

Abstract
Three new N-methyl-4-hydroxy-2-pyridinone analogs, 6-epi-oxysporidinone (3), the dimethyl ketal of oxysporidinone (4), and N-demethylsambutoxin (5), along with the known compounds, (−)-oxysporidinone (1), (−)-sambutoxin (2), wortmannin (6), enniatin A (7), enniatin A1 (8), and enniatin B1 (9) were isolated from Fusarium oxysporum (N17B) by bioassay-guided fractionation. Compounds 1 and 3 showed selective fungistatic activity against Aspergillus fumigatus and wortmannin had selective potent activity against Candida albicans. Moderate activity was observed with the enniatins 7–9 against C. albicans, Cryptococcus neoformans, and Mycobacterium intracellulare. Compounds 1–5 had no activity against the agriculturally important fungi Fusarium verticillioides (syn. F. moniliforme) and Aspergillus flavus.
Opportunistic fungal infections constitute a major cause of morbidity and mortality in AIDS patients.1 The drugs available for the treatment of these infections are of limited utility due to their toxicity, adverse side reactions, and the frequent emergence of resistant strains.2 As a part of a program to identify new drug candidates for the treatment of opportunistic fungal infections, we have screened a number of extracts from various natural sources against the following common opportunistic fungal pathogens: Candida albicans, Cryptococcus neoformans, Mycobacterium intracellulare, and Aspergillus fumigatus. The ethyl acetate extract of the fungus Fusarium oxysporum (N17B) showed broad-spectrum antifungal activity. Previous studies on F. oxysporum (N17B) have shown that it produces a toxin which causes hemorrhaging and death in mice,3 and wortmannin has been identified as the compound responsible for this toxicity.4,5 Wortmannin, a powerful inhibitor of phosphatidylinositide 3-kinase,6 was shown to have antifungal properties.7
Bioassay-guided fractionation of the ethyl acetate extract of F. oxysporum (N17B) grown on rice medium gave two fractions with selective activity against A. fumigatus and C. albicans respectively, and another with broad activity against C. albicans, C. neoformans, and M. intracellulare. Further purification of the fraction with selective activity against A. fumigatus led to the isolation of compounds 1 and 3 as the constituents responsible for the activity. Compound 1 had identical spectroscopic data including 1H-13C NMR correlations as those reported for oxysporidinone, which was previously isolated from a different strain of F. oxysporum.8 However, the optical rotation observed for compound 1 ([α]D −68.8) had the opposite sign of that reported for the previously reported compound ([α]D +97).8 (+)-Oxysporidine was shown to be active against several agriculturally important pathogenic fungi including A. niger.8 Compound 3 was identified as the 6′-hydroxy epimer of oxysporidinone. From the inactive fractions, (−)-sambutoxin (2) and two further 4-hydroxy-2-pyridinone analogs (4 and 5) were isolated. Sambutoxin, a hemorrhagic mycotoxin, has been isolated from F. sambucinum.9 This is the first report of compounds 3–5 in nature. From the fraction with selective activity against C. albicans, wortmannin (6) was isolated as the active constituent. Separation of the fraction with broad activity against C. albicans, C. neoformans, and M. intracellulare led to the isolation of enniatins A (7), A1 (8), and B1 (9) as the compounds responsible for this activity. The current study thus led to the isolation of three classes of compounds with different activity profiles against human pathogenic fungi. However, various other activities4 or toxicities5,6,9,10 associated with these classes would preclude them as viable leads for the treatment of human fungal infections.
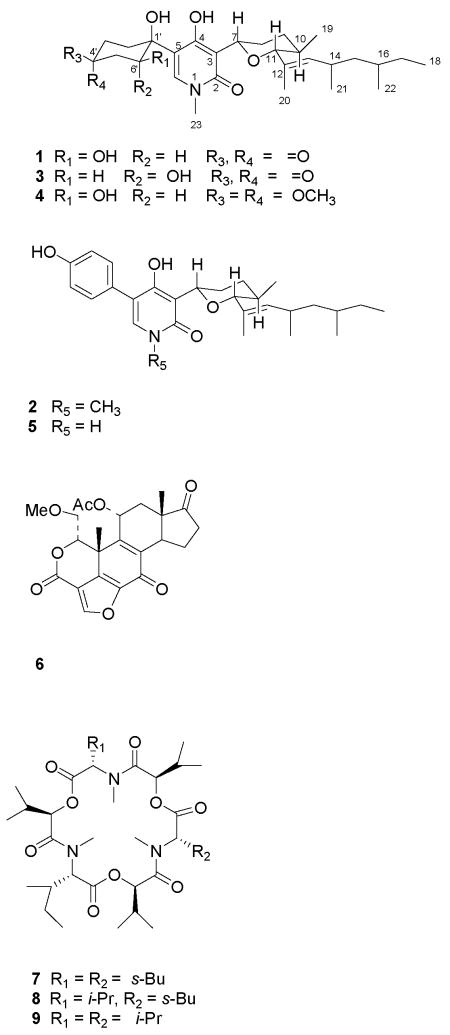
The 1H NMR data of 3 were similar to those of oxysporidinone (1) except for the signals in the cyclohexanone moiety. The major difference was the appearance of the H-6′ signal in 3 as a triplet (δ 4.90) with a coupling constant of 4.8 Hz. This is in contrast to the dd (J = 10.8, 5.4 Hz) that appeared at δ 4.65 for the same proton in oxysporidinone (1), suggesting an equatorial orientation for H-6′ in 3. The mass spectrum did not afford a molecular ion but gave the base peak at m/z 472.3067 [M+H-H2O]+ (calcd for C28H42NO5, 472.3063) indicating ready elimination of the 6′-hydroxyl group to generate a stable ion with a α,β-unsaturated carbonyl. This evidence suggested that compound 3 is the 6′-hydroxy epimer of oxysporidinone. COSY, HMBC, and HMQC NMR spectroscopic correlations (Table 1) further supported this structure.
Table 1.
1H NMR and 1H-13C NMR Correlation Data (600 MHz, CDCl3) of Compounds 3–5.
| 3 | 4 | 5 | ||
|---|---|---|---|---|
| position | δ (mult. J in Hz) [HMBC correlations] | |||
| 6 | 7.24 (s) [1′,2,4,5,23] | 7.33 (s) [1′,2,5,6,23] | 6.61 (s) [1′,2,4,5] | |
| 7 | ax | 4.61 (brd, 11.4) [2,3,4] | 4.94 (brd, 11.2) [2,4,8] | 5.07 (dd,, 11.4, 2.4) [3,4] |
| 8 | ax | 2.12 (qd, 12.6, 3.0) | 2.02 (brd, 12.0) | 2.14 (dd, 12.6, 1.8) |
| eq | 1.42 (brd, 12.6) | 1.54 (brq, 12.6) | 1.64a | |
| 9 | ax | 1.25 (qd, 12.3, 3.0) | 1.44 (qd, 12.4, 3.0) | 1.48 (qd, 12.6, 3.0) |
| eq | 1.84 (dd, 12.6, 3.0) | 1.87a | 1.94 (dd, 13.2, 3.0) | |
| 10 | ax | 1.60 (m) | 1.66 (m) | 1.69 (m) |
| 11 | ax | 3.34 (d, 10.2) [9,12,13,19] | 3.48 (d, 10.0) [10,12,13] | 3.57 (d, 10.0) [9,12,13,20] |
| 13 | 5.12 (d, 9.0) [11,21] | 5.18 (d. 9.6) [11] | 5.23 (d, 9.0) [11,14,20,21] | |
| 14 | 2.46 (heptet, 7.8) | 2.47 (heptet, 7.6) | 2.48 (heptet, 7.2) [13] | |
| 15 | 1.03a [16,22] | 1.04a [13,14,16,17,22] | 1.06 a [13,16,17,22] | |
| 1.17 (pentet 6.0) [16,22] | 1.19 a [13,14,16,17,22] | 1.22 a [13,16,17,22] | ||
| 16 | 1.31a | 1.30 a(20,21] | 1.31 a [17] | |
| 17 | 1.37a [18,22] | 1.33 a [14,16] | 1.37 a [18,22] | |
| 1.03a [16,18,22] | 1.02 a [[15,16] | 1.07 a [18,15] | ||
| 18 | 0.82 (t, 7.0) [16,17] | 0.87 (t, 7.2) [17] | 0.85 (t, 7.5) [17] | |
| 19 | 0.69 (d, 6.6) [10,11] | 0.78 (d, 6.4) [9,10,11] | 0.76 (d, 6.6) [10,11] | |
| 20 | 1.60 (s) [11,12,13] | 1.62 (s) [11,12,13] | 1.64 (s) [11,12,13] | |
| 21 | 0.87 (d, 6.6) [13,14,15] | 0.90 (d, 6.4) [13,14,15] | 0.92 (d, 6.5) [13,14,15] | |
| 22 | 0.81 (d, .6.6) [15,16,17] | 0.87 (d, 6.4) [15,17] | 0.84 (d, 6.5) [2′,6′,15,16] | |
| 23 | 3.30 (s) [2,6] | 3.43 (s) [2,6] | ||
| 2′ | 7.17 (d, 8.0) [4′,5] | |||
| ax | 2.32 (ddd, 15.0, 12.0, 4.2) | 2.00a | ||
| eq | 2.19 (dt, 14.4, 4.2) [1′,4′,6′] | 1.80a | ||
| 3′ | 6.98 (d, 8.0) [1′,3′,4′,5′] | |||
| ax | 2.07 (ddd, 18.0, 12.0, 4.2) [4′] | 1.77a | ||
| eq | 2.40 (dt, 18.0, 4.2) [1′,2′] | 1.89a [2′] | ||
| 5′ | 6.98 (d, 8.0) [1′,3′,4′,5′] | |||
| ax | 2.78 (dd, 16.8, 4.8) [1′,4′,6′] | 1.79a [3′,4′,5′,6′] | ||
| eq | 2.91 (dd, 16.8, 4.8) [1′,4′,6′] | 2.22 (dd, 14.0, 4.4) [4′,5′,6′] | ||
| 6′ | 7.17 (d, 8.0) [4′,5] | |||
| ax | 4.35 (dd, 11.0, 4.4) | |||
| eq | 4.90 (t, 4.8) [1′,4′] | |||
| OH | 10.24 [3,4,5] | 10.29 | ||
| OMe | 3.17. 3.22, [4′] | |||
Multiplicity cannot be determined due to overlapping
The lack of NOESY correlations between the protons of the cyclohexanone ring and the rest of the molecule of 3 prevented the establishment of the relative configuration of the C-1′ hydroxyl group. Both H-7 and H-11 were determined to be axial on the basis of coupling constants (11.4 and 10.2 Hz respectively). The large coupling constant of H-11 showed that H-10 is also in an axial configuration, indicating that CH3-10 is in an equatorial position. The E-configuration for the 12,13 double bond was established based on the chemical shift of CH3-2011 and was further supported by the absence of a NOE correlation between CH3-20 and H-13. The difference in the chemical shifts of C-21 and C-22 (1.2 ppm) indicated that they are in an anti arrangement.12 Based on this evidence, the chemical structure of compound 3 was established as 6-epi-oxysporidinone.
The molecular formula of compound 4 was determined to be C30H49NO7 on the basis of HRMS data. The 1H NMR spectrum of 4 was similar to that of compound 1,8 except for the presence of two additional methoxy groups resonating at δ 3.22 and 3.17 ppm in the former. The additional methoxy groups signals were also present in the 13C NMR spectrum of compound 4.8 Furthermore, the 13C NMR spectrum of 4 showed a high-field signal at 100.7 ppm instead of a carbonyl signal in 1. This information, in combination with the molecular formula, suggested that compound 4 is the dimethyl ketal of oxysporidinone (1). COSY, HMQC, and HMBC NMR spectroscopic correlations (Table 1) confirmed this structure for compound 4. The possibility that compound 4 could be formed as an artifact during the chromatographic process was eliminated by confirming its presence in the original ethyl acetate extract.
The relative configuration of compound 4 was determined by 1H NMR coupling constants and NOESY correlations. Large coupling constants observed for H-6′ (11.0, 4.4) indicated that this proton is in an axial configuration. Strong NOE interactions between H-6′ in the cyclohexane ring and H-6 in the pyridinone ring suggested that the pyridinone moiety is above the plane of the cyclohexane ring. This would be possible only if the pyridinone ring is in equatorial position. Both H-7 and H-11 exhibited large coupling constants (11.2 and 10.0 Hz, respectively), indicating that these two protons, as well as H-10, are in axial configurations. The configuration of the C12,13-double bond was assigned the E configuration based on the chemical shift value of CH3-2010 and the lack of NOE interactions between CH3-20 and H-13. The small difference in the 13C NMR chemical shifts suggested an anti arrangement for CH3-21 and CH3-22.11 This combined evidence was used to establish the structure of compound 4 as the dimethyl ketal of oxysporidinone (1).
The 1H and 13C NMR data of compound 5 were very similar to those of sambutoxin.9 The major differences were the lack of a N-CH3 signal and small changes in the chemical shifts of the protons and carbons in the pyridinone moiety. This evidence indicated that this compound is the N-demethyl analog of sambutoxin. The molecular formula, C27H37NO4, suggested by HRMS was in agreement with this observation. COSY, HMQC, and HMBC NMR spectroscopic correlations confirmed this structure.
The 1H NMR coupling constants of 5 showed that H-7 (J = 11.4, 2.4 Hz) is in an axial configuration. The large coupling constant (10.0 Hz) of H-11 implied that both H-11 and H-10 are also in axial configurations. The 13C chemical shift value of CH3-2011 and the lack of NOE correlations between CH3-20 and H-13 were suggestive of an E-configuration for the 12,13 double bond. The 13C NMR chemical shift difference of CH3-21 and CH3-22 (1.1 ppm) showed that they are in an anti arrangement, as in sambutoxin.12 This evidence established compound 5 as the N-demethyl analog of sambutoxin.
(−)-Oxysporidinone (1) suppressed the growth of A. fumigatus at low concentrations (Table 3); however, its inability to eliminate the fungus completely, suggested fungistatic rather than fungicidal activity. Compound 3, the 6′-hydroxy epimer of oxysporidinone, had only marginal activity against A. fumigatus. Compounds 2, 4, and 5 were inactive against all test organisms. These results indicated that the functional groups in the cyclohexyl ring are critical for the anti-A. fumigatus activity of this class of compounds. Change of configuration of the 6′-hydroxyl group resulted in reduction of activity and the removal of the carbonyl or aromatization of the cyclohexyl ring led to complete loss of the antifungal activity. Wortmannin (6) exhibited potent selective activity towards C. albicans. Enniatin A (7), A1 (8), and B1 (9) showed moderate activity against C. albicans, C. neoformans, and M. intracellulare. Compounds 1–5 were also evaluated against agriculturally important fungi F. verticillioides and A. flavus using a disk assay but showed no activity up to a concentration of 1mg/mL.
Table 3.
Antifungal Activity of Compounds 1, 3, and 6–9a
| compound | Candida albicans | Cryptococcus neoformans | Mycobacterium intracellulare | Aspergillus fumigatus | ||||
|---|---|---|---|---|---|---|---|---|
| IC50 | MIC | IC50 | MIC | IC50 | MIC | IC50 | MIC | |
| 1 | -b | - | 35 | - | - | - | 2.0 | - |
| 3 | - | - | - | - | - | - | 35 | - |
| 6 | 0.25 | 0.78 | - | - | - | - | - | - |
| 7 | 2.0 | 3.13 | 3.5 | 12.5 | 5.0 | 50 | - | - |
| 8 | 2.0 | 6.25 | 4.5 | 12.5 | 9.0 | 50 | - | - |
| 9 | 2.0 | 6.25 | 9.0 | 25 | 15.0 | - | - | - |
| Amphotericin Bd | 0.35 | 1.25 | 0.45 | 1.25 | NT | NT | 0.91 | 1.25 |
| Ciprofloxacind | NTc | NT | NT | NT | 0.30 | 0.63 | NT | NT |
IC50 and MIC (minimum inhibitory concentration) values are in μg/mL.
-: not active at the highest test concentration of 50 μg/mL.
NT: not tested
Positive control
Experimental Section
General Experimental Procedures
Melting points (uncorrected) were recorded on an Electrothermal 9100 instrument. UV spectra were obtained in CHCl3, using a Hewlett-Packard 8452A spectrometer. 1H NMR and 13C NMR spectra were recorded on Varian Mercury-400BB (400 MHz for 1H NMR and 100 MHz for 13C NMR), Bruker Avance DRX-500 (500 MHz for 1H NMR and 125 MHz for 13C NMR), or Varian Inova-600 (600 MHz for 1H NMR and 150 MHz for 13C NMR) spectrometers, run in CDCl3 with TMS as an internal standard. HRTOFMS were measured on an Agilent Series 1100 SL mass spectrometer equipped with an ESI source. Preparative TLC was carried out using silica gel F 254 plates (thickness 1 mm). Preparative HPLC was preformed on an Agilent 1100 series instrument equipped with a photodiode array detector.
Organism and Fermentation
Isolation and identification of Fusarium oxysporum Schlecht. emend. Snyd. et Hans. N17B has previously been reported.4,5 The fungal isolate was grown on rice medium as previously described.5
Extraction and Isolation
Rice (2.3 kg) inoculated with F. oxysporum was ground and extracted three times with ethyl acetate at room temperature with sonication to give a thick gum (29 g). The hexane-soluble fraction (22 g) of this extract was chromatographed over silica gel and eluted with an increasing concentration of ethyl acetate in hexanes to give 19 fractions. Fractions 15 and 18 showed antifungal activity.
Fraction 15 was chromatographed over silica gel and eluted with CHCl3 and CHCl3-MeOH (90:10) to give four fractions. The first fraction was purified by preparative TLC on silica gel using CHCl3-MeOH (98:2) to give wortmannin as white crystals (16 mg). The identity of this compound was confirmed by comparison of the reported physical and spectroscopic data.13,14 The second fraction yielded a mixture of enniatins. This fraction was separated by preparative HPLC using a Luna 10 C18(2) (250 × 21 mm i.d., 10 micron particle size) column, with the mobile phase MeOH-H2O (80:20), to give enniatin A (7) (21 mg) and a fraction containing two compounds. This fraction was separated using the same column with the mobile phase CH3CN-H2O (80:20) to give enniatins A1 (8) (19 mg) and B1 (9) (11 mg) as white amorphous residues. The identity of enniatins A (7), A1 (8) and B1 (9) was confirmed by comparison with reported physical and spectroscopic data.15
Fraction 18 from the first column was chromatographed over silica gel and eluted with CHCl3-MeOH (95:5) to yield three fractions. Fraction 1 was purified using a Luna 10 C18(2) (250 × 21 mm i.d., 10 micron particle size) preparative column, with the mobile phase CH3CN-H2O (80:20), to give compound 4 as a white amorphous residue (16 mg). The second fraction was separated by HPLC using a Luna 10 C18(2) (250 × 21 mm i.d., 10 micron particle size) preparative column, with the mobile phase MeOH-H2O (80:20), to give 1 (21 mg) and 3 (17 mg). Sambutoxin was isolated from fraction 16 from the first column by preparative TLC on silica gel using CHCl3-MeOH (92:8) as solvent (31 mg). The identity of this compound was confirmed by comparison with physical and spectroscopic data previously reported.9 Fraction 17 from the first column was chromatographed over silica gel and elution with CHCl3-MeOH (95:5) yielded three fractions. Fraction 2 was purified using a Luna 10 C18(2) (250 × 21 mm i.d., 10 micron particle size) preparative column, with the mobile phase CH3CN-H2O (80:20), to give compound 5 as a white amorphous residue (19 mg).
Dimethyl ketal of oxysporidinone (4)
[α]26D −30.6 (c 0.1, CHCl3); UV (CHCl3) λmax (log ε) 216 (3.62), 290 (3.45); 1H NMR and 13C NMR data are presented in Table 1 and 2, respectively; HRESITOFMS m/z 536.3573 [M+H]+ (calcd for C30H50NO7, 536.3587).
Table 2.
13C NMR Chemical Shift Assignments (δ) (150 MHZ, CDCl3) of Compounds 3–5
| Position | 3 | 4 | 5 |
|---|---|---|---|
| 2 | 163.3 | 161.5 | 163.2 |
| 3 | 109.0 | 111.0 | 110.1 |
| 4 | 166.1 | 162.4 | 164.2 |
| 5 | 116.7 | 115.1 | 116.6 |
| 6 | 132.8 | 136.1 | 132.7 |
| 7 | 72.1 | 78.2 | 77.0 |
| 8 | 29.2 | 30.9 | 31.7 |
| 9 | 33.1 | 32.3 | 32.3 |
| 10 | 32.1 | 32.6 | 32.7 |
| 11 | 91.9 | 92.7 | 92.8 |
| 12 | 133.6 | 130.2 | 129.9 |
| 13 | 136.5 | 138.3 | 138.1 |
| 14 | 29.7 | 29.8 | 30.0 |
| 15 | 45.1 | 44.9 | 45.0 |
| 16 | 31.8 | 32.2 | 32.3 |
| 17 | 29.1 | 29.2 | 29.2 |
| 18 | 11.5 | 11.4 | 12.0 |
| 19 | 17.8 | 17.8 | 18.0 |
| 20 | 11.4 | 11.8 | 11.6 |
| 21 | 21.0 | 20.8 | 21.1 |
| 22 | 19.8 | 19.8 | 20.0 |
| 23 | 38.0 | 37.4 | |
| 1′ | 76.4 | 74.2 | 125.1 |
| 2′ | 33.5 | 32.0 | 130.5 |
| 3′ | 35.1 | 27.2 | 115.6 |
| 4′ | 207.6 | 100.7 | 156.4 |
| 5′ | 42.1 | 36.5 | 115.6 |
| 6′ | 90.3 | 69.3 | 130.5 |
(−)-Oxysporidinone (1)
[α]26D −68.8 (c 0.15, EtOH); spectroscopic data and 1H–13C NMR correlations were identical to those previously reported for (+)-oxysporidinone.8
6-epi-Oxysporidinone (3)
White needles (CH3OH); mp 176–78 °C; [α]26D −86.9 (c 0.1, CHCl3); UV (CHCl3) λmax (log ε) 220 (3.64), 296 (3.45); 1H NMR and 13C NMR data are presented in Table 1 and 2, respectively; HRESITOFMS m/z 472.3067 [M+H-H2O]+ (calcd for C28H42NO5, 472.3063).
N-Demethylsambutoxin (5)
[α]26D −98.6 (c 0.1, CHCl3); UV (CHCl3) λmax (log ε) 212 (4.32), 252 (4.03); 1H NMR and 13C NMR data are presented in Table 1 and 2, respectively; HRESITOFMS m/z 440.2787 [M+H]+ (calcd for C27H38NO4, 440.2800).
Antifungal Bioassay
All organisms were obtained from the American Type Culture Collection (Manassas, VA) and included Candida albicans ATCC 90028, Cryptococcus neoformans ATCC 90113, Aspergillus fumigatus ATCC 90906 and Mycobacterium intracellulare ATCC 23068. Susceptibility testing was performed using a modified version of the NCCLS methods.16–18 M. intracellulare was tested using a modified method of Franzblau, et al.19 Briefly, samples (dissolved in DMSO) were diluted serially using 20% DMSO/saline and transferred in duplicate (10 μL) to 96-well flat-bottom microplates. Inocula were prepared by diluting microbe suspensions with assay medium [RPMI 1640/2% dextrose/MOPS at pH 6.0 (Cellgro) for C. albicans, Sabouraud Dextrose (Difco) for C. neoformans, 5% Alamar Blue™/RPMI 1640 broth (2% dextrose buffered with 0.165 M MOPS at pH 7.3) for A. fumigatus, and 5% Alamar Blue™ in Middlebrook 7H9 broth with OADC enrichment (Difco) pH = 7.3 for M. intracellulare] to afford the following colony forming units/mL after addition to samples: C. albicans: 1.0 × 104, C. neoformans: 1.0 × 105, A. fumigatus: 3.0 × 104, and M. intracellulare: 2.0 × 106. The microbial inocula were added to the samples to achieve a final volume of 200 μl and final sample concentrations starting with 50 μg/mL. Drug controls [Ciprofloxacin (ICN Biomedicals, Solon, OH) for M. intracellulare and Amphotericin B (ICN Biomedicals) for fungi] were included. C. albicans and C. neoformans were read at 630 nm using the EL-340 Biokinetics Reader (Bio-Tek Instruments, Winooski, VT), and M. intracellulare and A. fumigatus were read at 544ex/590em using the Polarstar Galaxy Plate Reader (BMG LabTechnologies, Offenburg, Germany) prior to and after incubation: C. albicans at 37 °C for 18–24 h, C. neoformans and A. fumigatus at 30 °C for 68–72 h, and M. intracellulare at 37 °C and 10% CO2 for 68–72 h. Percent growth was calculated and plotted versus test concentration to afford the IC50 (sample concentration that affords 50% growth of the organism). The minimum inhibitory concentration (MIC) is defined as the lowest test concentration that allows no detectable growth.
Compounds 1–5 were assayed against F. verticillioides and A. flavus using disk assay described by Alam et al.20 using captan as the positive control.
Acknowledgments
This work was supported by the National Institute of Health (R21 A1061431-01) and in part by the United States Department of Agriculture, ARS, Specific Cooperative Agreement No. 58-6408-2-009. The authors sincerely thank Dr. Bharathi Avula for recording the mass spectra, and Bobbie J. Johnson and Mary Duke, USDA-ARS, for their technical assistance in this research.
Footnotes
Dedicated to Dr. Norman R. Farnsworth of the University of Illinois at Chicago for his pioneering work on bioactive natural products.
References and Notes
- 1.Ruhnke M. Drugs. 2004;64:1163–1180. doi: 10.2165/00003495-200464110-00002. [DOI] [PubMed] [Google Scholar]
- 2.Polak A. In: Antifungal Agents: Advances and Problems. Jucker E, editor. Chapter 4. Birkhauser Verlag; Basel: 2003. pp. 59–190. [Google Scholar]
- 3.Abbas HK, Mirocha CJ, Gunther R. Mycopathologia. 1989;105:143–151. doi: 10.1007/BF00437246. [DOI] [PubMed] [Google Scholar]
- 4.Abbas HK, Mirocha CJ. Appl Environ Microbiol. 1988;54:1268–1274. doi: 10.1128/aem.54.5.1268-1274.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbas HK, Mirocha CJ, Shier WT, Gunther R. JAOAC Int. 1992;75:474–480. [Google Scholar]
- 6.Woscholski R, Kodaki T, McKinnon M, Waterfield MD, Parker PJ. FEBS Lett. 1994;342:109–114. doi: 10.1016/0014-5793(94)80482-6. [DOI] [PubMed] [Google Scholar]
- 7.MacMillan J, Vanstone AE, Yeboah SK. Chem Commun. 1968:613–614. [Google Scholar]
- 8.Breinholt J, Ludvigsen S, Rassing BR, Rosendahl CN, Nielsen SE, Olsen CE. J Nat Prod. 1997;60:33–35. doi: 10.1021/np9605596. [DOI] [PubMed] [Google Scholar]
- 9.Kim JC, Lee YW, Tamura H, Yoshizawa T. Tetrahedron Lett. 1995;36:1047–1050. [Google Scholar]
- 10.Uhlig S, Gutleb AC, Thrane U, Flaoyen A. Toxicon. 2005;46:150–159. doi: 10.1016/j.toxicon.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Couperus PA, Clague ADH, Van Dongen JPCM. Org Magn Reson. 1976;8:426–431. [Google Scholar]
- 12.Organ MG, Bilokin YV, Bratovanov S. J Org Chem. 2002;67:5176–5183. doi: 10.1021/jo0201777. [DOI] [PubMed] [Google Scholar]
- 13.MacMillan J, Vanstone AE, Yeboah SK. J Chem Soc, Perkin Trans 1. 1972:2898–903. [Google Scholar]
- 14.Simpson TJ, Lunnon MW, MacMillan J. J Chem Soc, Perkin Trans1. 1979:931–934. [Google Scholar]
- 15.Blais LA, ApSimon JW, Blackwell BA, Greenhalgh R, Miller JD. Can J Chem. 1992;70:1281–1287. [Google Scholar]
- 16.NCCLS. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard. NCCLS Document M27-A. 2. National Committee on Clinical Laboratory Standards; Wayne, PA: 2002. [Google Scholar]
- 17.NCCLS. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi; Approved Standard. NCCLS Document M38-A. National Committee on Clinical Laboratory Standards; Wayne, PA: 2002. [Google Scholar]
- 18.NCCLS. Susceptibility Testing of Mycobacteria, Nocardia, and Other Aerobic Actinomycetes; Tentative Standard. NCCLS Document M24-T2. 2. National Committee on Clinical Laboratory Standards; Wayne, PA: 2000. [PubMed] [Google Scholar]
- 19.Franzblau SG, Witzig RS, Mclaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, Gilman RH. J Clin Microbiol. 1998;36:362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alam S, Miah MAJ, Islam A. J Biol Sci. 2004;44:527–531. [Google Scholar]