Fas and Daxx in the Western blots indicated the presence of the adducts of 4-HNE with Fas and Daxx in the immunoprecipitates. The immunoprecipitates obtained with Fas antibodies were also probed with anti-Daxx antibodies. Results of these experiments showed that Daxx was immunoprecipitated with Fas (Fig.6) confirming the previously reported binding of Daxx to Fas (14-18). Increased binding of Daxx to Fas was observed with the time of exposure of cells to 4-HNE (not shown). FADD was not detected in the Fas-immunoprecipitates indicating no interaction of Fas with FADD during 4-HNE induced apoptosis (Fig. 6). Collectively, the results of these experiments indicate that 4-HNE-induced, Fas- mediated apoptosis of Jurkat cells is independent of FADD and procaspase8, and that it is accompanied by i) binding of 4-HNE to Fas and Daxx; ii) induction of both Fas and Daxx; iii) translocation of Daxx to the cytoplasm; iv) increased binding of Daxx to Fas; and v) a rapid and sustained activation of ASK1 and JNK. It should be, however, noted that these results demonstrate a correlation but not necessarily a cause-effect relationship between binding of Daxx to Fas and the activation of downstream signaling kinases ASK1 and JNK.
Fig.5. 4-HNE-mediated induction of Daxx and its cytoplasmic export.
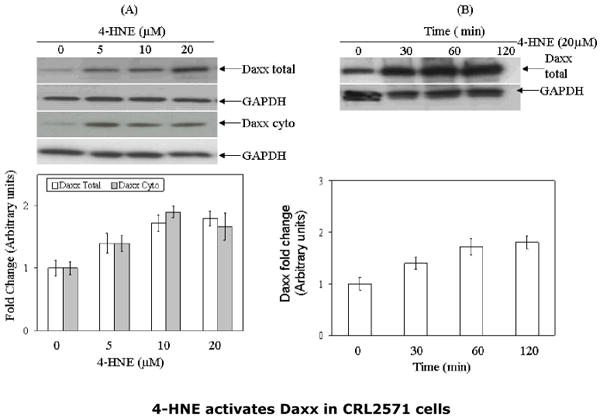
(A) CRL2571(5×105) cells were treated with 4-HNE (0-20μM) at 37°C for 2h and 4-HNE-mediated export of Daxx from nucleus to cytoplasm (B) Cells were treated with 4-HNE (20μM) for indicated time periods. Cells were washed, pelleted and extracted in RIPA buffer. Cell extracts (50μg protein) were resolved on 4-20% gel, immunoblotted on PVDF membranes and immunoblots were probed with anti-Daxx polyclonal antibodies. To assess the 4-HNE-mediated cytoplasmic export of Daxx, cytoplasmic and nuclear extracts of the cells were prepared by using Imgenex cell processing kit, subjected to Western blot analysis and immunoblots were probed with anti-Daxx polyclonal antibodies. Blots were developed by West Pico-chemiluminescence reagent (Pierce). Blots were probed with GAPDH used as loading controls. (C) and (D) Bar charts showing the results of the densitometric analysis (Kodak 1D 3.6 software) of bands in Panels (A) and (B).
Fig.6. Analysis of immunoprecipitates obtained with Fas and Daxx antibodies.
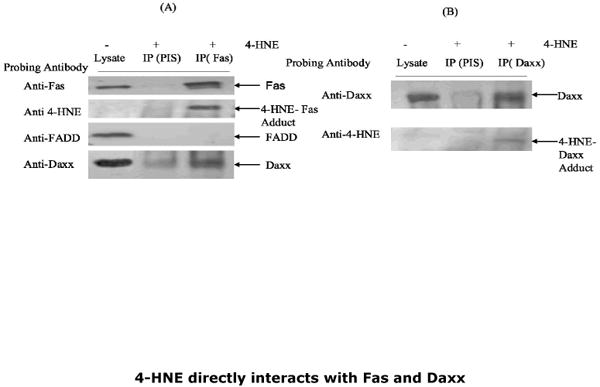
The whole cell lysates of control and 4-HNE (20μM, 2h) treated cells were immunoprecipitated with anti-Fas and anti-Daxx antibodies as described in the Methods section. Immunoprecipitates with preimmune serum (PIS) were included as controls. (A) Immunoprecipitate of Fas (IP Fas) probed with antibodies indicated on panel shows the presence of Fas, 4-HNE-Fas adduct, and Daxx and absence of FADD (B) Immunoprecipitate of Daxx (IP Daxx) probed with antibodies indicated on panel shows the presence of Daxx and 4-HNE-Daxx adduct.
Effect of 4-HNE on Daxx-deficient CRL2571 cells
Since 4-HNE-induced binding of Daxx to Fas was accompanied by signaling for apoptosis, we anticipated that deletion of Daxx will prevent 4-HNE-triggered apoptosis. Furthermore, if binding of Daxx to Fas contributed to the activation of ASK1 and JNK, then these kinases should not be activated upon 4-HNE treatment in Daxx-depleted cells. To test these predictions, we silenced the expression of Daxx by siRNA. As shown in Fig 7 (Panel A), in CRL2571 cells transfected with Daxx siRNA, the level of Daxx was only about 5% of cells transfected with scrambled siRNA. Contrary to our expectations, treatment of Daxx-depleted cells with 4-HNE resulted in a remarkable potentiation of cytotoxic (Fig. 7 Panels B and C) and apoptotic effects of 4-HNE as compared to control cells (Fig 8 Panels A and B). Also, 4-HNE caused a more pronounced activation of ASK1 and JNK in Daxx-depleted than in control cells (Fig. 9). Moreover, CRL2571 cells that were resistant to apoptosis triggered by CH11 Fas antibodies, became sensitized to these antibodies by Daxx depletion, as indicated by the onset of apoptosis in Daxx-deficient cells which was absent in the control cells with normal Daxx expression (Fig. 8 Panel A). Together, these results suggest that the binding of Daxx to Fas is not necessary for either activation of ASK1, JNK and caspase3, or for the onset of 4-HNE induced apoptosis. Increased sensitivity of Daxx-deficient cells to 4-HNE-induced apoptosis along with comparatively more facile activation of ASK1, JNK and caspase3 suggest that at least in Jurkat cells, the binding of Daxx to Fas plays an inhibitory role rather than that of the propagator of proapoptotic signals by 4-HNE. This inhibitory role of Daxx in the mechanisms of signaling for apoptosis is further indicated by abrogation of the resistance of Daxx-depleted CRL2571 cells to apoptosis by CH11 Fas antibodies. It is to be noted that activation of caspase3 and hence apoptosis is not completely inhibited in Daxx deficient 4-HNE treated cells (Fig. 7). This may be expected because in parallel to the extrinsic pathway, 4-HNE also causes activation of p53-mediated intrinsic apoptotic pathway as described later in this section.
Fig.7. Potentiation of the 4-HNE-cytotoxicity to CRL2571 cells upon silencing of Daxx with siRNA.
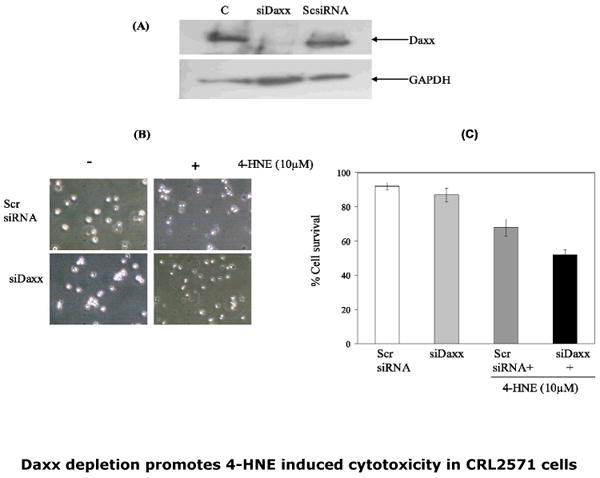
(A) Silencing of Daxx: Cells were transfected with Daxx siRNA duplex (50pmol) as per manufacturer's instructions (SantaCruz Biotech) while controls were treated with scrambled siRNA duplex in a similar manner. The treated cells were harvested 48h post transfection and silencing of Daxx expression was examined by Western blot analysis of the cell extracts using anti-Daxx antibodies. Daxx siRNA (middle lane) caused almost complete silencing of Daxx expression. After ascertaining silencing of Daxx expression, Daxx-depleted cells (2×105) were then grown in six well plates and treated with 10μM 4-HNE for 2h. (B) Phase contrast micrographs showing the effect of 4-HNE (100×) and (C) bar graph showing the percent cell survival determined by Trypan blue dye exclusion, of Daxx depleted and scrambled siRNA transfected cells in control and 4-HNE treated cells. Values are mean ±SD of two separate experiments done in triplicate (* significant differences observed in control and treated cells p<0.01)
Fig.8. Potentiation of apoptosis by 4-HNE and CH-11 through caspase3 activation in Daxx-depleted CRL2571 cells.
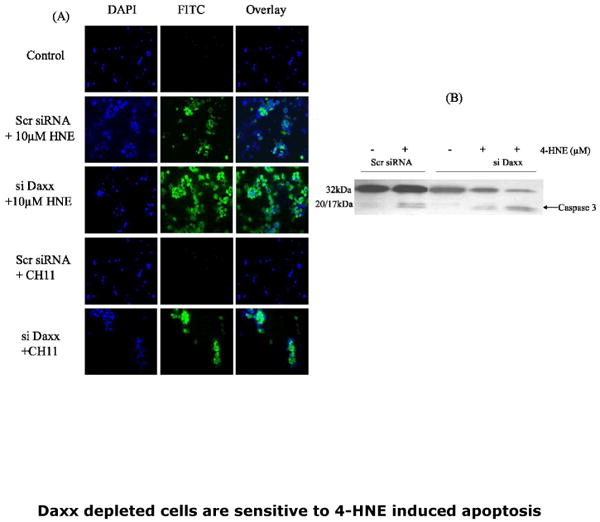
(A) In-situ immunofluorescence analysis: Cells (2×105) were grown in six well plates and treated with either 50ng/well Fas agonistic antibodies or 10uM 4-HNE for 2h at 37°C in complete growth medium. The activation of caspase3 in these cells was examined by staining with 10 μM CaspACE FITC-VAD-FMK in situ marker as per the manufacturer's instructions. After treatment with the in situ marker, the cells were washed twice with PBS and cytospun on polylysine coated slides at 500 rpm for 5min. Cell were fixed in 4% paraformaldehyde, washed 3× with PBS, mounted with Vectashield DAPI mounting medium and observed under fluorescence microscope (Nikon). The photographs were taken with 40× objective lens. Respective labels of DAPI and FITC stained cells in control and 4-HNE treated or Fas CH11 antibodies are shown against different panels. (B) Western blots showing the caspase3 activation in control and Daxx depleted cells exposed to 10 and 20μM 4-HNE.
Fig.9. Potentiation of 4-HNE induced activation of ASK1, JNK and caspase3 in Daxx depleted CRL2571 cells.
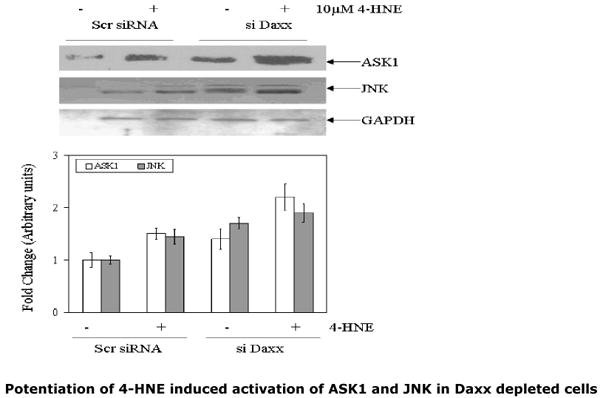
(A) Scrambled siRNA and Daxx-si RNA transfected cells (2×105) were separately incubated with or without 10μM 4-HNE for 2h at 37°C. The extracts of the cells (60μg of protein) were subjected to Western blot analysis. Immunoblots were probed with anti-ASK1 and anti-JNK antibodies and developed by West Pico-chemiluminescence's reagent (Pierce). (B) Bar charts showing the results of the densitometric analysis (Kodak 1D 3.6 software) of bands of immunoblots obtained from three different experiments (mean ± SD, n=3)
4-HNE causes translocation of HSF1 to nucleus and up regulation of expression of Hsp70
Previous studies (27) have suggested that stress-induced export of Daxx from nucleus to cytoplasm leads to the activation of HSF1. Therefore, we examined whether or not 4-HNE-induced transport of Daxx to cytoplasm had any effect on HSF1. Results presented in Fig.10 showed that concomitant with the cytoplasmic export of Daxx in 4-HNE treated CRL2571 cells, enhanced accumulation of HSF1 in the nucleus was observed (Fig. 10 Panels A, B and C) in a time dependent manner suggesting an increase in the transcription of stress responsive heat shock proteins. This was confirmed by the results showing that the treatment of cells with 4-HNE caused a significant increase in the expression of Hsp70 in cells (Fig. 10 Panel D)
Fig.10. 4-HNE-induced cytoplasmic export of nuclear Daxx in CRL2571 cells is accompanied with the activation of HSF1 and up regulation of Hsp70.
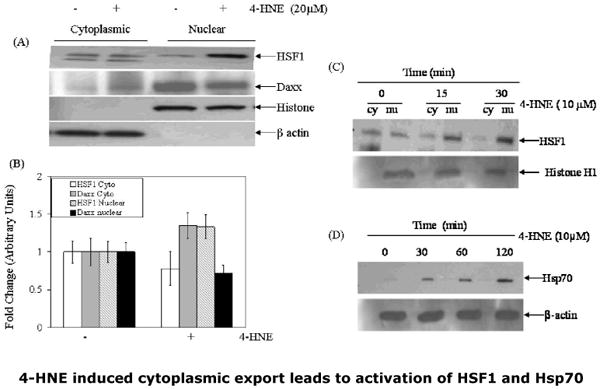
(A) Cells (2×105) were treated with 20μM 4-HNE for 2h at 37°C. After completion of incubation, the cells were washed 2× in PBS and pelleted by centrifugation. Nuclear and cytoplasmic extracts of the cell pellets were prepared by using the Imgenex cell processing kit. Extracts (50μg protein) were subjected to Western blot analysis by using anti-HSF1 and anti-Daxx antibodies as described in the Methods section. Immunoblots were also probed with β-actin and histone H1 antibodies as loading controls and to ascertain purity of cytoplasmic and nuclear fractions. (B) Bar chart showing the densitometric analysis of HSF1 and Daxx bands of immunoblots obtained from three different experiments expressed as fold change (mean ± SD, n=3). (C) Time course of 4-HNE induced nuclear accumulation and activation of HSF1 (D) Time course of 4-HNE induced activation of Hsp70 in CRL2571 cells.
Effect of 4-HNE on p53
The contribution of 4-HNE to apoptosis through the Fas-dependent extrinsic pathway and its ability to interact with the nuclear protein Daxx in a parallel manner prompted us to investigate any possible effect of 4-HNE on p53-mediated intrinsic pathway for apoptosis (28, 29). We therefore measured p53 in the cytoplasmic and nuclear compartments of CRL2571 cells treated with different concentrations of 4-HNE for 2h. Results of these experiments, presented in Fig 11 (Panel A) indicated a dose-dependent increase in p53 expression in both cytosolic and nuclear fractions. Induction of Bax has been reported during the p53-mediated intrinsic apoptotic pathway. We therefore compared the expression of Bax in control and 4-HNE-treated CRL2571 cells. Similar to p53, a consistent increase in the expression of Bax was observed in 4-HNE-treated cells (Fig.11 Panel B). These results suggest that, besides activating the Fas-linked pathway, 4-HNE may also contribute to apoptosis via the p53-dependent intrinsic pathway, thus supporting the existence of multiple pathways for 4-HNE-induced apoptosis, similar to mechanisms reported for UV-induced apoptosis in HeLa cells (30).
Fig.11. Effect of 4-HNE on the expression of p53 and Bax in CRL2571 cells.
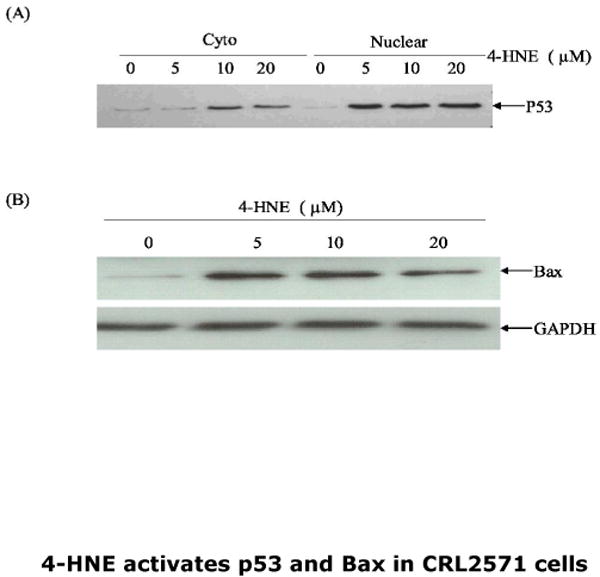
Cells (2×105) were treated with 4-HNE (0-20μM) for 2h at 37°C in complete growth medium. (A) Nuclear and cytoplasmic extracts of the cells were prepared by the Imgenex cell processing kit and 50μg of protein from each extract was subjected to the Western blot analysis by using anti-p53 antibodies. (B) The immunoblots of whole cell extract (50μg protein) prepared in RIPA lysis buffer probed with anti- Bax antibodies. Blots probed with anti-GAPDH antibodies are shown as loading control.
Discussion
The results of our present work are consistent with previous studies suggesting that 4-HNE acts as an important signaling molecule and that it can induce apoptosis in a wide variety of cells through the activation of JNK and caspase3 (2-5, 30-34). We have now demonstrated that in Jurkat cells, 4-HNE induces apoptosis through a pathway which is independent of Fas-ligand and of DISC formation. CRL2571 cells express an inactive mutant of procaspase8 (19) and are resistant to apoptosis caused by FasL and by toxic-Fas antibodies CH11 (19) because of an inability to form DISC. Since 4-HNE causes apoptosis in both wild type and procaspase8 mutant CRL2571 Jurkat cells, 4-HNE-induced apoptosis in these cells is independent of the canonical extrinsic pathway initiated by the interaction of Fas with FasL. Our present studies suggest that 4-HNE-induced Fas-mediated apoptosis is not limited to a particular cell type because we have earlier shown that 4-HNE causes Fas-dependent apoptosis in HLE B-3 cells (13).
The presence of 4-HNE-Fas adducts in the immunoprecipitates of the lysates from 4-HNE treated cells obtained with Fas antibodies indicates that 4-HNE does indeed modify Fas. The formation of 4-HNE adducts on Fas is required to transduce signaling for apoptosis, because when Fas is masked by coating with non-apoptotic, antagonistic anti-Fas antibodies, the cells acquire resistance to 4-HNE-induced apoptosis. Though not investigated in detail, the present studies suggest that 4-HNE also induces p53- mediated apoptosis in Jurkat cells. Further studies are underway to explore this possibility. Our results showing modification of Fas by 4-HNE and subsequent activation of the Fas-L independent pathway for apoptosis lend support to the idea that 4-HNE can affect cellular signaling by mimicking the effects of receptor ligands (13, 35). It has been demonstrated that 4- HNE can activate membrane tyrosine kinase receptors (RTK) and mimic the effect of their ligands on the downstream signaling events (8, 35-37). Classically, the epidermal growth factor receptor (EGFR) is activated by binding its ligand EGF. This interaction leads to dimerization of the receptor and activation of its intrinsic cytosolic tyrosine kinase resulting in the trans-auto-phosphorylation of tyrosine residues and activation of down stream signaling events (35-37). It has been shown that reaction of 4-HNE with EGFR leads to its aggregation (38), which can affect the EGFR-mediated signaling events somewhat similar to EGF (35-37). The induction of Fas-mediated signaling for apoptosis by 4-HNE reported here indicates that membrane receptors of different gene families (e.g., death receptor, RTK) can be modulated by 4-HNE. Therefore, the possibility of 4-HNE interacting with other membrane receptors should be vigorously explored. In addition to its interactions with membrane receptors, 4-HNE can also exert a regulatory role on cellular signaling through its interactions with nuclear proteins as indicated by its anti-apoptotic effect on the transcription repressor Daxx and a simultaneous pro-apoptotic effect on p53. Such interactions of 4-HNE may explain a wide spectrum of effects of 4-HNE on cellular events reported over the years (1, 24, 30-32).
Earlier studies, including our own (13-18), have suggested the involvement of Daxx in a DISC-independent pathway for apoptosis such as that seen in the present studies. Binding of Daxx to Fas is accompanied by the activation of ASK1 and subsequent downstream signals for the execution of apoptosis (14). Our present studies reaffirm the binding of Daxx to Fas and demonstrate further that 4-HNE enhances this binding by facilitating the export of Daxx from the nucleus to the cytoplasm. However, while the binding of Daxx to Fas has been conclusively demonstrated (14-18), there is no strong evidence that this interaction is required for downstream signals for apoptosis. In Fas- mediated apoptosis caused by UV, the suggested role of Daxx is controversial. While some studies indicate that the binding of Daxx to Fas is required for the activation of ASK1, JNK, and subsequent apoptosis (14-18, 30), in other studies an inhibitory role of Daxx has been suggested (39). Our studies of procaspase-8-deficient Jurkat cells clearly demonstrate that the silencing of Daxx leads to a potentiation of 4-HNE-induced apoptosis in these cells, as well as to the activation of ASK1, JNK, and caspase3. These results indicate that binding of Daxx to Fas inhibits 4-HNE-triggered signaling from Fas to ASK1 and JNK that is required for apoptosis in these cells. CRL2571 cells are resistant to apoptosis caused by the ‘toxic’ Fas antibodies (CH11) which can induce apoptosis in the wild type cells. Upon silencing of Daxx, CRL2571 cells become sensitive to apoptosis by toxic Fas antibodies. This is consistent with the interpretation that binding of Daxx to Fas is not only unnecessary for Fas-mediated signaling for apoptosis, but that such binding is actually inhibitory to Fas-mediated apoptosis. This inhibitory role of Daxx in Fas-mediated apoptosis upon exposure to 4-HNE agrees with some of the previous studies (30, 39) showing that Daxx silencing promotes Fas-mediated apoptosis caused by UV or oxidative stress causing agents (e.g. doxorubicin) in HeLa cells (30, 39).
Our results showing ASK1, JNK and caspase3 activation in CRL2571 cells by 4-HNE are also consistent with numerous other studies on various cell types in which a sustained activation of JNK has been demonstrated during 4-HNE or stress (UV, H2O2, superoxide anion, heat, DOX)-induced apoptosis (2, 3, 24). It has been suggested that activation of ASK1 is dependent on the cellular redox status regulated by thioredoxin and thioredoxin reductase system (40). The reduced form of thioredoxin binds to ASK1 leading to inhibition of its kinase activity. However, during oxidative stress dissociation of thioredoxin from ASK1 causes the activation of ASK1 by oligomerization which leads to the activation of JNK and apoptosis (41). The inhibition of thioredoxin reductase through direct interaction with 4-HNE has also been shown both in vitro and in vivo (42). ASK1-independent activation of JNK during oxidative stress has also been shown to be regulated by GST-pi (43). It is possible that the 4-HNE induced activation of ASK1 and JNK observed during present studies in CRL2571 may be due to the modulation of thioredoxin -thioredoxin reductase system by 4-HNE. Further studies are required to explore this possibility. 4-HNE-induced apoptosis in CRL-2571 cells can be blocked by inhibiting JNK which indicates that the activation of JNK is an obligatory step in the process. Since 4-HNE is almost always formed in cells subjected to stress through UV, chemicals, heat, starvation etc., 4-HNE is likely to be a major player in signaling initiated by these agents, in agreement with several earlier studies (2-5) where 4-HNE has been shown to be a common denominator in signaling for apoptosis by oxidants and UV radiation in various cell types.
An interesting parallel can be drawn between 4-HNE signaling, and signaling mediated by hydrogen peroxide (H2O2) (44, 45). Both, H2O2 and 4- HNE cause proliferation at lower concentrations and apoptosis at higher concentrations. Both these messengers are reactive small molecules with a biological half-life comparable to the time scale of many biological processes. This allows for diffusibility over distances needed to transmit a signal, but also assures a physiologically appropriate rate of signal termination. The moderate reactivity of both H2O2 and 4-HNE provides a balance between signaling efficiency and selectivity. However, these similarities of H2O2 and 4-HNE signaling do not necessarily mean that the mechanisms are redundant. Sulfhydryl groups are the main targets of H2O2-mediated oxidation whereas modification by 4-HNE is characterized by a broader set of targets, including amino groups in addition to sulfhydryls, and a wider range of reaction types, including Michael addition and Schiff base formation. Therefore, during signaling H2O2 and 4-HNE are likely to target different proteins, and the resulting modifications are chemically and functionally distinct from each other.
Our present studies show that 4-HNE causes a dose and time-dependent up regulation of Fas and Daxx. Moreover, we demonstrate for the first time that 4-HNE also promotes the translocation of Daxx to the cytosol where it binds to Fas. Such translocation has been previously documented (14-18) for cells stressed with UV or DOX exposure, i.e., under conditions that trigger 4-HNE generation (24). These findings are consistent with a hypothetical model in which 4-HNE would act as one of the chemical pro-apoptotic signals elicited by a variety of different stressors.
The effects of 4-HNE on the expression of Fas and on the expression and compartmentalization of Daxx suggest the presence of a self-limiting regulatory circuit. The physiologic significance and the mechanistic details of the inhibitory Daxx-Fas interaction still need to be clarified, particularly in terms of concentration dependence and time course relative to that of the activation of Fas by 4-HNE. However, according to our proposed model (Fig.12), moderate levels of 4-HNE could trigger the translocation of Daxx to the cytoplasm where Daxx would inhibit Fas-mediated apoptosis. During severe stress, elevated 4-HNE could cause both an over-expression of Fas and its modification by 4-HNE, leading to the onset of Fas-mediated signaling. This signaling could reach the threshold necessary to overcome the Daxx inhibition, and lead to massive apoptosis.
Fig.12. Schematic representation of a model showing the role of 4-HNE in Fas-mediated signaling.
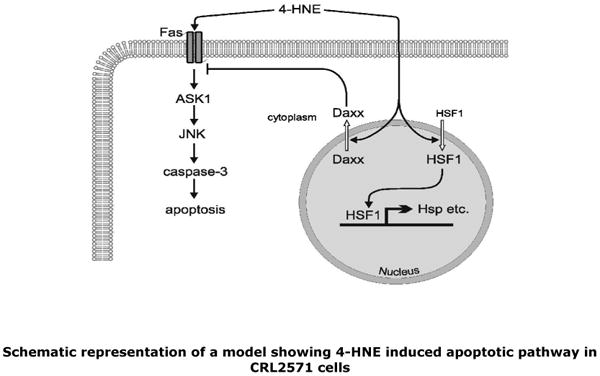
4-HNE promotes apoptosis by directly interacting with Fas resulting in the activation of down stream kinases ASK1 and JNK leading to the proteolytic cleavage of caspase3 culminating in apoptosis. On the other hand, a direct interaction of 4-HNE with Daxx facilitates its export from the nuclear compartment to cytosol where it binds to Fas and inhibits Fas-mediated apoptosis. Translocation of Daxx also leads to the activation of transcription factor HSF1 to transcribe stress responsive heat shock proteins.
Because of the diffusibility of 4-HNE, a pro-apoptotic signal could spread beyond the initial site of the insult to adjacent cells or tissues. Therefore, a 4-HNE-triggered export of Daxx to the cytoplasm that serves to control and limit a runaway apoptosis process would be of special importance to adjacent “bystander” cells. Such a protective role of 4-HNE may be augmented by 4-HNE-linked translocation of HSF1 into the nucleus as observed during present studies. In a complex with remaining nuclear Daxx (27), trimeric HSF1 would activate the transcription of several heat shock proteins and thus contribute to protection of the cells. Such seemingly opposite effects of 4-HNE on cellular signaling have been reported previously in studies showing that 4-HNE can either activate or inhibit PKC in a concentration dependent manner (46, 47).
It is noteworthy that the already mentioned similarities in H2O2 and 4-HNE signaling extend to the logic of their respective regulatory circuits (albeit not to the underlying biochemical reactions). Specifically, we postulate that the inhibitory action of Daxx dampens the apoptotic response to 4-HNE at low (sub-threshold) but not at high (above-threshold) 4-HNE concentrations, and that the system becomes self-limiting, at least spatially, because diffusion of 4-HNE into adjacent cells elicits Daxx-mediated inhibition of apoptosis. This non-linear response of the 4-HNE circuit parallels that of H2O2. (48): below a certain threshold, peroxiredoxin efficiently removes any H2O2 that is generated and inhibits signaling; when H2O2 levels rise above that threshold, peroxiredoxin is oxidatively inactivated, thus opening the “floodgates” and allowing H2O2 signaling to proceed (48). Similarly, it is possible that the inhibition of thioredoxin-thioredoxin reductase system by 4-HNE above a certain threshold concentration may profoundly affect the apoptotic signal transduction through ASK1-JNK-caspase3 pathway. The striking functional similarity of mechanisms that utilize different signaling molecules, different chemistry and different target proteins suggests that the resulting non-linear regulatory circuits are robust and biologically useful.
Stress induced activation and nuclear translocation of the heat shock transcription factor 1 (HSF1) resulting in enhanced expression of heat shock proteins is one of the major mechanisms for cells to recover from oxidative damage. It has been suggested that in unstressed cells the activation of HSF1 is inhibited through its association with heat shock proteins such as Hsp70 Hsp90 and other co-chaperons (49, 50). Under the conditions of oxidative stress this inhibition of HSF1 has been suggested to be abrogated/attenuated by the direct interaction of 4-HNE with Hsp70 and/or Hsp90 (51,52). Recently, through immunoprecipitation and c-myc tagged Hsp70 data, it has been shown that 4-HNE treatment inhibits the interaction of Hsp70 and HSF1 leading to enhanced accumulation of HSF1 and increased transcription of heat shock proteins (53). Our results showing 4-HNE-induced translocation of Daxx from the nucleus, and HSF1 into the nucleus may have major implications for the mechanisms of stress-mediated signaling, particularly by stressors such as exposure to UV radiation, heat shock, or oxidants which invariably increase intracellular concentration of 4-HNE in cells. Daxx is a transcription repressor which can regulate the expression of stress responsive genes. It has been also shown that release of Daxx from nuclear compartment to cytoplasm leads to the activation of transcription factor HSF1 that up regulates the expression of HSF1-associated genes (27 Likewise, it is known that interaction of Daxx with other transcription factors such as ETS1, Pax 3, Pax5, smad4 and p53 also affects the expression of associated genes (46, 54-57). The involvement of 4-HNE in promoting the cytoplasmic export of Daxx with concomitant activation and accumulation of HSF1 and the activation of Hsp70 in CRL2571 demonstrated during present studies suggests a novel mechanism in stress mediated signaling through which 4-HNE can influence a large set of fundamental cellular processes including proliferation, cell cycle events and apoptosis, in part by profoundly affecting the expression of numerous genes (10, 34). For example, 4-HNE can modulate the expression of genes including p53, c-myc, AKT, PKC, TGFβ, ASK1, JNK, Nrf2, integrins, connexins, ERK pathway genes, aldose reductase, glycogen synthase kinase, gamma GGT, adenylate cyclase, RalBP-1, and heat shock proteins among others, and affect cellular processes associated with these genes (1,33,34,58). The present studies, demonstrating the effects of 4-HNE on the transcription repressor, Daxx, may provide a clue to the mechanism(s) through which 4-HNE could affect such a multitude of cellular events.
Abbreviations
- 4-HNE
4-hydroxynonenal
- DISC
death-inducing signaling complex
- ASK1
apoptosis signal regulating kinase
- JNK
c-jun N-terminal kinase
- DOX
doxorubicin
- TNFR
tumor necrosis factor receptor
- RTK
receptor tyrosine kinase
- Daxx
death domain- associated protein
- PARP
poly(ADP-ribose) polymerase
- PBS
phosphate buffered saline
- HSF
heat shock factor 1
- PKC
protein kinase C
Footnotes
Support for this study was provided in part by NIH grants EY 04396, ES021171 (YCA), CA77495 (SA), and ES 07804 (PZ).
References
- 1.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;23:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 2.Cheng JZ, Sharma R, Yang Y, Singhal SS, Sharma A, Saini MK, Singh SV, Zimniak P, Awasthi S, Awasthi YC. Accelerated metabolism and exclusion of 4-hydroxynonenal through induction of RLIP76 and hGST5.8 is an early adaptive response of cells to heat and oxidative stress. J Biol Chem. 2001;276(44):41213–41223. doi: 10.1074/jbc.M106838200. [DOI] [PubMed] [Google Scholar]
- 3.Yang Y, Sharma A, Sharma R, Patrick B, Singhal SS, Zimniak P, Awasthi S, Awasthi YC. Cells preconditioned with mild, transient UVA irradiation acquire resistance to oxidative stress and UVA-induced apoptosis: role of 4-hydroxynonenal in UVA-mediated signaling for apoptosis. J Biol Chem. 2003;278(42):41380–41388. doi: 10.1074/jbc.M305766200. [DOI] [PubMed] [Google Scholar]
- 4.Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17(13):5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Awasthi YC, Sharma R, Cheng JZ, Yang Y, Sharma A, Singhal SS, Awasthi S. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Mol Aspects Med. 2003;24(45):219–230. doi: 10.1016/s0098-2997(03)00017-7. [DOI] [PubMed] [Google Scholar]
- 6.Barrera G, Pizzimenti S, Dianzani MU. 4-hydroxynonenal and regulation of cell cycle: effects on the pRb/E2F pathway. Free Radic Biol Med. 2004;37(5):597–606. doi: 10.1016/j.freeradbiomed.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 7.West JD, Ji C, Duncan ST, Amarnath V, Schneider C, Rizzo CJ, Brash AR, Marnette LJ. Induction of apoptosis in colorectal carcinoma cells treated with 4-hydroxynonenal and structurally related aldehydic products of lipid peroxidation. Chem Res Toxicol. 2004;17(4):453–462. doi: 10.1021/tx034248o. [DOI] [PubMed] [Google Scholar]
- 8.Negre-Salvayre A, Vieira O, Escarguil-Blane I, Salvayre R. Oxidized LDL and 4-hydroxynonenal modulate tyrosine kinase receptor activity. Mol Apects Med. 2003;24(45):251–261. doi: 10.1016/s0098-2997(03)00020-7. [DOI] [PubMed] [Google Scholar]
- 9.Nakashima I, Liu W, Akhand AA, Takeda K, Kawamoto Y, Kato M, Suzuki H. 4-hydroxynonenal triggers multistep signal transduction cascades for suppression of cellular functions. Mol Aspects Med. 2003;24(45):231–238. doi: 10.1016/s0098-2997(03)00018-9. [DOI] [PubMed] [Google Scholar]
- 10.Sharma R, Brown D, Awasthi S, Yang Y, Sharma A, Patrick B, Saini MK, Singh SP, Zimniak P, Singh SV, Awasthi YC. Transfection with 4-hydroxynonenal-metabolizing glutathione S-transferase isozymes leads to phenotypic transformation and immortalization of adherent cells. Eur J Biochem. 2004;271(9):1690–1701. doi: 10.1111/j.1432-1033.2004.04067.x. [DOI] [PubMed] [Google Scholar]
- 11.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 12.Nagata S. Apoptosis by death factor. Cell. 1997;88(3):355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Sharma R, Patrick B, Sharma A, Jeyabal PV, Reddy PM, Saini MK, Dwivedi S, Dhanani S, Ansari NH, Zimniak P, Awasthi S, Awasthi YC. Regulation of CD95 (Fas) expression and Fas-mediated apoptotic signaling in HLE B-3 cells by 4-hydroxynonenal. Biochemistry. Biochemistry. 2006;45(40):12253–12264. doi: 10.1021/bi060780+. [DOI] [PubMed] [Google Scholar]
- 14.Yang X, Khosravi-Far R, Chang HY, Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell. 1997;89(7):1067–1076. doi: 10.1016/s0092-8674(00)80294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khelifi AF, D'Alcontres MS, Salomoni P. Daxx is required for stress-induced cell death and JNK activation. Cell Death Differ. 2005;12(7):724–733. doi: 10.1038/sj.cdd.4401559. [DOI] [PubMed] [Google Scholar]
- 16.Chang CC, Lin DY, Fang HI, Chen RH, Shih HM. Daxx mediates the small ubiquitin-like modifier-dependent transcriptional repression of Smad4. J Biol Chem. 2005;280(11):64–73. doi: 10.1074/jbc.M409161200. [DOI] [PubMed] [Google Scholar]
- 17.Boehrer S, Nowak D, Hochmuth S, Kim SZ, Trepohl B, Afkir A, Hoelzer D, Mitrou PS, Weidmann E, Chow KU. Daxx overexpression in T-lymphoblastic Jurkat cells enhances caspase-dependent death receptor- and drug-induced apoptosis in distinct ways. Cell Signal. 2005;17(5):581–595. doi: 10.1016/j.cellsig.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 18.Salomoni P, Khelifi AF. Daxx: death or survival protein? Trends Cell Biol. 2006;16(2):97–104. doi: 10.1016/j.tcb.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Juo P, Kuo CJ, Yuan J, Blenis J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr Biol. 1998;8(18):1001–1008. doi: 10.1016/s0960-9822(07)00420-4. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, Shimizu S, Sakamaki K, Yonehara S, Tsujimoto Y. A caspase-8 independent signaling pathway activated by Fas ligation leads to exposure of Bak N-terminus. J Biol Chem. 2004;279(32):33865–33874. doi: 10.1074/jbc.M403499200. [DOI] [PubMed] [Google Scholar]
- 21.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(12):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 22.Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activation of stress signaling pathways by thge end product of lipid peroxidation. 4-Hydroxy-2-nonenal is a potential inducer of intracellular peroxide formation. J Biol Chem. 1999;274(4):2234–2242. doi: 10.1074/jbc.274.4.2234. [DOI] [PubMed] [Google Scholar]
- 23.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 24.Awasthi YC, Ansari GA, Awasthi S. Regulation of 4-hydroxynonenal mediated signaling by glutathione S-transferases. Methods Enzymol. 2005;401:379–407. doi: 10.1016/S0076-6879(05)01024-4. [DOI] [PubMed] [Google Scholar]
- 25.Fadeel B, Thorpe CJ, Yonehara S, Chiodi F. Anti-Fas IgG1 antibodies recognizing the same epitope of Fas/APO-1 mediate different biological effects in vitro. Int Immunol. 1997;9(2):201–209. doi: 10.1093/intimm/9.2.201. [DOI] [PubMed] [Google Scholar]
- 26.Raoul C, Barthelemy C, Couzinet A, Hancock D, Pettmann B, Hueber AO. Expression of a dominant negative form of Daxx in vivo rescues motoneurons from Fas (CD95)-induced cell death. J Neurobiol. 2005;62(2):178–188. doi: 10.1002/neu.20086. [DOI] [PubMed] [Google Scholar]
- 27.Boellmann F, Guettouche T, Guo Y, Fenna M, Mnayer L, Voellmy R. DAXX interacts with heat shock factor 1 during stress activation and enhances its transcriptional activity. Proc Nat l Acad Sci U S A. 2004;101(12):4100–41005. doi: 10.1073/pnas.0304768101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang XW. Role of p53 and apoptosis in carcinogenesis Anticancer Res. Anticancer Res. 1999;19(6A):4759–4771. [PubMed] [Google Scholar]
- 29.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2(8):594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 30.Chen LY, Chen JD. Daxx silencing sensitizes cells to multiple apoptotic pathways. Mol Cell Biol. 2003;23(20):7108–7121. doi: 10.1128/MCB.23.20.7108-7121.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parola M, Robino G, Marra F, Pinzano M, Bellomo G, Leonarduzzi G, Chiarugi P, Camandola S, Poli G, Waeg G, Gentilini P, Dianzani MU. HNE interacts directly with JNK isoforms in human hepatic stellate cells. J Clin Invest. 1998;102(11):1942–1950. doi: 10.1172/JCI1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickinson DA, Iles KE, Watanabe N, Iwamoto T, Zhang H, Karzywanski DM, Foreman HJ. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic Biol Med. 2002;33(7):974–987. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 33.Awasthi YC, Yang Y, Tiwari NK, Patrick B, Sharma A, Li J, Awasthi S. Regulation of 4-hydroxynonenal-mediated signaling by glutathione S-transferases. Free Radic Biol Med. 2004;37(5):607–619. doi: 10.1016/j.freeradbiomed.2004.05.033. [DOI] [PubMed] [Google Scholar]
- 34.Dianzani MU. 4-hydroxynonenal from pathology to physiology. Mol Aspects Med. 2003;24(45):263–272. doi: 10.1016/s0098-2997(03)00021-9. [DOI] [PubMed] [Google Scholar]
- 35.Liu W, Akhand AA, Kato, Yokoyama I, Miyata T, Kurokawa K, Uchida K, Nakashima IM. 4-hydroxynonenal triggers an epidermal growth factor receptor-linked signal pathway for growth inhibition. J Cell Sci. 1999;112(Pt14):2409–2417. doi: 10.1242/jcs.112.14.2409. [DOI] [PubMed] [Google Scholar]
- 36.Escargueil-Blanc I, Salvayre R, Vacaresse N, Jurgens G, Darblade B, Arnal JF, Parthasarathy S, Negre-Salvayre A. Mildly oxidized LDL induces activation of platelet-derived growth factor beta-receptor pathway. Circulation. 2001;104(15):1814–1821. doi: 10.1161/hc4001.097179. [DOI] [PubMed] [Google Scholar]
- 37.Vacaresse N, Vieira O, Robbesyn F, Jurgens G, Salvayre R, Negre-Salvayre A. Phenolic antioxidants trolox and caffeic acid modulate the oxidized LDL-induced EGF-receptor activation. Br J Pharmacol. 2001;132(8):1777–1788. doi: 10.1038/sj.bjp.0703981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uchida K, Stadtman ER. Selective cleavage of thioether linkage in proteins modified with 4-hydroxynonenal. Proc Natl Acad Sci U S A. 1992;89(12):5611–5615. doi: 10.1073/pnas.89.12.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michaelson JS, Leder P. RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J Cell Sci. 2003;116(Pt 2):345–352. doi: 10.1242/jcs.00234. [DOI] [PubMed] [Google Scholar]
- 40.Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40(6):928–39. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 41.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17(9):2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang J, Holmgren A. Inhibition of thioredoxin and thioredoxin reductase by 4-hydroxy-2-nonenal in vitro and in vivo. J Am Chem Soc. 2006;128(6):1879–1885. doi: 10.1021/ja057358l. [DOI] [PubMed] [Google Scholar]
- 43.Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z. Regulation of JNK signaling by GSTp. EMBO J. 1999;18(5):1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8(9):722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- 45.Karin M, Shaulian E. AP-1: linking hydrogen peroxide and oxidative stress to the control of cell proliferation and death. IUBMB Life. 2001;52(12):17–24. doi: 10.1080/15216540252774711. [DOI] [PubMed] [Google Scholar]
- 46.Marinari UM, Nitti M, Pronzato MA, Domenicotti C. Role of PKC-dependent pathways in HNE-induced cell protein transport and secretion. Mol Aspects Med. 2003;24(45):205–211. doi: 10.1016/s0098-2997(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 47.Numazawa S, Ishikawa M, Yoshida A, Tanaka S, Yoshida T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am J Physiol Cell Physiol. 2003;285(2):C334–342. doi: 10.1152/ajpcell.00043.2003. [DOI] [PubMed] [Google Scholar]
- 48.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300(5619):650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 49.Mosser DD, Duchaine J, Massie B. The DNA-binding activity of the human heat shock transcription factor is regulated in vivo by hsp70. Mol Cell Biol. 1993;13(9):5427–38. doi: 10.1128/mcb.13.9.5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94(4):471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 51.Carbone DL, Doorn JA, Kiebler Z, Sampey BP, Petersen DR. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem Res Toxicol. 2004;17(11):1459–1467. doi: 10.1021/tx049838g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Exp Ther. 2005;315(1):8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- 53.Jacobs AT, Marnett LJ. Heat shock factor-1 attenuates 4-hydroxynonenal-mediated apoptosis: Critical role for HSP70 induction and stabilization of BC L-XL. J Biol Chem. 2007 doi: 10.1074/jbc M706799200. [DOI] [PubMed] [Google Scholar]
- 54.Perlman R, Schiemann WP, Brooks MW, Lodish HF, Weinberg RA. TGF-beta-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat Cell Biol. 2001;3(8):708–714. doi: 10.1038/35087019. [DOI] [PubMed] [Google Scholar]
- 55.Hollenbach AD, Sublett JE, McPherson CJ, Grosveld G. The Pax3-FKHR oncoprotein is unresponsive to the Pax3-associated repressor hDaxx. EMBO J. 1999;18(13):3702–3711. doi: 10.1093/emboj/18.13.3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li R, Pei H, Watson DK, Papas TS. EAP1/Daxx interacts with ETS1 and represses transcriptional activation of ETS1 target genes. Oncogene. 2000;19(6):745–753. doi: 10.1038/sj.onc.1203385. [DOI] [PubMed] [Google Scholar]
- 57.Emelyanov AV, Kovac CR, Sepulveda MA, Birshtein BK. The interaction of Pax5 (BSAP) with Daxx can result in transcriptional activation in B cells. J Biol Chem. 2002;277(13):11156–11164. doi: 10.1074/jbc.M111763200. [DOI] [PubMed] [Google Scholar]
- 58.Patrick B, Li J, Jeyabal PV, Reddy PM, Yang Y, Sharma R, Sinha M, Luxon B, Zimniak P, Awasthi S, Awasthi YC. Biochem Biophys Res Commun. 2005;334(2):425–432. doi: 10.1016/j.bbrc.2005.06.099. [DOI] [PubMed] [Google Scholar]