

The crystal structure of apo nicotinic acid mononucleotide adenylyltransferase (NaMNAT) from B. anthracis was determined to 2.3 Å resolution and compared with other bacterial NaMNAT structures.
Keywords: nicotinic acid mononucleotide adenylyltransferase, Bacillus anthracis, self-interaction chromatography, second virial coefficient
Abstract
Nicotinic acid mononucleotide adenylyltransferase (NaMNAT; EC 2.7.7.18) is the penultimate enzyme in the biosynthesis of NAD+ and catalyzes the adenylation of nicotinic acid mononucleotide (NaMN) by ATP to form nicotinic acid adenine dinucleotide (NaAD). This enzyme is regarded as a suitable candidate for antibacterial drug development; as such, Bacillus anthracis NaMNAT (BA NaMNAT) was heterologously expressed in Escherichia coli for the purpose of inhibitor discovery and crystallography. The crystal structure of BA NaMNAT was determined by molecular replacement, revealing two dimers per asymmetric unit, and was refined to an R factor and R free of 0.228 and 0.263, respectively, at 2.3 Å resolution. The structure is very similar to that of B. subtilis NaMNAT (BS NaMNAT), which is also a dimer, and another independently solved structure of BA NaMNAT recently released from the PDB along with two ligated forms. Comparison of these and other less related bacterial NaMNAT structures support the presence of considerable conformational heterogeneity and flexibility in three loops surrounding the substrate-binding area.
1. Introduction
NAD+ is a ubiquitous and essential cofactor that is involved in multiple oxidation–reduction reactions in all living cells (Berger et al., 2004 ▶). NAD+ is biosynthesized de novo or through the pyridine-salvage pathway; these two pathways converge at the penultimate enzyme in NAD+ biosynthesis, nicotinic acid mononucleotide adenylyltransferase (NaMNAT). Bacterial NaMNAT catalyzes the transfer of the adenylyl moiety of ATP to nicotinic acid mononucleotide (NaMN) to form nicotinic acid adenine dinucleotide (NaAD). NaAD is catalytically transformed to NAD+ by the enzyme NAD+ synthetase. Since NaMNAT is a conserved enzyme and is essential to the survival of every bacterium studied to date, it is regarded as a potential target for the development of antibacterial drugs.
The crystallization conditions for Bacillus anthracis NaMNAT (BA NaMNAT) were optimized in part with the aid of self-interaction chromatography (SIC), a technique which is growing in popularity for its ability to identify and predict solution conditions that can improve protein solubility (Tessier et al., 2002 ▶). The structure of BA NaMNAT is described and compared with other deposited bacterial NaMNAT structures.
2. Methods
2.1. Expression and purification of the BA NaMNAT nadD gene
The nadD gene (gi:9187226) was amplified from B. anthracis genomic DNA. The sequence was inserted into expression vector pET21b (Novagen Inc). This construction added a 12-residue tag to the C-terminus of the recombinant protein with sequence LAAALEHHHHHH; the formula weight of the protein was 23 347 Da. The protein was overexpressed in Escherichia coli BL21 (DE3) pLysS cells. The cells were grown at 310 K to an OD600 of 0.6 in Luria–Bertani medium containing 50 µg ml−1 ampicillin. Protein expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and growth took place for 4 h at 301 K after induction. Cells were harvested and purified at 277 K using affinity and size-exclusion chromatography (SEC) with Ni–NTA and Superdex 75 columns (Amersham-Pharmacia). The SEC buffer composition optimized with the aid of SIC was 50 mM Tris pH 7.0, 100 mM NaCl, 100 mM Arg, 100 mM Glu, 1% glycerol, 1 mM DTT. The homogeneity of the purified protein was assessed by SDS–PAGE and dynamic light scattering.
2.2. K m determination
The K m values for ATP and NaMN were determined using a discontinuous HPLC assay. The reaction conditions were 50 mM HEPES pH 7.5, 10 mM MgCl2 and 0.25 µg ml−1 BA NAMNAT at 295 K. For determination of K m, ATP was varied from 6 to 600 µM with NaMN held at 350 µM (14-fold K m) and NaMN was varied from 5 to 250 µM with ATP held at 600 µM (14-fold K m). The reaction was stopped at various time points by the addition of 1.2 M guanidine–HCl. The reactants and product were separated on a 4.6 × 100 mm Synergi Polar-RP column. The peak area of the product NaAD was used to calculate the production of NaAD. The reaction-progress curves were linear under these conditions. The apparent initial velocity V 0 (µM min−1) was equivalent to the slope of the reaction-progress curve. V 0 was plotted against the substrate concentration. The K m and V max values were determined by a nonlinear fit of the curve to the Michaelis–Menten equation using Origin software (Microcal Software Inc., Northampton, Massachusetts, USA). The K m and V max reported error terms are the standard errors in the calculated fit.
2.3. Self-interaction chromatography (SIC)
The second virial coefficient (B 22) is an indicator of protein aggregation under certain conditions (George et al., 1997 ▶). SIC is an affinity-chromatography system used to measure B 22 under a variety of conditions; a description of the technique can be found in Tessier et al. (2002 ▶). SIC was carried out for BA NaMNAT with of Arg, Glu or trehalose additives in order to search for solution conditions that would improve protein solubility. SIC was also used to monitor crystallization optimization.
2.4. Crystallization and X-ray data collection
BA NaMNAT was concentrated to 4 mg ml−1 in SEC buffer containing 200 mM trehalose. Initial crystallization screening was carried out by the vapor-diffusion method at 297 K. 1 µl protein solution mixed with 1 µl reservoir liquor was equilibrated against 1 ml reservoir screening solution in 24-well Nextal plates (Qiagen). Bar-shaped crystals appeared within one week using Hampton Crystal Screen kit I condition No. 39. Optimization was conducted and the final refined reservoir condition was 2.1 M ammonium sulfate, 100 mM HEPES pH 7.0, 2% PEG 400 and 10 mM MgCl2. Crystals were soaked in artificial mother liquor containing 25% glycerol and flash-frozen before data collection. X-ray diffraction data were collected on the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory using a MAR 300 CCD detector. A total of 360 frames with 0.5° oscillation angle were collected at 100 K using wavelength of 1 Å and a crystal-to-detector distance of 200 mm. Data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶).
2.5. Structure determination and refinement
The structure of BA NaMNAT was determined by molecular replacement using the MOLREP program in the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). The structure of the B. subtilis NaMNAT (BS NaMNAT) dimer (PDB code 1kam) was used as a search model. The molecular-replacement solution was further refined in the Crystallography & NMR System (Brünger et al., 1998 ▶) and interactive sessions of model building were performed in Coot (Emsley & Cowtan, 2004 ▶). The structure was validated using the ADIT validation server (http://deposit.pdb.org/validate/; Yang et al., 2004 ▶) and deposited in the PDB under code 3dv2.
3. Results
3.1. Characterization and crystallization of NaMNAT
Purified B. anthracis NaMNAT exhibits the K m and V max values shown in Table 1 ▶, which are similar to those found for BS NaMNAT (Olland et al., 2002 ▶). The protein had an initial concentration of 2.7 mg ml−1 after nickel-affinity chromatography. However, after overnight dialysis against the SEC buffer (50 mM Tris–HCl pH 7.0, 100 mM NaCl, 1% glycerol, 1 mM DTT) a precipitant was observed and only 0.4 mg ml−1 protein remained in solution. Differential scanning calorimetry suggested that the protein was highly aggregated. A second purification was performed: the nickel column was followed by SEC. The latter was made possible by the incorporation of 50 mM Arg and 50 mM Glu into the SEC buffer, which improved the solubility. SIC experiments were performed on the protein obtained from SEC with various combinations and concentrations of additives in order to search for conditions to eliminate precipitation. As shown in Appendix A , Arg, Glu and trehalose improved the protein solubility. The final optimized gel-filtration buffer contained 100 mM Arg and 100 mM Glu. Trehalose was added at a concentration of 200 mM during protein concentration and BA NaMNAT could be concentrated to more than 12 mg ml−1 with no precipitation.
Table 1. K m and V max for BA NaMNAT.
| Substrate | Km (mM) | Vmax (µM min−1) |
|---|---|---|
| NaMN | 0.025 ± 0.004 | 4.7 ± 0.2 |
| ATP | 0.044 ± 0.003 | 4.3 ± 0.1 |
4 mg ml−1 protein was used for crystallization screening using the sitting-drop and hanging-drop vapor-diffusion methods. Several crystals with a variety of shapes were found in conditions from a 360-condition in-house screen (Lisa screen360), as well as from Hampton Crystal Screens. An SIC experiment was performed on the best hit, which was from Hampton Crystal Screen I condition No. 39. As shown in Appendix A , the calculated B 22 value of this condition resided within the range of values known to be conducive to crystallization (George et al., 1997 ▶). This condition was then refined and the final optimized crystallization condition was 2.1 M ammonium sulfate, 100 mM HEPES pH 7.0, 2% PEG 400 and 10 mM MgCl2. Bar-shaped crystals grew to dimensions of 0.05 × 0.1 × 0.2 mm after one week in the optimized condition.
A summary of the data-collection and refinement statistics is shown in Table 2 ▶. The structure was validated and deposited in the PDB with code 3dv2. During the course of the preparation of this manuscript, coordinates were released for the independently solved structure of apo BA NaMNAT (PDB code 2qtm) and two ligated forms complexed with NMN (2qtn) and NXX (2qtr), respectively; the latter ligand is apparently the product NaAD (V. C. Sershon, B. D. Santarsiero & A. D. Mesecar, unpublished work).
Table 2. BA NaMNAT crystallographic data-collection and refinement statistics.
Values in parentheses are for the outer shell.
| Data collection | |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 63.07, b = 100.59, c = 73.07, α = 90, β = 110.42, γ = 90 |
| Matthews coefficient (Å3 Da−1) | 2.33 |
| Solvent content (%) | 47 |
| Unique reflections | 37491 |
| Rmerge† | 5.9 (28.5) |
| Redundancy | 3.7 (3.3) |
| Completeness (%) | 97.6 (88.9) |
| I/σ(I) | 10.5 (3.2) |
| Refinement | |
| Resolution (Å) | 50.0–2.3 |
| R factor/Rfree‡ (%) | 0.228/0.263 |
| No. of residues | 751 |
| No. of water molecules | 131 |
| No. of sulfate ions | 8 |
| R.m.s. deviations from ideality | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.2 |
| Ramachandran plot, residues in | |
| Most favored regions (%) | 89.2 |
| Additional allowed regions (%) | 10.4 |
| Generously allowed regions (%) | 0.5 |
| Average B values (Å2) | |
| Main-chain atoms | 41.99 |
| Side-chain atoms | 42.90 |
| Solvent atoms | 40.22 |
R
merge = 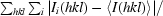
 , where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the average value over multiple measurements.
, where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the average value over multiple measurements.
R factor = 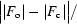
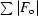 , where F
o and F
c are the observed and calculated structure factors, respectively. R
free was calculated for 5% of the reflections removed randomly from the refinement.
, where F
o and F
c are the observed and calculated structure factors, respectively. R
free was calculated for 5% of the reflections removed randomly from the refinement.
3.2. Overall structure of BA NaMNAT
BA NaMNAT belongs to the α and β protein (α/β) class. There are four molecules in the asymmetric unit, forming two dimers AB and CD. The four molecules form a distorted tetrahedron in which the primary dimer-to-dimer interface is between residues 135 and 139 from monomers A and C, which form a local pseudo-twofold symmetry axis. The dimer interfaces are stabilized by main-chain interactions between residues 151–153 from monomers A and B or C and D, which form distorted antiparallel β-strands with close hydrogen-bond contacts about a twofold-symmetric dimer axis. Reciprocal side-chain hydrogen bonds involved in holding the dimer together are Asn23–Thr168, Glu24–Arg162, Glu67–Lys170 and Glu151–Arg133. These residues are mostly conserved between BA and BS NaMNAT. Size-exclusion chromatography and dynamic light scattering also indicate that the protein is a dimer in solution (data not shown). As expected, the dimer structure (Fig. 1 ▶) is very similar to PDB entry 2qtm (V. C. Sershon, B. D. Santarsiero & A. D. Mesecar, unpublished work) as well as to that observed for apo BS NaMNAT (PDB code 1kam; Olland et al., 2002 ▶). The monomer is composed of a classic Rossmann fold consisting of a right-handed β/α/β super secondary-structure element formed by six twisted parallel β-strands with order 321456 in the center flanked by α-helices A and B on one side and α-helices C/C′ and D/D′ on the other side of the central β-sheet. After the Rossmann fold is another domain composed of α-helices E and F. The C-terminal His-containing tag was not observed on any of the monomers in the crystal structure.
Figure 1.

Dimer structure of BA NaMNAT (chain A in green and chain B in cyan). Secondary structures are labelled for chain A.
3.3. Structural comparison with BS NaMNAT
BA NaMNAT and BS NaMNAT share 56% sequence identity and 76% similarity. The present apo BA NaMNAT monomer structure (PDB code 3dv2) is shown in Fig. 2 ▶(a) superimposed with apo BS NaMNAT (1kam), the BS NaMNAT–NaAD complex (1kaq) and apo BA NaMNAT (2qtm). The r.m.s.d.s of the respective Cα positions are 1.13 Å (3dv2:2qtm) and 1.22 Å (3dv2:1kam), indicating high conservation of the protein structure between the two species, although it is surprising that the r.m.s.d. between the two BA apo structures is not lower. It can be seen from the superpositioned structures that there are three regions of greater conformational diversity that are not only different from one apo structure to the next, but also change position upon ligation, thus suggesting a degree of flexibility, viz. residues 40–53 (between β-strands 2 and 3), residues 104–124 (between β-strands 4 and 5) and residues 132–143 (between β-strands 5 and 6). After removing the above three flexible regions, the Cα r.m.s.d.s were 0.71 Å (3dv2:2qtm) and 0.97 Å (3dv2:1kam).
Figure 2.

(a) Overlay of apo BA NaMNAT (PDB code 3dv2, green; 2qtm, blue), apo BS NaMNAT (1kam, yellow) and BS NaMNAT–NaAD complex (1kaq, red; NaAD is shown in stick representation and colored by element). Secondary structures, N- and C-termini are labelled. (b–d) The three regions with the greatest conformational flexibility. Each region is enlarged and rotated to show most clearly the differences in conformation between the proteins. All structures shown in (a) plus BA NaMNAT–NaAD (PDB code 2qtr; magenta) are included. The first and last residues of each region are labelled for BA.
In Figs. 2 ▶(b), 2 ▶(c) and 2 ▶(d), these three respective regions are enlarged to show detail and the BA NaMNAT–NaAD complex (PDB code 2qtr) is included. These overlays as well as comparisons with other published NaMNAT structures lend support to the proposal that conformational diversity is an intrinsic property of the three regions. Published NaMNAT structures derived from E. coli display flexibility in these regions dependent on the presence or absence of ligand, while for Staphylococcus aureus there is no apo NaMNAT structure but two different NaAD structures derived from crystals exhibiting different space groups that exhibit different conformations in the N-terminal flexible region. In contrast, the Pseudomonas aeruginosa and human enzymes show little to no change upon ligation in these regions. An alignment of bacterial NaMNAT sequences is shown in Fig. 3 ▶.
Figure 3.

Sequence alignment of NaNMATs from B. anthracis (BA), B. subtilis (BS), S. aureus (SA), E. coli (EC) and P. aeruginosa (PA). The structure of BA NaMNAT was used to generate the structural annotations. The alignment was performed with ClustalW (Chenna et al., 2003 ▶) and the figure was prepared with ESPript (Gouet et al., 1999 ▶)
The apo BS NaMNAT structure is the only published apo NaMNAT structure that lacks electron density for the region containing residues 40–53, whereas in apo BA NaMNAT these residues were easily modeled (Fig. 4 ▶); they are also present in apo BA NaMNAT (2qtm). In the BS NaMNAT–NaAD (1kaq) and BA NaMNAT–NaAD (2qtr) structures, the conserved 42PPHK45 sequence in this region is observed to interact with the nicotinic ring of NaAD through Pro43 and His44 and is also involved in the interaction with ATP through His44-Lys45 in the P. aeruginosa NaMNAT–ATP complex structure (PDB code 1yun). When substrate is bound, those residues are pulled in towards the binding site. However, residues 40–53 of both apo BA NaMNAT structures are stabilized away from the substrate-binding site; this might be caused by an interaction with a symmetry-related molecule as shown in Fig. 4 ▶. In 2qtm, which is derived from a different space group, a similar intermolecular interaction is made with a symmetry-related molecule. It is also possible that this region is naturally stable and provided a crystal contact that enabled crystallization. This intermolecular contact is not present in the crystals of apo BS NaMNAT. Residues 47–50 involved in the contact exhibit high variation in sequence comparisons. This could also contribute to the variation in conformation.
Figure 4.

Residues 43–51 in apo BA NaMNAT are shown in green with a 2F o − F c electron-density map at 1σ in blue. Part of a symmetry-related molecule is shown in yellow. Crystal packing between the two chains is obvious and could account for the stability of the loop 43–51. These residues are not visible in the BS apo structure, which does not have the same packing.
Residues 102–124 in the apo BA NaMNAT structure have a conformation that is intermediate between the BS NaMNAT–NaAD complex and the apo BS NaMNAT structure. In the BS NaMNAT–NaAD complex this region is pulled close to the NaAD product. In the apo BS NaMNAT structure these residues reside far from the binding site. The conformation of residues 132–143 in apo BA NaMNAT is almost the same as in the apo BS NaMNAT structure. However, in the BS NaMNAT–NaAD complex structure this region moves significantly toward the adenyl-diphosphate part of NaAD, covering the binding site. In the BS and BA apo structures, the substrate-binding site is not covered by residues 132–143.
There are two sulfate ions in each monomer of apo BA NaMNAT (3dv2) and they are located at the ATP-binding site of the protein. The two sulfate ions form hydrogen bonds with residues Thr10, His15, His18 and Arg160. In comparison, the three ATP phosphates in the PA NaMNAT–ATP complex form hydrogen bonds with Thr11, His16, His19 and Arg183. These sets of residues involved in hydrogen bonding to the sulfate ions and the ATP phosphates roughly superpose onto each other and are conserved as shown in the sequence alignment. However, it does not appear that the sulfate positions correspond exactly to any of the phosphate positions. There is also a water molecule found in all four monomers that bridges the two sulfates together and hydrogen bonds to the main-chain N atoms of Ser156 and Ser157.
4. Discussion
B. anthracis is a Gram-positive spore-forming bacterial pathogen which can cause anthrax and belongs to the class A priority bacterial pathogens. The existence of strains resistant to present antibiotics provides an incentive to develop new drug classes for treatment. The present structure has been solved to facilitate the discovery of therapeutics against this target.
The crystal structures of other NaMNAT/NMNATs (nicotinamide mononucleotide adenylyltransferases) from E. coli, B. subtilis, P. aeruginosa, S. aureus and Francisella tularensis (EC NaMNAT, BS NaMNAT, PA NaMNAT, SA NaMNAT and FT NMNAT, respectively) and two isoforms of human NMNAT termed PNAT-1 and PNAT-3 have been resolved in various forms (Zhang et al., 2002 ▶; Olland et al., 2002 ▶; Yoon et al., 2005 ▶; Han et al., 2006 ▶; Werner et al., 2002 ▶; Garavaglia et al., 2002 ▶; Zhou et al., 2002 ▶; Huang et al., 2008 ▶). Detailed comparative descriptions can be found in the citations provided. These structures along with that of BA NaMNAT may be useful to understand the differential binding of potential new inhibitors.
Supplementary Material
PDB reference: nicotinic acid mononucleotide adenylyltransferase, 3dv2, r3dv2sf
Acknowledgments
The authors would like to thank Ms Tasha Kane for protein purification and crystallization and Dr Irina I. Protasevich is thanked for purification advice. DNA sequencing was carried out by the CFAR DNA Sequencing Core at UAB with NIH CFAR Core grant P30A127767; mass spectrometry of proteins was carried out by the Mass Spectrometry Shared Facility within the UAB Comprehensive Cancer Center Core (Support Grant P30 CA-13148) using a Waters/Micromass Q-Tof mass spectrometer (purchased from an NCRR Shared Instrumentation grant; S10 RR13795). Some X-ray diffraction was carried out by the X-ray Crystallography Shared Facility within the UAB Comprehensive Cancer Center Core (Support Grant P30 CA-13148). Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Science under Contract No. W-31-109-Eng-38. This work was funded by NIH U01 AI 070386.
Appendix A. Self-interaction chromatography (SIC) of BA NaMNAT
The solubility of BA NaMNAT was limited in Tris gel-filtration buffer. SIC was performed to search for suitable additives to improve protein solubility. Table 3 ▶ shows that 50/100 mM Arg and/or Glu and/or trehalose affected the B 22 values of protein solutions dramatically. The more positive the B 22 value, the greater the protein solubility. 100 mM Arg and 100 mM Glu were added to the optimized gel-filtration buffer and 200 mM trehalose was added during protein concentration. The protein solubility was increased from 0.3–0.4 mg ml−1 to more than 12 mg ml−1.
Table 3. Experimental B 22 values for Hampton Crystal Screen condition No. 39 (100 mM HEPES pH 7.5, 2 M ammonium sulfate, 2% PEG 400).
The protein buffer was 50 mM Tris pH 7.0, 100 mM NaCl, 1% glycerol, 1 mM DTT, 100 mM Arg, 100 mM Glu and 200 mM trehalose. On increasing the percentage of the crystallization condition, the B 22 values became more negative and closer to the ‘crystallization slot’.
| Solution (%) | B22 × 104 (mol ml g−2) | Std dev. × 104 | |
|---|---|---|---|
| 1 | 12.5 | 3.8 | 0.61 |
| 2 | 25 | 2.8 | 0.75 |
| 3 | 50 | −0.46 | 0.045 |
| 4 | 75 | −0.95 | 0.075 |
The second virial coefficient (B 22) has been shown to predict protein-crystal growth (George et al., 1997 ▶). A good protein solution for crystallization normally has a B 22 value between −0.5 and −8 × 10−4 mol ml g−2, the so-called ‘crystallization slot’. SIC was used to measure protein B 22 values in our best crystal hit, Hampton Crystal Screen condition No. 39. Table 4 ▶ shows that with different dilutions of the crystallization solution from 12.5% to 75%, the B 22 value shifted to the crystallization slot. This confirmed that the optimized condition was a good one for BA NaMNAT crystallization.
Table 4. Experimental B 22 values of the original gel-filtration buffer (50 mM Tris pH 7, 100 mM NaCl, 10% glycerol, 1 mM DTT) with various additives.
Higher B 22 values indicate better protein solubility.
| Solution | B22 × 104 (mol ml g−2) | Std. dev. × 104 | |
|---|---|---|---|
| 1 | Buffer only | 0.24 | 0.54 |
| 2 | Buffer + 50 mM Arg | 7.0 | 1.2 |
| 3 | Buffer + 100 mM Arg | 11 | 0.24 |
| 4 | Buffer + 50 mM Glu | 9.7 | 0.67 |
| 5 | Buffer + 100 mM Glu | 6.1 | 1.9 |
| 6 | Buffer + 50 mM trehalose | 0.83 | 0.46 |
| 7 | Buffer + 100 mM trehalose | 2.6 | 0.35 |
| 8 | Buffer + 50 mM Arg + 50 mM Glu | 9.3 | 0.93 |
| 9 | Buffer + 50 mM Arg + 50 mM trehalose | 9.2 | 0.32 |
| 10 | Buffer + 50 mM Glu + 50 mM trehalose | 3.8 | 0.24 |
| 11 | Buffer + 50 mM Arg + 50 mM Glu + 50 mM trehalose | 14 | 0.53 |
References
- Berger, F., Ramirez-Hernandez, M. H. & Ziegler, M. (2004). Trends Biochem. Sci.29, 111–118. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T. J., Higgins, D. G. & Thompson, J. D. (2003). Nucleic Acids Res.31, 3497–3500. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Garavaglia, S., Dangelo, I., Emanuelli, M., Carnevali, F., Pierella, F., Magni, G. & Rizzi, M. (2002). J. Biol. Chem.277, 8524–8530. [DOI] [PubMed]
- George, A., Chiang, Y., Guo, B., Arabshahi, A., Cai, Z. & Wilson, W. W. (1997). Methods Enzymol.276, 100–110. [DOI] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Han, S., Forman, M. D., Loulakis, P., Rosner, M. H., Xie, Z., Wang, H., Danley, D. E., Yuan, W., Schafer, J. & Xu, Z. (2006). J. Mol. Biol.360, 814–825. [DOI] [PubMed]
- Huang, N., Sorci, L., Zhang, X., Brautigam, C. A., Li, X., Raffaelli, N., Magni, G., Grishin, N. V., Osterman, A. L. & Zhang, H. (2008). Structure, 16, 196–209. [DOI] [PMC free article] [PubMed]
- Olland, A. M., Underwood, K. W., Czerwinski, R. M., Lo, M. C., Aulabaugh, A., Bard, J., Stahi, M. L., Somers, W. S., Sullivan, F. X. & Chopra, R. (2002). J. Biol. Chem.277, 3698–3707. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Tessier, P. M., Lenhoff, A. M. & Sandler, S. I. (2002). Biophys. J.82, 1620–1631. [DOI] [PMC free article] [PubMed]
- Werner, E., Ziegler, M., Lerner, F., Schweiger, M. & Heinemann, U. (2002). FEBS Lett.516, 239–244. [DOI] [PubMed]
- Yang, H., Guranovic, V., Dutta, S., Feng, Z., Berman, H. M. & Westbrook, J. D. (2004). Acta Cryst. D60, 1833–1839. [DOI] [PubMed]
- Yoon, H. J., Kim, H. L., Mikami, B. & Suh, S. W. (2005). J. Mol. Biol.351, 258–265. [DOI] [PubMed]
- Zhang, H., Zhou, T., Kurnasov, O., Cheek, S., Grishin, N. V. & Osterman, A. (2002). Structure, 10, 69–79. [DOI] [PubMed]
- Zhou, T., Kurnasov, O., Tomchick, D. R., Binns, D. D., Grishin, N. V., Marquez, V. E., Osterman, A. L. & Zhang, H. (2002). J. Biol. Chem.277, 13148–13154. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: nicotinic acid mononucleotide adenylyltransferase, 3dv2, r3dv2sf