

The radixin FERM domain was shown to bind the MT1-MMP cytoplasmic peptide and crystals of the complex were obtained.
Keywords: radixin FERM domain, ERM proteins, membrane-type 1 matrix metalloproteinase
Abstract
ERM proteins play a role in the cross-linking found between plasma membranes and actin filaments. The N-terminal FERM domains of ERM proteins are responsible for membrane association through direct interaction with the cytoplasmic tails of integral membrane proteins. During cell migration and movement, membrane-type 1 matrix metalloproteinase (MT1-MMP) on plasma membranes sheds adhesion molecule CD44 in addition to degrading the extracellular matrix. Here, the interaction between the radixin FERM domain and the MT1-MMP cytoplasmic tail is reported and preliminary crystallographic characterization of crystals of the radixin FERM domain bound to the cytoplasmic tail of MT1-MMP is presented. The crystals belong to space group P6122, with unit-cell parameters a = b = 122.7, c = 128.3 Å, and contain one complex in the crystallographic asymmetric unit. The diffraction data were collected to a resolution of 2.4 Å.
1. Introduction
ERM (ezrin/radixin/moesin) proteins have been found in a variety of eukaryotic cells ranging from Caenorhabditis elegans to humans (Tsukita et al., 1997 ▶; Tsukita & Yonemura, 1999 ▶; Bretscher et al., 2002 ▶) and function as a cross-linker between actin filaments and plasma membranes. ERM proteins (about 580 amino-acid residues) consist of three domains: an N-terminal globular domain, a central helical domain and a C-terminal domain. The N-terminal globular domain (∼300 residues) is highly conserved (∼80% identity) in ERM proteins and shows 32% identity to the equivalent domain in protein 4.1 (Takeuchi et al., 1994 ▶). The FERM domain associates with the plasma membrane, whereas the C-tail domain binds filamentous actin (F-actin) through conserved actin-binding sites which consist of 34 residues (550–583 for radixin; Funayama et al., 1991 ▶; Tsukita et al., 1994 ▶; Turunen et al., 1994 ▶; Hirao et al., 1996 ▶).
The FERM domain interacts directly with phosphatidylinositol-4,5-bisphosphate (PIP2) and the cytoplasmic tails of integral membrane proteins that play key roles in cell adhesion and cell–cell communication, including CD44, CD43, intercellular adhesion molecules (ICAMs) 1, 2 and 3 and P-selectin glycoprotein ligand-1 (PSGL-1) (Tsukita et al., 1994 ▶; Yonemura et al., 1998 ▶; Legg & Isacke, 1998 ▶; Heiska et al., 1998 ▶; Alonso-Lebrero et al., 2000 ▶). Furthermore, the FERM domain binds the cytoplasmic adaptor protein Na+/H+-exchanger regulatory factor (NHERF), which is a PDZ-containing adaptor protein (Reczek et al., 1997 ▶; Yun et al., 1998 ▶). Crystallographic studies of the radixin FERM domain bound to inositol-1,4,5-trisphosphate (IP3) and the cytoplasmic tails of ICAM-2, PSGL-1 and CD43 and the C-terminal region of NHERF have been reported (Hamada, Shimizu et al., 2000 ▶; Hamada et al., 2003 ▶; Takai et al., 2007 ▶, 2008 ▶; Terawaki et al., 2006 ▶), revealing two signature sequences RxxTYxVxxA (motif 1) and MDWxxxxx(L/I)Fxx(L/F) (motif 2) for FERM binding.
Transmembrane-type proteases play an important role in cell-surface proteolysis, which is involved in a broad range of biological activity via the processing of extracellular proteins or peptides that mediate a diverse range of cellular functions. Recent studies have shown that the cellular localization of one such protease, neutral endopeptidase 24.11 (NEP), is regulated by binding to the FERM domain of ERM proteins through the cytoplasmic region (Iwase et al., 2004 ▶; Terawaki et al., 2007 ▶). Membrane-type 1 matrix metalloproteinase (MT1-MMP) is another membrane-integrated proteinase that is responsible for degradation of the extracellular matrix (ECM) and activation of MMP-2 during cell movement (Seiki, 2002 ▶). At the front of migrating cells, this protease co-localizes with CD44, which is a binding partner for ERM proteins, and sheds the CD44 ectodomain. It has been shown that CD44 directly mediates MT1-MMP co-localization with actin cytoskeletons (Mori et al., 2002 ▶; Suenaga et al., 2005 ▶). However, it is unknown whether the cytoplasmic tail of MT1-MMP, comprising 20 residues, binds directly to ERM proteins. Here, we reveal a direct interaction between the MT1-MMP cytoplasmic tail and the radixin FERM domain and report on the crystallographic characterization of the radixin FERM domain complexed with the MT1-MMP cytoplasmic tail (the FERM–MT1-MMP complex).
2. Material and methods
2.1. Protein preparation and binding assay
The FERM domain of mouse radixin (residues 1–310) was expressed in Escherichia coli BL21 (DE3) RIL containing plasmid pGEX4P-3 as a fusion protein with glutathione S-transferase. The amino-acid sequence of this domain has 100% identity to that of the corresponding human protein. Details of the purification scheme of this domain have been described previously (Hamada, Matsui et al., 2000 ▶). In addition to the scheme, heparin Sepharose column chromatography was applied as the final step. Purified samples were verified using matrix-assisted laser desorption/ionization time-flight mass spectroscopy (MALDI–TOF MS; PerSeptive Inc.) and N-terminal sequence analysis (M492, Applied Biosystems). Synthetic peptides corresponding to the full-length cytoplasmic tail (residues 563–582) of human MT1-MMP were purchased from Toray Research Center (Tokyo, Japan). For crystallization, the peptide was dissolved in a buffer containing 50 mM NaCl, 10 mM Na MES pH 6.8 and 1 mM DTT.
A binding assay utilizing the MT1-MMP cytoplasmic tail peptide and the FERM domain was performed by surface plasmon resonance measurements using a BIAcore Biosensor instrument (BIAcore 3000, Pharmacia Biosensor/GE Healthcare). The biotinylated peptide was immobilized on the surface of an SA (streptavidin) sensor chip. The purified FERM domain was injected onto the peptide surface. All binding experiments were performed at 298 K with a flow rate of 10 µl ml−1 in HBS-P buffer (10 mM Na HEPES pH 7.4, 150 mM NaCl, 0.05% surfactant P20). The kinetic parameters were evaluated using the BIA Evaluation software (Pharmacia/GE Healthcare).
2.2. Crystallization
For the crystallization screening, a protein–peptide solution (1:5 molar ratio) was prepared by mixing 0.6 mM FERM-domain solution with 3 mM MT1-MMP peptide solution in a 1:1 volume ratio. Crystallization conditions were examined by employing the sitting-drop vapour-diffusion method at 277 K using a Hydra II-Plus-One crystallization robot (Matrix Technology) with a commercial crystallization solution kit. Optimization of the crystallization conditions was performed using the hanging-drop vapour-diffusion method. Crystals were transferred stepwise into cryoprotective solution and then flash-frozen at 100 K.
2.3. X-ray data collection
Initial diffraction tests of the FERM–MT1-MMP complex were performed using a Rigaku FR-E X-ray generator equipped with a Rigaku R-AXIS VII detector at 100 K. X-ray diffraction data for structure determination were collected with a Jupiter 315 detector installed on the BL38B1 beamline at SPring-8 (Hyogo, Japan) using flash-frozen crystals. The data collection was performed using a total oscillation range of 180° with a step size of 1.0° and an exposure time of 15 s. The camera distance was 225 mm. The diffraction data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
A quantitative analysis of peptide binding to the radixin FERM domain was performed with the MT1-MMP peptide, which corresponds to the full-length cytoplasmic region (563-RRHGTPRRLLYCQRSLLDKV-582). We found that the radixin FERM domain binds the MT1-MMP peptide with a dissociation constant (K d) of 0.55 µM (Fig. 1 ▶). This result encouraged us to try crystallization of the FERM domain complexed with the peptide. The best crystals of the FERM–MT1-MMP complex were obtained in two weeks by combining 1 µl protein solution with 1 µl reservoir solution containing 12% polyethylene glycol 4000 (PEG 4K), 100 mM MOPS pH 7.0 and 100 mM NaCl. The crystals grew to maximum dimensions of 0.3 × 0.3 × 0.2 mm (Fig. 2 ▶).
Figure 1.

Binding of the FERM domain to the MT1-MMP cytoplasmic peptide-coated sensor chip as measured by surface plasmon resonance (BIAcore). (a) Sensorgrams for each concentration of the FERM domain. The signals from the control surface were subtracted. (b) Binding isotherms for the FERM domain and the MT1-MMP cytoplasmic peptide from equilibrium SPR measurements. A solid line represents a theoretical curve constructed from R max and K d values determined by nonlinear least-squares analysis of the isotherm using the equation R = R max/(1 + K d/[P]), where R is the saturated (equilibrated) resonance and [P] is the protein concentration.
Figure 2.

A pyramidal crystal of the complex formed by the radixin FERM domain with the MT1-MMP cytoplasmic tail peptide. The scale bar indicates 0.1 mm.
Crystals were transferred stepwise into a cryoprotective solution containing 25% PEG 200, 12% PEG 4K, 100 mM NaCl and 100 mM MOPS pH 7.0 for flash-cooling. Crystals were found to diffract to a resolution of 2.4 Å and to belong to space group P6122, which was determined by molecular replacement as described below, with unit-cell parameters a = b = 122.7, c = 128.3 Å. The total number of observed reflections was 480 219 and included 22 917 unique reflections (Table 1 ▶). The resulting data gave an R merge of 4.5% (27.5% for the outer shell, 2.49–2.40 Å) with a completeness of 100% (100% for the outer shell). The estimated mosaicity of the crystal was estimated as 0.30–0.5°. Assuming the presence of one complex in the asymmetric unit, the calculated value of the crystal volume per protein weight (V M; Matthews, 1968 ▶) was 3.45 Å3 Da−1. This value corresponds to a solvent content of approximately 65%.
Table 1. X-ray diffraction data of the FERM–MT1-MMP complex.
Values in parentheses are for the outer resolution shell.
| Beamline | BL38B1, SPring-8 |
| Detector | Jupiter 315 detector |
| Wavelength (Å) | 1.00 |
| Temperature (K) | 100 |
| Oscillation range (°) | 180 (180 × 1° images) |
| Exposure time (s) | 15 |
| Space group | P6122 |
| Unit-cell parameters (Å, °) | a = b = 122.7, c = 128.3, γ = 120 |
| Resolution (Å) | 20–2.4 (2.47–2.40) |
| Reflections (total/unique) | 48021/22917 |
| Completeness (%) | 100.0 (100.0) |
| Mosaicity | 0.3–0.5 |
| Multiplicity | 21.0 (19.6) |
| 〈I/σ(I)〉 | 15.6 (9.0) |
| Rmerge† (%) | 4.5 (27.5) |
R
merge = 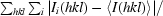
 , where Ii(hkl) is the ith observed intensity of reflection hkl and 〈I(hkl)〉 is the average intensity over symmetry-equivalent measurements.
, where Ii(hkl) is the ith observed intensity of reflection hkl and 〈I(hkl)〉 is the average intensity over symmetry-equivalent measurements.
Structural analysis of the FERM–MT1-MMP complex crystal was performed using the molecular-replacement method with the free radixin FERM domain as a search model (Hamada, Shimizu et al., 2000 ▶). Using the solution obtained from the molecular-replacement analysis, an initial model was refined by the simulated-annealing method in CNS (Brünger et al., 1998 ▶). The R value of the current structure was 25.2% (R free = 29.2%). In the F o − F c map, an electron density thought to correspond to the MT1-MMP peptide was found to be located at the molecular surface of the FERM domain. Efforts are currently being directed toward solving the complex structure by structural modelling and refinement.
Acknowledgments
We would like to thank J. Tsukamoto for technical support in performing the MALDI–TOF MS analysis. We gratefully acknowledge Sh. Tsukita and Sa. Tsukita for providing the mouse radixin cDNA. This work was supported by a Grant-in-Aid for Scientific Research (A) and Grants-in-Aid for Scientific Research on Priority Areas, Cancer, Macromolecular Assembly, Membrane Interface and Nano-systems in Cells from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. The early stage of this work was supported by Core Research for Evolutional Science and Technology (CREST) from the Japan Science and Technology Corporation (JST). ST was supported by a postdoctoral research fellowship from a Grant-in-Aid for the 21st Century COE Research from MEXT. We also acknowledge K. Hasegawa, H. Sakai and M. Yamamoto at SPring-8 for help with data collection on synchrotron beamline BL38B1.
References
- Alonso-Lebrero, J. L., Serrador, J. M., Domínguez-Jiménez, C., Barreiro, O., Luque, A., del Pozo, M. A., Snapp, K., Kansas, G., Schwartz-Albiez, R., Furthmayr, H., Lozano, F. & Sánchez-Madrid, F. (2000). Blood, 95, 2413–2419. [PubMed]
- Bretscher, A., Edwards, K. & Fehon, R. G. (2002). Nature Rev. Mol. Cell Biol.3, 586–599. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Funayama, N., Nagafuchi, A., Sato, N., Tsukita, S. & Tsukita, S. (1991). J. Cell Biol.115, 1039–1048. [DOI] [PMC free article] [PubMed]
- Hamada, K., Matsui, T., Tsukita, S., Tsukita, S. & Hakoshima, T. (2000). Acta Cryst. D56, 922–923. [DOI] [PubMed]
- Hamada, K., Shimizu, T., Matsui, T., Tsukita, S. & Hakoshima, T. (2000). EMBO J.19, 4449–4462. [DOI] [PMC free article] [PubMed]
- Hamada, K., Shimizu, T., Yonemura, S., Tsukita, S., Tsukita, S. & Hakoshima, T. (2003). EMBO J.22, 502–514. [DOI] [PMC free article] [PubMed]
- Heiska, L., Alfthan, K., Grönholm, M., Vilja, P., Vaheri, A. & Carpén, O. (1998). J. Biol. Chem.273, 21893–21900. [DOI] [PubMed]
- Hirao, M., Sato, N., Kondo, T., Yonemura, S., Monden, M., Sasaki, T., Takai, Y., Tsukita, S. & Tsukita, S. (1996). J. Cell Biol.135, 37–51. [DOI] [PMC free article] [PubMed]
- Iwase, A., Shen, R., Navarro, D. & Nanus, D. M. (2004). J. Biol. Chem.279, 11898–11905. [DOI] [PubMed]
- Legg, J. W. & Isacke, C. M. (1998). Curr. Biol.8, 705–708. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Mori, H., Tomari, T., Koshikawa, N., Kajita, M., Itoh, Y., Sato, H., Tojo, H., Yana, I. & Seiki, M. (2002). EMBO J.21, 3949–3959. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Reczek, D., Berryman, M. & Bretscher, A. (1997). J. Cell Biol.139, 169–179. [DOI] [PMC free article] [PubMed]
- Seiki, M. (2002). Curr. Opin. Cell Biol.14, 624–632. [DOI] [PubMed]
- Suenaga, N., Mori, H., Itoh, Y. & Seiki, M. (2005). Oncogene, 24, 859–868. [DOI] [PubMed]
- Takai, Y., Kitano, K., Terawaki, S., Maesaki, R. & Hakoshima, T. (2007). Genes Cells, 12, 1329–1338. [DOI] [PubMed]
- Takai, Y., Kitano, K., Terawaki, S., Maesaki, R. & Hakoshima, T. (2008). J. Mol. Biol.381, 634–644. [DOI] [PubMed]
- Takeuchi, K., Kawashima, A., Nagafuchi, A. & Tsukita, S. (1994). J. Cell Sci.107, 1921–1928. [DOI] [PubMed]
- Terawaki, S., Kitano, K. & Hakoshima, T. (2007). J. Biol. Chem.282, 19854–19862. [DOI] [PubMed]
- Terawaki, S., Maesaki, R. & Hakoshima, T. (2006). Structure, 14, 777–789. [DOI] [PubMed]
- Tsukita, S., Oishi, K., Sato, N., Sagara, J., Kawai, A. & Tsukita, S. (1994). J. Cell Biol.126, 391–401. [DOI] [PMC free article] [PubMed]
- Tsukita, S. & Yonemura, S. (1999). J. Biol. Chem.274, 34507–34510. [DOI] [PubMed]
- Tsukita, S., Yonemura, S. & Tsukita, S. (1997). Trends Biochem. Sci.22, 53–58. [DOI] [PubMed]
- Turunen, O., Wahlström, T. & Vaheri, A. (1994). J. Cell Biol.126, 1445–1453. [DOI] [PMC free article] [PubMed]
- Yonemura, S., Hirao, M., Doi, Y., Takahashi, N., Kondo, T., Tsukita, S. & Tsukita, S. (1998). J. Cell Biol.140, 885–895. [DOI] [PMC free article] [PubMed]
- Yun, C. H., Lamprecht, G., Forster, D. V. & Sidor, A. (1998). J. Biol. Chem.273, 25856–25863. [DOI] [PubMed]