

Abstract
Microarray technology was utilized to isolate disease-specific changes in gene expression by sampling across inferior parietal lobes of patients suffering from late onset AD or non-AD-associated dementia and non-demented controls. Primary focus was placed on understanding how inflammation plays a role in AD pathogenesis. Gene ontology analysis revealed that the most differentially expressed genes related to nervous system development and function and neurological disease followed by genes involved in inflammation and immunological signaling. Pathway analysis also implicated a role for chemokines and their receptors, specifically CXCR4 and CCR3, in AD. Immunohistological analysis revealed that these chemokine receptors are upregulated in AD patients. Western analysis demonstrated an increased activation of PKC, a downstream mediator of chemokine receptor signaling, in the majority of AD patients. A very specific cohort of genes related to amyloid beta accumulation and clearance were found to be significantly altered in AD. The most significantly downregulated gene in this data set was the endothelin converting enzyme 2 (ECE2), implicated in amyloid beta clearance. These data were subsequently confirmed by real-time PCR and Western blot analysis. Together, these findings open up new avenues of investigation and possible therapeutic strategies targeting inflammation and amyloid clearance in AD patients.
Keywords: Alzheimer's disease, Endothelin converting enzyme, Inflammation, Aging, Chemokine
Introduction
Alzheimer's disease (AD) affects over 4 million people in the United States each year [1]. The molecular mechanisms that underlie the pathogenesis of this disease have not yet been elucidated, and it is likely that these changes may begin well before detectable cognitive impairment occurs. Determining the biological and molecular alterations that occur prior to the onset of the disease may provide valuable insight into the early diagnosis and management of AD. Moreover, it is also important to identify the molecular targets that will assist in the treatment of later stage disease as many patients who present with cognitive impairment will already exhibit advanced pathology associated with this disease.
Microarray technology is an efficient means by which the molecular differences between control and diseased tissues can be identified and compared in a high throughput fashion. Several studies using this technique have already been reported examining the changes in AD patients with mild to moderate disease [2–5]. In one such study, the gene expression differences between AD samples and control tissues utilized pooled RNA from 5 patients and demonstrated differences in the expression of genes associated with amino acid metabolism and neurotransmitter release [3]. Similarly, using pooled RNA preparations, Colangelo and colleagues specifically examined the hippocampal cornu ammonis 1 and demonstrated a very general overall repression of transcription in this region in AD patients [5]. In a third study, gene expression differences were observed between the affected hippocampus to that of the unaffected parietal cortex of the same patient [4]. RNA from only one patient was utilized to perform the microarray analysis, and the data verification was expanded to 6 samples during real-time PCR validation of the genes identified. The overall focus of this latter study was the identification of the protein calcineurin as being one of the key players in AD progression. While all of these data are intriguing, the results are somewhat disparate and little detailed ontology was shown in these reports.
In an effort to better understand the global changes in gene transcription in AD compared to control tissues, we examined the inferior parietal cortex of four highly defined AD patients using microarray analysis and subsequently compared these data to the results obtained from the same tissues of patients with non-AD-related dementia and using pooled, non-demented controls as a reference. These experiments were performed on whole-genome Agilent 44k oligomer arrays and were subjected to z-transformation followed by analysis using distance-based gene selection criteria. Using this analysis, we were able to identify changes specific to AD and not simply those that result from the acquisition of changes characteristic to neurodegeneration. All of the AD patient tissues utilized were matched for similar levels of neuritic plaques (P) and neurofibrillatory tangles (T), indicative of their levels of neurodegeneration. Contrary to other studies, the tissues from the AD and dementia patients were examined as individual samples and not as pooled RNA. The results from this small pool of AD patients were expanded to a larger set of ten AD patients using real-time PCR, immunohistochemical (IHC) and Western blot analyses. Gene ontology analysis revealed that the most significant group of differentially expressed genes related to nervous system development and function and the regulation of amyloid clearance. In addition, several genes associated with immune cell and chemokine signaling were also found to be differentially expressed by AD tissues, further supporting the involvement of immune cells and various inflammatory mediators in the AD process. Together, these findings may open up some new avenues of investigation in AD research as well as elucidating potential therapeutic strategies for targeting inflammation and regulating amyloid clearance in AD patients.
Results
Cluster analysis reveals distinct differences in the expression of immune-related genes between AD and non-AD-related dementia
AD patients were matched for similar levels of neurodegeneration (with Braak scores of 5 or more) with other dementia samples (with Braak scores of 3 or less). Non-AD dementia patients were comprised of those suffering from Parkinson's disease, hippocampal sclerosis, and patients in whom the dementia had no distinct histology (Table 1, italics). RNA was hybridized to commercial 44K oligomer-based arrays, using pooled RNA from normal age-matched patients as a reference. Data from all these experiments were normalized, averaged and then z-transformed. These data were subsequently subjected to distance-based gene selection analysis, from which a multidimensional scaling diagram (Fig. 1A) and a weighted gene list were obtained (Figs. 1B and C) comprised of genes that were significant to a p value of 0.02 or less. The number next to each gene name is indicative of the significance of that gene as a discriminator of AD samples as compared to the others in the set. This MDS analysis demonstrated that the AD samples maintained a pathology distinct from the other non-AD dementia. Given that limited numbers of Parkinson's and other dementia samples were available, the Alzheimer's samples were compared against all of these samples as a group, as opposed to each one as a distinct pathology. The object of this exercise was to extricate the AD-specific changes from those associated with other forms of neurodegeneration. The cohort of non-AD dementia patients were of a mean younger age (70.6) than the AD patients (82.3) or unaffected controls (84.9), and it is possible that the specific changes identified could reflect this difference. However, the gene expression profile of the youngest AD patient analyzed by microarray (AD4, age 76) did not differ significantly from the other AD patients (Fig. 1) and was quite different from the patients with non-AD dementia.
Table 1. Pathological summary of AD, control and dementia IPL samples.
| Case | Age | Sex | PMI | Braak | P/T | APOE |
|---|---|---|---|---|---|---|
| C1 | 74 | M | 2.5 | 2 | 0/3.5 | e3/e3 |
| C2 | 82 | F | 2 | 2 | 0.25/4.25 | e3/e3 |
| C3 | 91 | F | 2.5 | 3 | 1/4 | e2/e2 |
| C4 | 78 | M | 1.66 | 1 | 0/0 | e3/e3 |
| C5 | 85 | M | 3.16 | 2 | 0/4.25 | e3/e3 |
| C6 | 86 | F | 2.5 | 3 | 0/5 | e3/e3 |
| C7 | 88 | F | 3 | 2 | 0.25/2 | e3/e4 |
| C8 | 90 | F | 3 | 3 | 6.5/4.5 | e3/e3 |
| C9 | 88 | F | 2 | 4 | 0/4.5 | e3/e3 |
| C10 | 87 | F | 2.83 | 2 | 0/1.75 | e3/e3 |
| AD1 | 84 | M | 2 | 5 | 12/11.83 | e3/e4 |
| AD2 | 81 | M | 3 | 5 | 14/15 | e3/e4 |
| AD3 | 81 | M | 3 | 5 | 12.5/11 | e3/e4 |
| AD4 | 76 | M | 2.33 | 6 | 12.5/15 | e3/e3 |
| AD5 | 72 | M | 2.5 | 6 | 14/15 | e2/e3 |
| AD6 | 79 | M | 2 | 5 | 14.5/15 | e3/e3 |
| AD7 | 96 | F | 2.75 | 5 | 12.5/9.6 | e3/e3 |
| AD8 | 89 | F | 3 | 5 | 14/14.5 | e3/e4 |
| AD9 | 80 | F | 2.25 | 6 | 12.5/14.5 33 | e3/e3 |
| AD10 | 85 | F | 1.66 | 5 | 12.25/12 | e3/e3 |
| HS1 | 55 | M | 2 | 3 | 0.5/1.5 | e3/e4 |
| HS2 | 72 | M | 4.5 | 1 | 0/0 | e3/e3 |
| PD1 | 73 | F | 2.16 | 3 | 1.25/3.5 | e3/e4 |
| PD2 | 70 | M | 1.83 | 3 | 0/3.25 | e3/e3 |
| DLDH1 | 83 | F | 1.5 | 1 | 6.5/4.5 | e3/e3 |
| DLDH2 | 71 | M | 5 | 1 | 5.25/0 | e3/e3 |
C=control; AD=Alzheimer's disease; HS=hippocampal sclerosis; PD=Parkinson's dementia; DLDH=dementia lacking distinct histology; PMI=postmortem interval; P/T=plaque/tangle count.
Fig. 1.
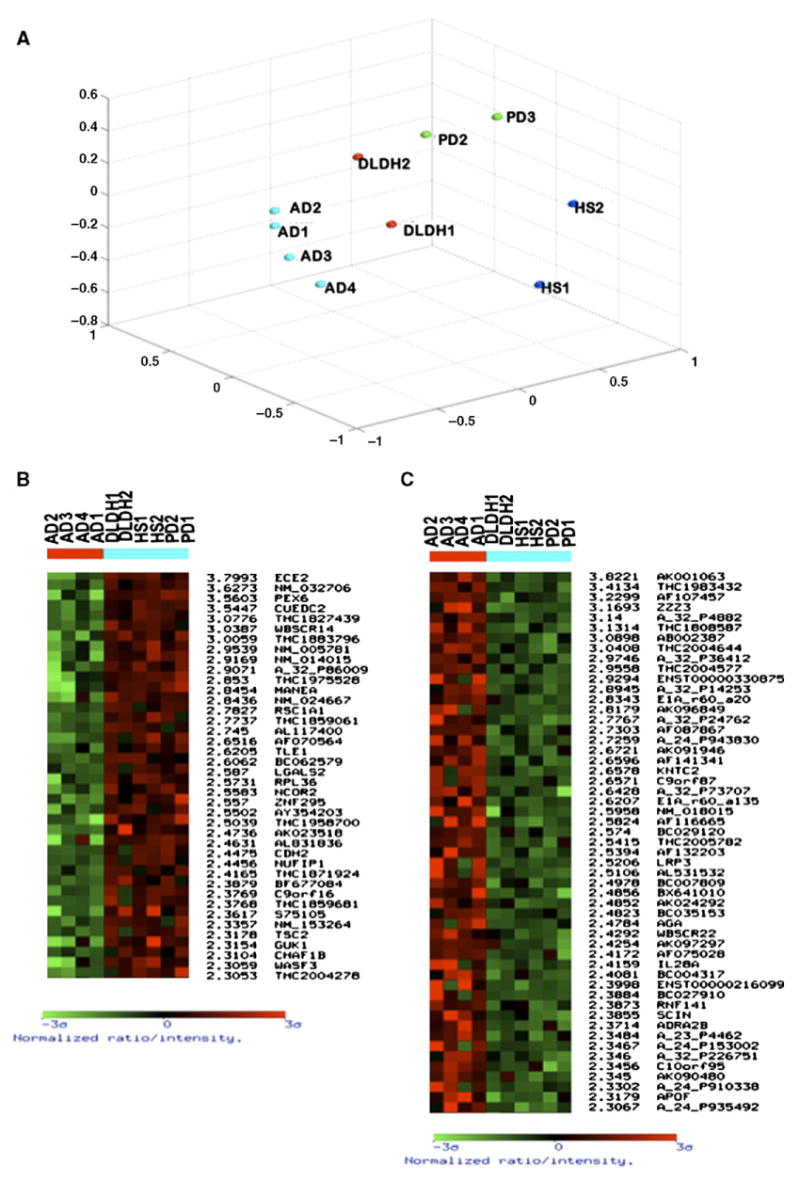
Microarray analysis of the IPL of AD versus non-AD-related dementia patients. The IPLs of four AD patients and six patients representing other non-AD-related dementias were subjected to array analysis using whole genome chips (Agilent, 44K). (A) Multidimensional scaling analysis demonstrates that the AD patients segregate together, demonstrating that they have a specific gene expression profile distinct from the other types of dementia. The weighted gene lists shows genes with a significance of 2.3 or higher including the top 40 downregulated genes (B) and the top 50+ upregulated genes (C) as compared to both control and other non-AD-related dementia samples.
Genes from this analysis were then subjected to pathway analysis using the Ingenuity pathway program (www.ingenuity.com). The program was asked to identify major signaling pathways and also to group the genes by ontology. A large cohort of genes relating to cytokine and immune signaling pathways were identified (Table 2). Of these genes, it was apparent that both interleukin and chemokine signaling pathways are activated in AD tissue, reflected by the presence of CXCR2 (IL-8Rβ) and IL-28A on the weighted gene lists. The presence of these two genes was verified using real-time RT-PCR. CXCR2 was upregulated in 8 out of 10 AD patients as compared to controls (Fig. 2A). IL-28A was also upregulated in 8 out of 10 AD patients (Fig. 2B). In order to determine if other such genes that were found to be upregulated by array, but did not meet the significance level we required for the weighted gene list, could also be validated by qRT-PCR, an interleukin receptor, IL-6R, and the chemokine CCL27 (CTAK) were examined and verified by real-time RT-PCR analysis. IL-6R was not as highly upregulated as IL-28A but was modestly upregulated in the majority of the AD samples, but CCL27 was dramatically increased in 7 out of 10 AD patients (Figs. 2C and D).
Table 2. Immune signaling pathways in AD.
| Canonical pathway | Significance | # Genes | |
|---|---|---|---|
| B-Cell receptor signaling |

|
3.25E−5 | 14 |
| Chemokine signaling | 3.85E−3 | 9 | |
| ERK/MAPK signaling | 1.33E−2 | 13 | |
| IGF-1 signaling | 2.73E−2 | 8 | |
| T-Cell receptor signaling | 4.04E−2 | 8 | |
| JAK/STAT signaling | 5.01E−2 | 6 | |
| IL-2 signaling | 5.95E−2 | 5 | |
| NF-kB | 7.15E−2 | 10 | |
Fig. 2.
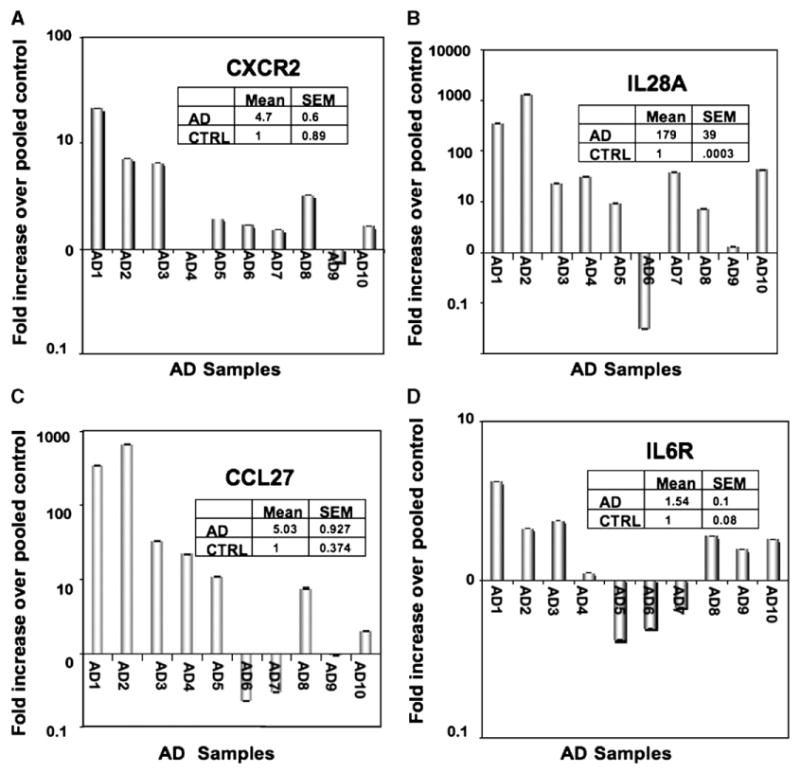
Real-time RT-PCR validation of immune-related genes. Based on the gene ontogeny analysis, several genes associated with immune signaling and function were selected from the top 100 genes on the weighted gene list. Real-time PCR was performed on ten AD samples and compared to pooled controls. The genes upregulated in the majority of AD patients included CCL7 (A), IL-28A (B), IL-6R (C) and CXCR2 (D).
CXCR4 is upregulated in Alzheimer's disease and corresponds to the levels of activated PKC
The upregulation of CCL27 and the chemokine receptor CXCR2 were in keeping with the pathway identification of enhanced chemokine signaling in AD tissues (Figs. 2A, C). Specifically, the pathway analysis program, Ingenuity, identified the chemokine receptors CXCR4 and CCR3 as potentially important molecules in AD pathogenesis (Fig. 3A). We performed real-time PCR on the AD samples and found that CXCR4 was indeed highly elevated in the AD compared to control samples (Fig. 3D). Furthermore, in the majority of the AD samples, a significant elevation in the levels of phospho-PKC could be noted in each tissue sample compared to control samples (Fig. 3E), suggesting active cytokine and chemokine signaling within the AD IPL tissues. The activation of PKC is an important feature of chemokine signaling as well as other G-protein coupled receptors (GPCRs) [6]. However, there were exceptions in the data, where, in samples AD3, 5, 9 and 10, CXCR4 expression was not increased, but PKC was activated. This speaks to the redundancy of chemokine receptor signaling as the majority of these tissues have increases in other chemokines, including CCR3 and CCR5. In two other samples, CXCR4 was elevated, but did not correlate to PKC increases (AD 1 and 6) and may speak to a lack of available ligand for receptor activation in these particular samples. To confirm that the expression of CXCR4 was indeed increased in AD tissues, immunohistochemical analysis was performed using an antibody specific to CXCR4. While normal control brain tissues exhibited a low level of staining for CXCR4 (Figs. 4A and C, arrows), the AD tissues demonstrated significantly stronger CXCR4 immunoreactivity (Figs. 4B, D and F, arrows). An AD sample with no primary antibody is shown in Fig. 4E as a negative control. This pattern of staining was also observed for CCR3 (Supplementary Fig. 1) and supports previous data implicating these two chemokines in Alzheimer's disease [7].
Fig. 3.
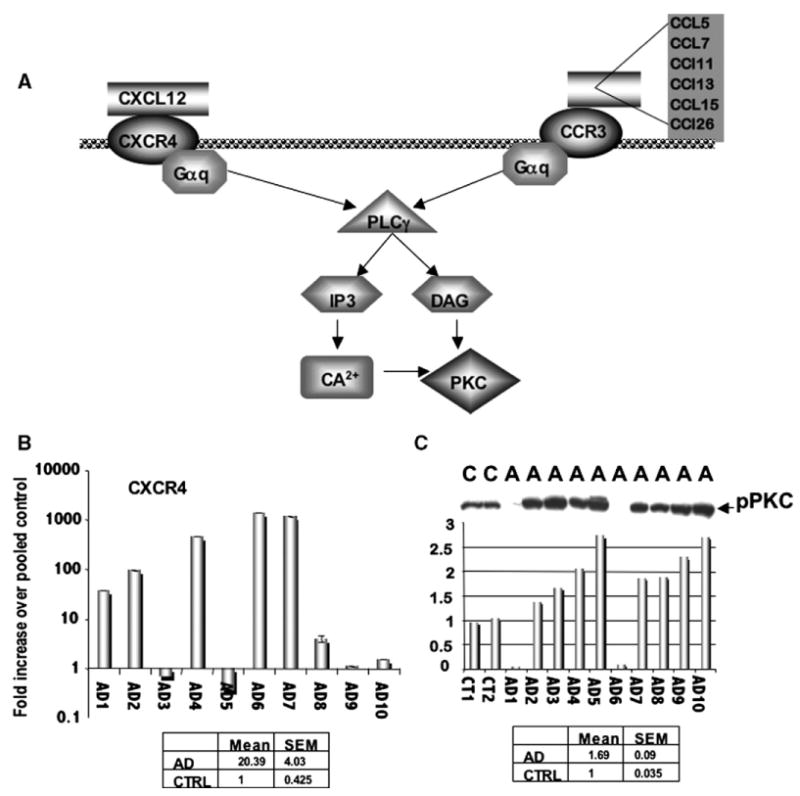
Genes associated with the chemokine pathway are upregulated in AD tissues. (A) Pathway analysis revealed the increased expression of CXCR4 and CCR3 in AD tissue. (B) Real-time RT-PCR analysis confirmed that CXCR4 was highly upregulated in the majority of AD patients, and (C) this upregulation correlated with the activation of PKC in the majority of these patients. “A” or “AD” denotes AD patient samples, while “C” denotes non-demented controls. Densitometry shows that the elevation of PKC in these samples ranges from 1.5- to almost 3-fold increases over the normal samples (first two bars).
Fig. 4.
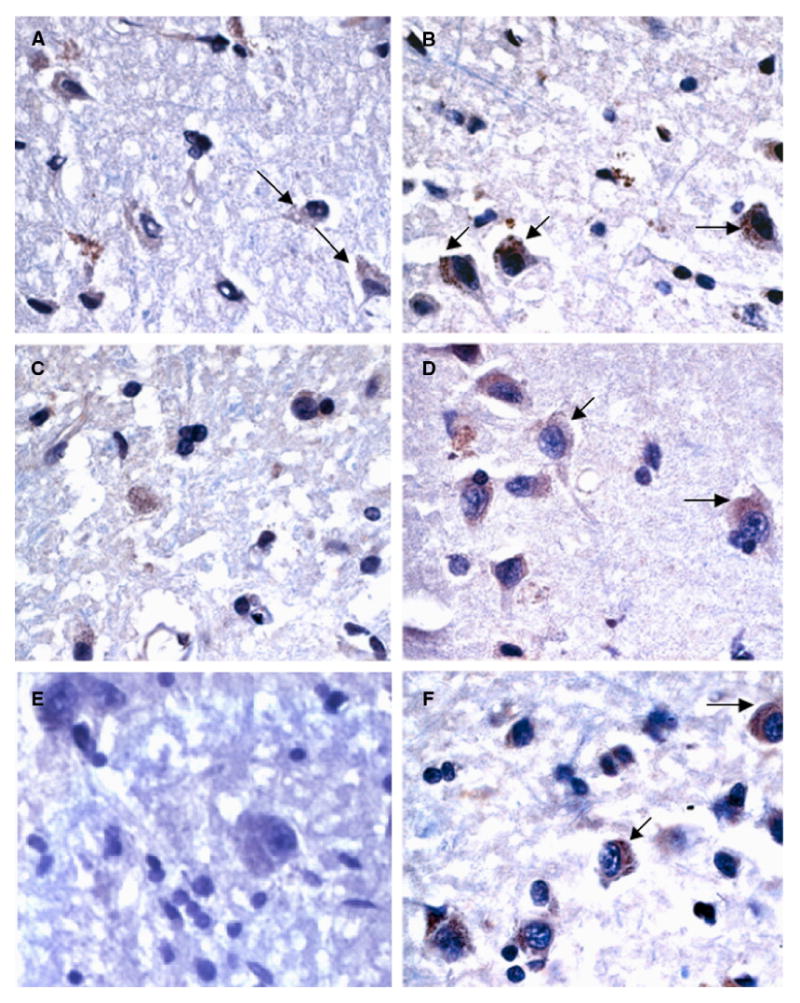
CXCR4 is overexpressed in AD. Immunohistochemistry was performed on AD tissues and non-demented controls. CXCR4 is expressed at low to moderate levels in the normal tissues (A, C, arrows), but expression is highly upregulated in AD patients (B, D, F, arrows). An AD patient sample (same patient as in panel F) was incubated with no primary antibody as a negative control for CXCR4 staining (E).
Genes identified by profiling of AD tissues are largely involved in neuronal development and disease and highlight the central theme of amyloid deposition
Although a number of genes associated with immune signaling were identified in AD pathogenesis, the largest groups of genes identified by pathway analysis were those involving nervous system development and function (Table 3). An example of such a gene is aquaporin 1, which is known to be important in paracellular transport of water across cell membranes. In the brain, aquaporin 1 has been suggested to be important in the build-up of interstitial fluid in brain tumors and is also key in the production of cerebrospinal fluid [8]. Interestingly, while the full microarray analysis of four AD donors revealed that aquaporin 1 is upregulated compared to control age-matched samples, this gene was found to be only upregulated in approximately half of the AD samples using real-time RT-PCR (Fig. 5A). This is not unexpected as donor to donor variations are often noted upon verification of array data. But perhaps the most interesting neuronal gene data set examined were the differentially expressed genes coding for proteins involved in the deposition of amyloid. One of the most upregulated genes on the weighted gene list was the gene LRP3, a member of the low-density lipid receptor family. LRPs are the receptors for apoliproteins, and apolipoprotein E (APOE) and LRP1 have been shown to specifically interact and aggregate in the senile plaques of AD patients [9]. Polymorphisms in LRP1 also correlate to the onset of AD [10–12] and, in the presence of the e4 allele of APOE, LRP1 polymorphisms correspond to increased risk of AD [13]. It has also been shown that increases in apolipoproteins can stimulate LRP1 receptor expression [13]. These receptors have been implicated in AD in a dichotomous fashion. For example, LRP receptors are important for the clearance of amyloid beta [14] and thus one could speculate that loss of this protein would contribute to AD pathogenesis. However, in several studies, LRP1 proteins are upregulated in AD tissues [15] and aggregate in senile plaques [9]. In addition, overexpression of LRP1 is also associated with increases in amyloid deposition in mouse models [16]. In our present study, we have found by real-time RT-PCR that the LRP family member LRP3 was slightly upregulated in the majority of the AD patients examined (Fig. 5B). Little is known about this member of the LRP family or its role in Alzheimer's disease, but it is likely that it may play a similar role to its more famous LRP cousins. Interestingly, although APOE was not identified by the gene array analysis, another member of this family, APOF, was also upregulated on the array. However, the expression of this gene by real-time widely varied among controls as well as AD patients (data not shown). Thus, it was not possible to confirm this array observation. It will be of great interest to determine if this APOF/LRP3 combination can act in the same manner as the APOE/LRP ligand–receptor pair.
Table 3. Gene ontology analysis of major protein families involved in AD.
| Function | Significance | # Genes | |
|---|---|---|---|
| Nervous system development/function |

|
5.83E−5–4.86E−2 | 87 |
| Organ development | 5.83E−5–2.32E−2 | 9 | |
| Cancer | 2.07E−4–4.49E−2 | 47 | |
| Cell cycle | 2.07E−4–4.77E−2 | 58 | |
| Neurological disease | 2.07E−4–4.63E−2 | 19 | |
| Tissue development | 2.07E−4–4.49E−2 | 40 | |
| Cell-to-cell signaling & interaction | 9.31E−4–4.49E−2 | 52 | |
| Cell morphology | 3.27E−3–4.41E−2 | 40 | |
Fig. 5.
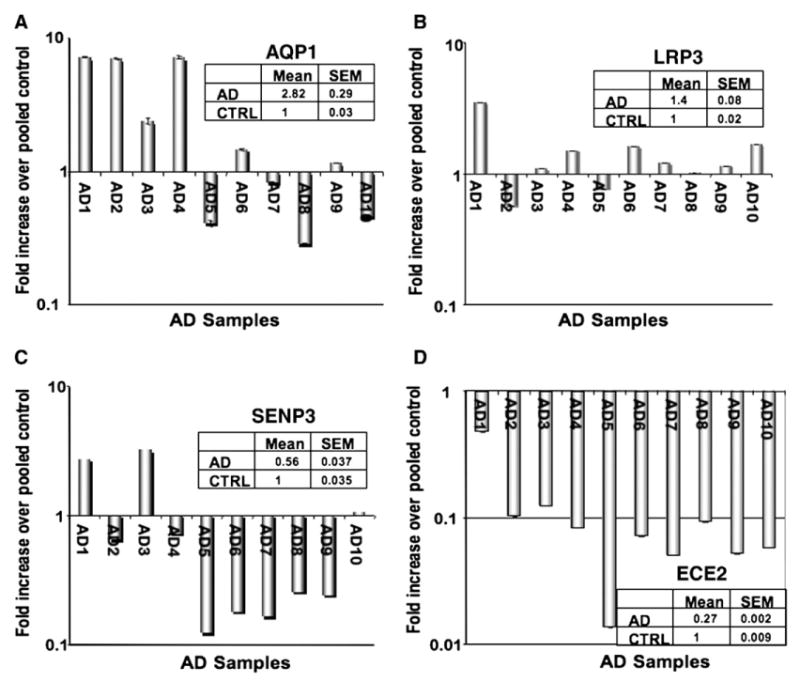
Real-time RT-PCR verification of neuronal-specific genes. Based on the gene ontogeny analysis, several neuronal genes were identified to be differentially expressed in AD tissues. (A) Aquaporin 1 (A) and LRP3 (B) were found to be slightly elevated to elevated in the majority of AD patients. (C) SENP3, a sumoylation related protein, was significantly downregulated in Ad tissues versus control tissues. (D) ECE2 was dramatically underexpressed in every AD patient compared to the controls.
In addition to the upregulation of genes associated with amyloid deposition, we also observed the downregulation of several genes involved in neuronal survival and amyloid clearance. One of these genes, sentrin P3 (SENP3), is a SUMO protease that reverses the sumoylation of the myocyte enhancement factor MEF2 [17], which in turn promotes neuronal survival. Thus, loss of this protease would putatively lead to increased neuronal apoptosis. However, as with LRP, the role of SENP3 is dichotomous. It has been shown that amyloidogenesis of APP can be dramatically decreased in the presence of sumoylation factors such as SUMO-3 and dramatically increased when this process is reversed [18]. SENP3 can reverse sumoylation, making its loss in AD patients paradoxical from the standpoint of amyloid clearance, albeit significant from the standpoint of increased neuronal apoptosis. Real-time PCR analysis indicates that 7 out of 10 AD tissue samples have lost SENP3 (Fig. 5C).
A gene that is indeed important in amyloid clearance but has no record of being involved in increased amyloid deposition is the gene endothelin converting enzyme 2 (ECE2), which we have found was extremely downregulated in AD samples compared to all other patients and controls. ECE2 is an endothelin converting enzyme responsible for the degradation of amyloid beta [19,20]. ECEs are important in Aβ clearance, and mice deficient in ECE1 have increased steady state levels of Aβ [20]. Loss of these enzymes in human tissue has not yet been demonstrated, but this gene was the most significantly downregulated in our arrays of human patients. Expansion of the array results to a larger cohort of patients by real-time PCR showed that this gene was indeed markedly downregulated in all of the Alzheimer's patients (Fig. 5D), but not in patients exhibiting other types of dementia (Fig. 1B, top row, ECE2). We then performed immunohistochemical analysis and show here, for the first time, that there is a dramatic loss of ECE2 immunoreactivity in AD tissue samples (Figs. 6B and D, arrows) as compared to normal controls (Figs. 6A and C, arrows).
Fig. 6.
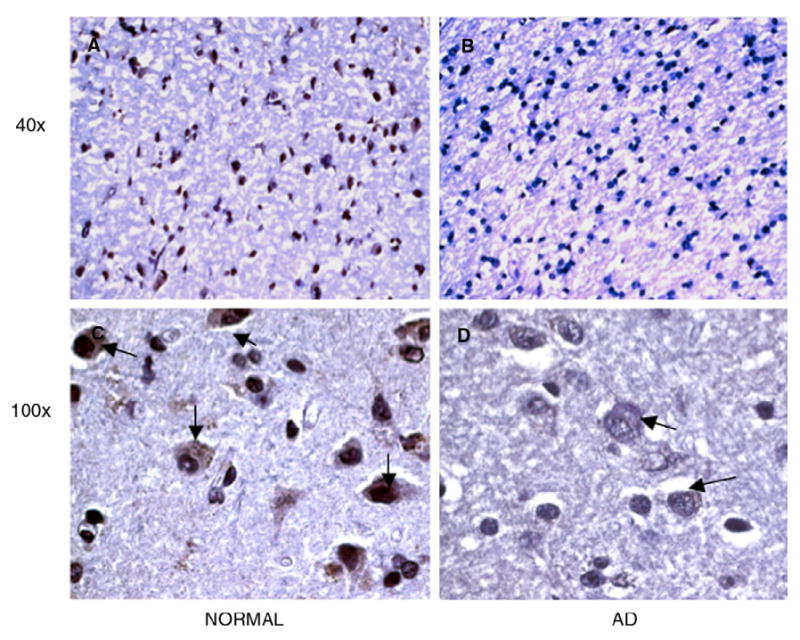
The expression of the ECE2 protein is lost in AD. Similar to the real-time RT-PCR described in Fig. 5, immunohistochemical analysis revealed abundant expression of ECE2 in control normal IPL tissues (A, C, arrows) and little to no staining for ECE2 in AD tissues (B, D, arrows). Sections are shown at 40× and 100×.
Discussion
In this study, we used both non-diseased patients and patients with non-AD-related dementia to identify AD-specific changes using gene expression profiling. The present study is intended to be an overview of the data generated by this analysis. We find that large cohorts of immune-related genes are upregulated, specifically several chemokines and chemokine receptors. Chemokine receptors are known to be expressed in CNS cells, and the upregulation of many of these receptors has previously been found in AD tissues, not only on microglial cells [21], but also on glia and neurons [7]. What was interesting in our data set of 44K genes is that these same receptors were identified as some of the most highly upregulated genes in AD when compared to other forms of dementia. In certain reports, chemokine receptors are expressed by activated microglia and some, such as CCR5, may actually play a role in microglia recruitment to senile plaques [22], possibly due to the presence of increased levels of amyloid beta. It has been suggested that chemokine receptor antagonists may be of great import in reducing the level of neuroinflammation associated with Alzheimer's disease and may even slow its onset [22]. In the current report, we demonstrate, for the first time, that CXCR4 is highly expressed in the IPLs and by glial cells in AD subjects when compared to age-matched controls. Similar to many G-protein coupled receptors (GPCR), chemokine receptors are known to activate PKC upon ligation with their respective ligands [6]. Our current data indicate that AD tissues as a whole tend to have much higher levels of activated, phosphorylated PKC, a hallmark of chemokine and GPCR signaling as well as a number of other signaling pathways. Whether this PKC activation is due to activation of the chemokine or other signaling pathways remains to be defined. However, there may be some potential clinical utility for the use of PKC inhibitors in AD patients, similar to therapeutic studies currently ongoing in the clinic for cancer therapy.
Surprisingly, the chemokine CCL27 was also found to be significantly increased in the examined AD lesions. There are several chemokines and their receptors that are associated with skin-homing CD4+ T cells, namely CCR4 and its ligands CCL17/TARC and CCL22/MDC, and CCR10 together with its ligand CCL27/CTACK. CCL27 has been shown to be expressed strongly and selectively in the skin, predominantly in keratinocytes [23]. It is thought that the CCL17–CCR4 pathway recruits lymphocytes into the dermis, whereas the CCL27–CCR10 pathway may guide them all the way up to the epidermis, thus suggesting that at least one of these chemokine-mediated pathways must be functional to effectively recruit T cells to inflammatory skin such as in atopic dermatitis, psoriasis and atopic eczema [23–28]. Moreover, there is a second form of the CCL27 protein known as PESKY, which lacks a signal peptide which has been replaced by a 40 aa segment that appears to function as a nuclear localization signal. This transcript shows an expression pattern that is very distinct from the secreted form of CCL27 with high levels of expression in brain and testis and weak expression in liver and kidney [23]. There is currently no known function for this nuclear form of CCL27. Given that T cells are not a predominant immune cell type within AD lesions, the functional relevance of CCL27 and/or its alternative form expression in AD tissues remains unclear but the high levels of expression do suggest a possible novel role for CCR27 in the neuronal system.
Another interesting gene shown to be increased in the AD tissues examined was one of the type I interferons, namely IFN-λ2 (IL-28A). There are three genes for IFN-λ and have been named IFN-λ1, IFN-λ2 and IFN-λ3 (also known as IL-29, IL-28A and IL-28B, respectively). These type I interferons have been shown to be induced by viral infection in the majority of nucleated cells and play a critical role in the innate and adaptive immune responses to viral infection [29]. While the relevance of increased IL-28A expression in AD lesions in the current work remains unclear, the significant levels of this cytokine in AD tissues may suggest the presence of a virus or virus-like infection. In 1999, Dobson and Itzhaki reported that herpes simplex type 1 virus (HSV1) is present in the brain of many elderly people and that it may be a risk factor for AD [30]. However, in earlier studies, no evidence of herpes virus RNA was observed in the hippocampus of demented individuals with extensive neuropathological changes of AD [31,32]. Additional studies will be required to discover the possible relevance of this cytokine in AD pathology and in viral infections in neuronal tissues.
In addition to the identification of various immune- and chemokine-related genes, the array analysis also highlighted the importance of genes associated with amyloidogenesis in AD patients. Interestingly, at least two of these genes, LRP3 and SENP3, were identified and have been previously reported to play possible dual roles in the process of amyloidogenesis. For example, LRP1 is known to be important for amyloid clearance [14], while in many other studies, its overexpression is associated with AD onset and progression [33]. It is possible that the overexpression of the specific family member LRP3 in our AD patients indicates that this form of the receptor may also be associated with progressive AD. Similarly, SENP3 is known to reverse sumoylation, a process which decreases APP deposition [18], yet this gene is downregulated in our AD patients. However, it should be noted that SENP3 is also important for the activity of MEF2 [17], a neuronal survival factor, and thus the loss of SENP3 may lead to the increased neuronal apoptosis often observed in the tissues of AD patients.
To date, only ten manuscripts have investigated the relationship between endothelin converting enzymes (ECEs) and AD, many of which are reviews speculating upon the potential importance of these enzymes. Only two reports to date have implicated ECEs in the clearance of β-amyloid in vitro [19] and in animal models [20], but no laboratory has demonstrated the loss of ECE mRNA or protein expression in human AD tissue. In our current report, we demonstrate that the ECE family member ECE2 is significantly lost in AD, but not in other types of dementia, suggesting that this is not merely a side-effect of neurodegeneration, but a specific AD-related event. This enzyme has great potential as a target for AD therapy as increasing its expression in AD patients may facilitate amyloid clearance. It is unknown how ECE2 degrades amyloid beta and whether or not it acts in concert with neprilysin, another major amyloid clearing peptidase [34]. Furthermore, if ECE is lost early in the disease, as its role in amyloid clearance suggests, it may be useful as a biomarker for AD, identifying patients who may benefit from treatment with compounds such as somatostatins to increase neprilysin levels or with artificially reconstituted levels of neprilysin and ECE2. Importantly, as pointed out by Eckman and coworkers, as ECE inhibitors are in preclinical development for use in the treatment of hypertension, there may be a risk that these drugs could increase amyloid deposits in the brain, leading to AD-like symptoms [34]. More detailed studies will indicate the likelihood of these setbacks and the potential utility of ECE as a marker and potential therapeutic target for AD.
Experimental procedures
Brain tissue
Human inferior parietal lobe (IPL) sections used in the study were obtained from Sun Health Research Institute Brain Donation Program (Sun City, AZ), with postmortem intervals (PMI) less than 3 h. IPLs from 6 Alzheimer's disease (AD) (age range 73–95 years), 6 demented with no AD (demented; age range 55–83 years) and 6 non-demented, age-matched control (control; age range 74–91 years) donors were utilized in this study. There were no significant age-associated differences between control vs. AD subjects (p=0.69) or control vs. demented subjects (p=0.07) (Table 1). AD tissue was obtained from individuals with Braak scores of 5 or greater, indicating maximal neurofibrillar and amyloid plaque deposition within the CNS [35,36]. On the other hand, demented and age-matched control brain tissues exhibited Braak scores of 3 or less.
Total RNA isolation
IPLs were frozen by sandwiching between sheets of dry ice to prevent ice-crystal artifact and preserve RNA quality. Total RNA was isolated using phenol-guanidinium thiocyanate reagent (Trizol, Invitrogen, Carlsbad, CA). Briefly, the gray matter from chunks of IPLs was dissected and homogenized in Trizol, incubated at room temperature in the presence of chloroform and centrifuged at maximum speed for 15 min at 4°C. The aqueous phase was removed, and an equal volume of 70% ethanol was added to the sample. Samples were then applied to Rneasy columns and purified according to manufacturer's protocols (Qiagen, Valencia, CA). RNA quality and quantity were analyzed using the Agilent Bioanalyzer platform (Agilent Technologies, Foster City, CA).
Agilent 44k microarray and data analysis
RNA was labeled and hybridized to the Agilent Human 44K oligo microarray kit according to manufacturer's protocols. Briefly, 500 ng of RNA was incubated with T7 promoter and reverse transcribed. Fluorescent cRNA was then generated and amplified by adding either Cy-3 or Cy-5 labeled dUTP to the cDNA and transcribing using T7 RNA polymerase. Experiments were set up so that each experimental condition was labeled with Cy-5 and control samples were labeled with Cy-3. Amplified, labeled RNA was then purified using Qiagen RNeasy mini columns, and the samples were quantitated using a NanoDrop spectrophotometer (Nano-Drop Technologies, Wilmington, DE). 750 ng of Cy-5 labeled probe and 750 ng of Cy-3 labeled control were combined with control targets and hybridization buffer and applied to the microarray slides. Slides were hybridized in a rotating chamber overnight at 60°C, washed the next day in 6× SSC, 0.005% Triton X-102 for 10 min and then washed in 0.1× SSC, 0.005% Triton X-102 for 5 min on ice. Slides were dried using a nitrogen-filled air gun and scanned using an Agilent scanner. Images were analyzed using the Agilent Feature Extractor Software, Version A.7.5.1, and the ratios for each spot were calculated. Data were normalized and preprocessed based on Z-score calculations. Gene selection among classes was performed, based on linear discriminant analysis, as previously described [37]. To evaluate the probability of a given gene achieving a significant weight by chance, a random permutation test using 1000 permutations was used to generate an empirical significance threshold for comparison with the calculated p value for each gene. Significant genes (weight greater than 2, p value<0.02) were considered for further validation and analysis.
Real-time SYBR-green PCR (RT-SYBR-PCR)
Genes of interest from microarray results were sequence-verified, and gene-specific primers (GSPs) were constructed using Primer Express software (Applied Biosystems, Foster City, CA). Ten micrograms of total RNA was reverse transcribed into cDNA as for microarray analysis, except that unlabeled dCTP was used instead of radiolabeled dCTP. One microliter of cDNA was then used in RT-SYBR-PCR reactions. The reaction mixture consisted of cDNA, 300 nM forward and reverse primers, and SYBR-green master mix PCR solution (Applied Biosystems, Foster City, CA). Amplification conditions were 2 min at 50°C followed by 10 min of initial denaturation at 95°C, and 40 cycles of denaturation of 15 s at 95°C and extension for 1 min at 60°C. cDNA from all samples was amplified on an ABI Prism 7700 Sequence Detector (Applied Biosystems. Foster City, CA) using GSPs or 18S in 96-well plates. Cycle thresholds obtained from AD, demented non-AD and control samples were averaged and normalized to the 18S control and subjected to a power analysis. The resulting values from AD and demented non-AD samples were then compared to control values resulting in fold-changes, as determined by the 2−ΔΔCT method as previously described [38]. All samples were run at least three times in duplicate. Primer sequences are shown in Supplementary Table 1.
Immunohistochemistry
IPL sections from the contralateral hemispheres of the same cases were previously frozen. Slices of these tissues were then immersion-fixed in 4% formaldehyde solution overnight at 4°C, after which they were paraffin-embedded and sliced into 5 μM thick sections. Antigen retrieval was performed by sequentially incubating the sections at room temperature for 3 min in each of the following: xylene, xylene, xylene, 100% ethanol, 100% ethanol, 95% ethanol, 85% ethanol and 70% ethanol. Sections were then steamed for 30 min in a sodium citrate buffer (DAKO target retrieval solution, DAKO Corp, Carpinteria, CA). Blocking was performed at room temperature for 30 min with a peroxidase block (Labvision, Fremont, CA) and for 5 min with a biotin block (Labvision, Fremont, CA). Tissues were stained overnight at 4°C with the antibodies as follows: CCR3, CXCR4 (both purchased from Axxora, San Diego, CA) 1:100, and ECE2 (R&D Systems, Minneapolis, MN) 1:100. Secondary staining was performed using the Labvision ABC system according to the manufacturer's protocol (Labvision, Fremont, CA) and using the DAB chromagen as a final step. Slides were counterstained using hematoxylin then dehydrated in a reverse ethanol/xylene series and cover-slipped using toluene based reagent. Slides were examined using a Zeiss (Carl Zeiss, Inc., Thornwood, NY) Axiovert-100 inverted microscope equipped with a 12-bit CCD camera system (Diagnostic Instruments, Inc., Sterling Heights, MI) connected to a Dell PC (Dell Computer Corp., USA).
Supplementary Material
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.yexcr.2006.10.028.
Acknowledgments
We are grateful to Ms. Angie Feehley for her assistance in the preparation of this document. We are grateful to the Sun Health Research Institute Brain Donation Program of Sun City, Arizona for the provision of human brain tissue. The Brain Donation Program is supported by the National Institute on Aging (P30 AG19610) Arizona Alzheimer's Disease Core Center, the Arizona Department of Health Services (contract 211002, Arizona Alzheimer's Research Center) and the Arizona Biomedical Research Commission (contract 0011, Arizona Parkinson's Disease Center). This work was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Baltimore, MD.
References
- 1.Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88:1337–1342. doi: 10.2105/ajph.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loring JF, Wen X, Lee JM, Seilhamer J, Somogyi R. A gene expression profile of Alzheimer's disease. DNA Cell Biol. 2001;20:683–695. doi: 10.1089/10445490152717541. [DOI] [PubMed] [Google Scholar]
- 3.Ho L, Guo Y, Spielman L, Petrescu O, Haroutunian V, Purohit D, Czernik A, Yemul S, Aisen PS, Mohs R, Pasinetti GM. Altered expression of a-type but not b-type synapsin isoform in the brain of patients at high risk for Alzheimer's disease assessed by DNA microarray technique. Neurosci Lett. 2001;298:191–194. doi: 10.1016/s0304-3940(00)01753-5. [DOI] [PubMed] [Google Scholar]
- 4.Hata R, Masumura M, Akatsu H, Li F, Fujita H, Nagai Y, Yamamoto T, Okada H, Kosaka K, Sakanaka M, Sawada T. Up-regulation of calcineurin Abeta mRNA in the Alzheimer's disease brain: assessment by cDNA microarray. Biochem Biophys Res Commun. 2001;284:310–316. doi: 10.1006/bbrc.2001.4968. [DOI] [PubMed] [Google Scholar]
- 5.Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res. 2002;70:462–473. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- 6.Huang MB, Bond VC. Involvement of protein kinase C in HIV-1 gp120-induced apoptosis in primary endothelium. J Acquired Immune Defic Syndr. 2000;25:375–389. doi: 10.1097/00042560-200012150-00001. [DOI] [PubMed] [Google Scholar]
- 7.Xia MQ, Hyman BT. Chemokines/chemokine receptors in the central nervous system and Alzheimer's disease. J NeuroVirol. 1999;5:32–41. doi: 10.3109/13550289909029743. [DOI] [PubMed] [Google Scholar]
- 8.Badaut J, Lasbennes F, Magistretti PJ, Regli L. Aquaporins in brain: distribution, physiology, and pathophysiology. J Cereb Blood Flow Metab. 2002;22:367–378. doi: 10.1097/00004647-200204000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Arelin K, Kinoshita A, Whelan CM, Irizarry MC, Rebeck GW, Strickland DK, Hyman BT. LRP and senile plaques in Alzheimer's disease: colocalization with apolipoprotein E and with activated astrocytes. Brain Res Mol Brain Res. 2002;104:38–46. doi: 10.1016/s0169-328x(02)00203-6. [DOI] [PubMed] [Google Scholar]
- 10.Baum L, Chen L, Ng HK, Chan YS, Mak YT, Woo J, Chiu HF, Pang CP. Low density lipoprotein receptor related protein gene exon 3 polymorphism association with Alzheimer's disease in Chinese. Neurosci Lett. 1998;247:33–36. doi: 10.1016/s0304-3940(98)00294-8. [DOI] [PubMed] [Google Scholar]
- 11.Beffert U, Arguin C, Poirier J. The polymorphism in exon 3 of the low density lipoprotein receptor-related protein gene is weakly associated with Alzheimer's disease. Neurosci Lett. 1999;259:29–32. doi: 10.1016/s0304-3940(98)00888-x. [DOI] [PubMed] [Google Scholar]
- 12.Sanchez L, Alvarez V, Gonzalez P, Gonzalez I, Alvarez R, Coto E. Variation in the LRP-associated protein gene (LRPAP1) is associated with late-onset Alzheimer disease. Am J Med Genet. 2001;105:76–78. [PubMed] [Google Scholar]
- 13.Zerbinatti CV, Bu G. LRP and Alzheimer's disease. Rev Neurosci. 2005;16:123–135. doi: 10.1515/revneuro.2005.16.2.123. [DOI] [PubMed] [Google Scholar]
- 14.Kang DE, Pietrzik CU, Baum L, Chevallier N, Merriam DE, Kounnas MZ, Wagner SL, Troncoso JC, Kawas CH, Katzman R, Koo EH. Modulation of amyloid beta-protein clearance and Alzheimer's disease susceptibility by the LDL receptor-related protein pathway. J Clin Invest. 2000;106:1159–1166. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu Z, Strickland DK, Hyman BT, Rebeck GW. Elevation of LDL receptor-related protein levels via ligand interactions in Alzheimer disease and in vitro. J Neuropathol Exp Neurol. 2001;60:430–440. doi: 10.1093/jnen/60.5.430. [DOI] [PubMed] [Google Scholar]
- 16.Zerbinatti CV, Wozniak DF, Cirrito J, Cam JA, Osaka H, Bales KR, Zhuo M, Paul SM, Holtzman DM, Bu G. Increased soluble amyloid-beta peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. Proc Natl Acad Sci U S A. 2004;101:1075–1080. doi: 10.1073/pnas.0305803101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gregoire S, Yang XJ. Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol Cell Biol. 2005;25:2273–2287. doi: 10.1128/MCB.25.6.2273-2287.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Wang H, Wang S, Quon D, Liu YW, Cordell B. Positive and negative regulation of APP amyloidogenesis by sumoylation. Proc Natl Acad Sci U S A. 2003;100:259–264. doi: 10.1073/pnas.0235361100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer's amyloid beta peptide by endothelin-converting enzyme. J Biol Chem. 2001;276:24540–24548. doi: 10.1074/jbc.M007579200. [DOI] [PubMed] [Google Scholar]
- 20.Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB. Alzheimer's disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem. 2003;278:2081–2084. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- 21.Albright AV, Shieh JT, Itoh T, Lee B, Pleasure D, O'Connor MJ, Doms RW, Gonzalez-Scarano F. Microglia express CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human immunodeficiency virus type 1 dementia isolates. J Virol. 1999;73:205–213. doi: 10.1128/jvi.73.1.205-213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosi S, Pert CB, Ruff MR, McGann-Gramling K, Wenk GL. Chemokine receptor 5 antagonist D-Ala-peptide T-amide reduces microglia and astrocyte activation within the hippocampus in a neuroinflammatory rat model of Alzheimer's disease. Neuroscience. 2005;134:671–676. doi: 10.1016/j.neuroscience.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 23.Homey B, Wang W, Soto H, Buchanan ME, Wiesenborn A, Catron D, Muller A, McClanahan TK, Dieu-Nosjean MC, Orozco R, Ruzicka T, Lehmann P, Oldham E, Zlotnik A. Cutting edge: the orphan chemokine receptor G protein-coupled receptor-2 (GPR-2, CCR10) binds the skin-associated chemokine CCL27 (CTACK/ALP/ILC) J Immunol. 2000;164:3465–3470. doi: 10.4049/jimmunol.164.7.3465. [DOI] [PubMed] [Google Scholar]
- 24.Campbell JJ, Butcher EC. Chemokines in tissue-specific and microenvironment-specific lymphocyte homing. Curr Opin Immunol. 2000;12:336–341. doi: 10.1016/s0952-7915(00)00096-0. [DOI] [PubMed] [Google Scholar]
- 25.Homey B, Alenius H, Muller A, Soto H, Bowman EP, Yuan W, McEvoy L, Lauerma AI, Assmann T, Bunemann E, Lehto M, Wolff H, Yen D, Marxhausen H, To W, Sedgwick J, Ruzicka T, Lehmann P, Zlotnik A. CCL27–CCR10 interactions regulate T cell-mediated skin inflammation. Nat Med. 2002;8:157–165. doi: 10.1038/nm0202-157. [DOI] [PubMed] [Google Scholar]
- 26.Reiss Y, Proudfoot AE, Power CA, Campbell JJ, Butcher EC. CC chemokine receptor (CCR)4 and the CCR10 ligand cutaneous T cell-attracting chemokine (CTACK) in lymphocyte trafficking to inflamed skin. J Exp Med. 2001;194:1541–1547. doi: 10.1084/jem.194.10.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vestergaard C, Deleuran M, Gesser B, Gronhoj Larsen C. Expression of the T-helper 2-specific chemokine receptor CCR4 on CCR10-positive lymphocytes in atopic dermatitis skin but not in psoriasis skin. Br J Dermatol. 2003;149:457–463. doi: 10.1046/j.1365-2133.2003.05505.x. [DOI] [PubMed] [Google Scholar]
- 28.Moed H, Boorsma DM, Tensen CP, Flier J, Jonker MJ, Stoof TJ, von Blomberg BM, Bruynzeel DP, Scheper RJ, Rustemeyer T, Gibbs S. Increased CCL27–CCR10 expression in allergic contact dermatitis: implications for local skin memory. J Pathol. 2004;204:39–46. doi: 10.1002/path.1619. [DOI] [PubMed] [Google Scholar]
- 29.Melchjorsen J, Siren J, Julkunen I, Paludan SR, Matikainen S. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-kappaB and IRF-3. J Gen Virol. 2006;87:1099–1108. doi: 10.1099/vir.0.81541-0. [DOI] [PubMed] [Google Scholar]
- 30.Dobson CB, Itzhaki RF. Herpes simplex virus type 1 and Alzheimer's disease. Neurobiol Aging. 1999;20:457–465. doi: 10.1016/s0197-4580(99)00055-x. [DOI] [PubMed] [Google Scholar]
- 31.Deatly AM, Haase AT, Fewster PH, Lewis E, Ball MJ. Human herpes virus infections and Alzheimer's disease. Neuropathol Appl Neurobiol. 1990;16:213–223. doi: 10.1111/j.1365-2990.1990.tb01158.x. [DOI] [PubMed] [Google Scholar]
- 32.Mattson MP. Infectious agents and age-related neurodegenerative disorders. Ageing Res Rev. 2004;3:105–120. doi: 10.1016/j.arr.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ulery PG, Strickland DK. LRP in Alzheimer's disease: friend or foe? J Clin Invest. 2000;106:1077–1079. doi: 10.1172/JCI11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eckman EA, Eckman CB. Abeta-degrading enzymes: modulators of Alzheimer's disease pathogenesis and targets for therapeutic intervention. Biochem Soc Trans. 2005;33:1101–1105. doi: 10.1042/BST20051101. [DOI] [PubMed] [Google Scholar]
- 35.Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 36.Braak H, Braak E. Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol (Berl) 1996;92:197–201. doi: 10.1007/s004010050508. [DOI] [PubMed] [Google Scholar]
- 37.Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, Sampas N, Dougherty E, Wang E, Marincola F, Gooden C, Lueders J, Glatfelter A, Pollock P, Carpten J, Gillanders E, Leja D, Dietrich K, Beaudry C, Berens M, Alberts D, Sondak V. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- 38.Weeraratna AT, Becker D, Carr KM, Duray PH, Rosenblatt KP, Yang S, Chen Y, Bittner M, Strausberg RL, Riggins GJ, Wagner U, Kallioniemi OP, Trent JM, Morin PJ, Meltzer PS. Generation and analysis of melanoma SAGE libraries: SAGE advice on the melanoma transcriptome. Oncogene. 2004;23:2264–2274. doi: 10.1038/sj.onc.1207337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.yexcr.2006.10.028.