

Abstract
Patients with Eisenmenger syndrome experience substantial morbidity and decreased survival rates. In advanced cases, lung transplantation with cardiac repair or heart–lung transplantation is often the only option. The efficacy of endothelin receptor antagonists in Eisenmenger syndrome, which has similar pathophysiology to idiopathic pulmonary hypertension, remains unknown.
We retrospectively studied adults with congenital heart disease and Eisenmenger syndrome who were treated with endothelin receptor antagonists. Analysis included chart reviews of clinical evaluations, oxygen saturation levels, functional class, 6-minute walk distances, and pulmonary artery pressures. In the 24 patients studied, Eisenmenger syndrome was caused by ventricular septal defect (6 patients), atrial septal defect (5), atrioventricular canal defect (3), complex congenital heart disease (9), and patent ductus arteriosus (1).
Eisenmenger syndrome was treated with bosentan (21 patients) and sitaxsentan (3 patients). On average, therapy lasted 19 ± 12 months. Subsequently, mean 6-minute walk distances improved from 226 ± 159 m to 351 ± 113 m (P = 0.004), and World Health Organization functional class improved ≥1 grade (P < 0.0001). Oxygen saturations increased on therapy from 80.5% to 87% (P < 0.0001). Pulmonary arterial systolic pressures decreased from 97 ± 21 mmHg to 78 ± 27 mmHg, and mean pressures from 59 ± 16 mmHg to 47 ± 17 mmHg (both P < 0.0001). Neither major complications from therapy nor changes in pulmonary capillary wedge pressure occurred.
Endothelin receptor antagonists may play an important role in improving 6-minute walk distance, oxygen saturation, pulmonary artery pressures, and symptoms in adults who have congenital heart defects and Eisenmenger syndrome.
Key words: Antihypertensive agents/administration & dosage/therapeutic use; bosentan; Eisenmenger complex/complications/drug therapy/mortality/physiopathology; endothelin-1/antagonists & inhibitors; exercise tolerance/drug effects; heart defects/congenital; hypertension, pulmonary/classification/complications/drug therapy/physiopathology; oximetry; patient selection; receptors, endothelin/antagonists & inhibitors/physiology; treatment outcome
In patients who have congenital heart defects, chronic left-to-right shunting may cause Eisenmenger syndrome, which is characterized by increased pulmonary vascular resistance and pulmonary artery hypertension. The congenital heart defects include systemic-to-pulmonary arterial communication as is seen in atrial septal defects, ventricular septal defects, patent ductus arteriosus, transposition of the great vessels, and surgical creation of aortopulmonary shunts. The likelihood of developing Eisenmenger syndrome depends on the size and location of the defect.1
Patients with Eisenmenger syndrome have a better life expectancy than patients with familial or idiopathic pulmonary hypertension who have similar hemodynamics, but life expectancy is still decreased.2 Eisenmenger syndrome is also associated with substantial long-term morbidity. Previously, the only treatment that could be offered to patients with advanced symptoms was lung transplantation with cardiac repair or heart–lung transplantation, but long-term survival was limited.3 Other management options include oxygen therapy when appropriate, avoidance of high-risk situations including pregnancy, caution when contemplating noncardiac surgery, and avoiding volume depletion.4,5
The use of pulmonary vasodilators in patients with Eisenmenger syndrome is conceptually appealing, but only sparse data confirm a benefit of this approach. Novel therapeutic agents that have emerged for the treatment of pulmonary hypertension are prostacyclin analogs, endothelin receptor antagonists, nitric oxide, and sildenafil.6-9 Although these agents have been used in patients who have idiopathic and familial pulmonary artery hypertension, data are limited regarding their benefit in patients who have Eisenmenger syndrome. Endothelin-1 (ET-1) is implicated in the pathogenesis of pulmonary hypertension.10-12 Histologic studies of the pulmonary arteries in patients who have Eisenmenger syndrome suggest that endothelin receptors are up-regulated in this disease.13
Bosentan is an oral endothelin-1A/1B receptor (ET-1A and ET-1B) antagonist that is approved for the treatment of idiopathic and secondary pulmonary hypertension.14 Selective blockers of the ET-1A receptors are also being investigated for the treatment of pulmonary artery hypertension.15,16 A recently reported randomized controlled trial (BREATHE-5) showed an improvement in 6-minute walk distance, with no harmful effects, in patients with Eisenmenger syndrome and simple intracardiac shunts (atrial or ventricular septal defects) who were treated with bosentan.17 An observational, prospective study from Germany showed in 2005 that bosentan was well tolerated and improved the functional classification in adult patients with congenital heart disease (CHD).18 No data are available on the use of endothelin receptor antagonists in patients who have more complex congenital defects or extracardiac shunts.
Herein, we describe the effect of the nonselective ET-1 receptor antagonist, bosentan, on symptoms, objective measures of exercise, oxygen saturations, and pulmonary artery pressures in adult patients with CHD and pulmonary hypertension.
Patients and Methods
We reviewed the charts of consecutive, symptomatic adult patients with a diagnosis of CHD and Eisenmenger syndrome who had been treated with ET-1 antagonists. For the purposes of this study, Eisenmenger syndrome was defined as an intracardiac or extracardiac systemic-to-pulmonary communication with reversal of the left-to-right shunt, resulting from severe pulmonary hypertension. Patients who had undergone late closure of an intracardiac communication with pulmonary hypertension were included in the study. All patients were monitored by cardiologists who were experienced in the care of adults with CHD. The study was approved by our institutional review board.
Twenty-five patients with Eisenmenger syndrome were treated with ET-1 antagonists (21 with bosentan and 4 with sitaxsentan). One patient treated with sitaxsentan was removed from the study because of disruption of therapy for > 1 month. Table I lists the baseline characteristics of the 24 study patients (10 men, 14 women), and Table II shows the causes of their Eisenmenger syndrome.
Table I. Baseline Characteristics of the Patients at Start of ET-1 Receptor Antagonist Therapy

Table II. Oxygen Saturation Levels, 6MWT Distances, and WHO Functional Classes in the Patients Before and After Endothelin-1 Receptor Antagonist Therapy
Twenty-one patients began taking 62.5 mg of oral bosentan twice daily. The dosage was increased to 125 mg twice daily after 4 weeks. All patients took the maximum dose of 125 mg twice daily, except for 1 patient of < 50 kg body weight who was maintained on 62.5 mg twice daily. Three patients who were enrolled in an open-label study of sitaxsentan began taking 50 mg daily for 4 weeks and then 100 mg per day. Liver-function tests were monitored monthly in all patients. Comprehensive clinical follow-up was performed at least every 3 months, and room-air oxygen saturations, 6-minute walk test (6MWT) distances, and World Health Organization functional classes (WHO FC) were recorded. Oxygen saturations were obtained by use of a finger pulse oximeter (Nonin Medical, Inc.; Plymouth, Minn) at rest and during the 6MWT. The 6MWTs were performed by an experienced practitioner on a level, straight, 250-foot hallway that was marked in feet. Patients on home oxygen walked with oxygen. For purposes of analysis, resting oxygen saturations were used. Echocardiography was performed at 6-month intervals by the same ultrasonographer, who used standard methods. Preliminary symptomatic improvement in 9 of these patients on bosentan has been previously reported.19
Data Collection and Analysis. Medical records, including electrocardiograms and echocardiograms, were reviewed for all patients. The type of congenital heart defect, medication history, surgical history, need for oxygen, baseline hemoglobin level, liver function tests, side effects from medication, pulmonary artery pressures (by Doppler echocardiography), pre- and post-treatment 6MWT distances, and pre- and post-treatment oxygen saturations were examined approximately 6 months after the start of therapy.
Statistical Analysis. Data are expressed as mean ± SD or as median and range, where appropriate. The paired 2-sample t test was used to compare oxygen saturations and 6MWT distances before and after therapy. The Wilcoxon rank-sum test was used to compare variables of WHO FC. Significance was determined at P < 0.05. The statistical analysis was performed with STATA® 6.0 (StataCorp LP; College Station, Tex).
Results
The average duration of therapy was 19 ± 12 months, and the average duration of follow-up was 22 months (range, 3–48 mo). Data collection was complete for 100% of oxygen saturations and functional class, for 58% of the pre- and post-treatment 6MWTs (14/24), and for 71% of the pulmonary artery pressures (17/24).
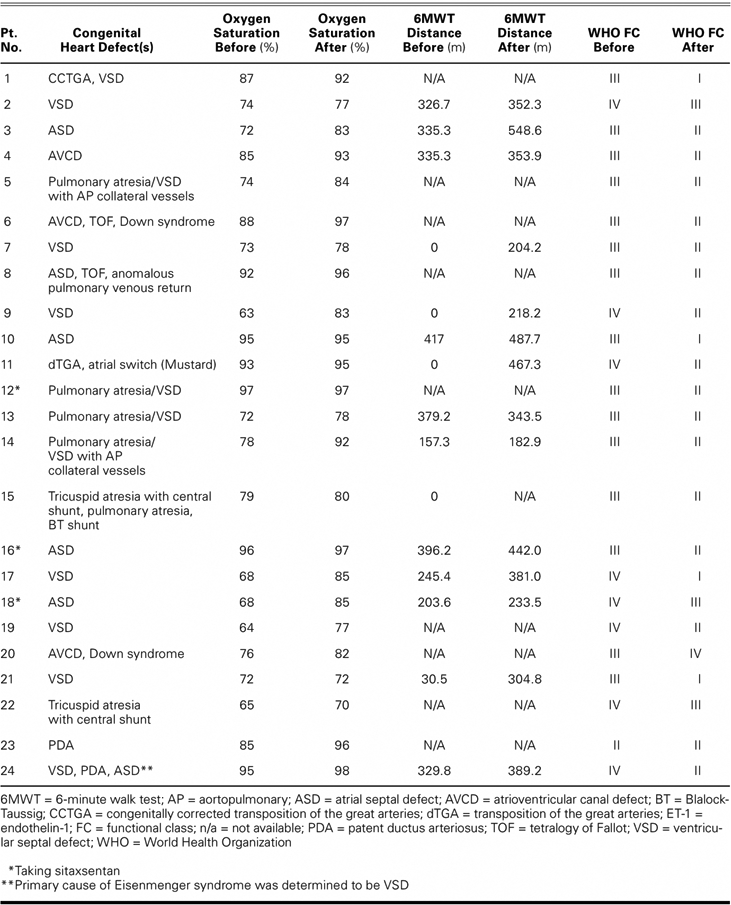
After treatment with ET-1 antagonists, 22 of the 24 patients showed improvement in WHO FC of ≥1 grade (P < 0.0001). Fourteen patients improved by 1 class and 8 patients improved by ≥2 classes (Table II; Fig. 1). The 6MWT distances improved on therapy from a mean of 226 ± 159 m to 351 ± 113 m (P < 0.004) (Fig. 2). Three patients were assigned a 0-ft walk distance before therapy because of their extremely limited functional capacity (wheelchair-bound, WHO FC IV) and an inability to walk. These 3 patients showed a dramatic improvement in their 6MWT distances (204 m, 218 m, and 467 m). With these 3 patients removed from the analysis, 6MWT distance improved a mean of 78 m (P < 0.019). Oxygen saturation improved on therapy from a mean of 80% to 87% (P < 0.0001) (Fig. 3). On therapy, pulmonary artery systolic pressures decreased from 97 ± 21 mmHg to 78 ± 27 mmHg, and mean pressures decreased from 59 ± 16 mmHg to 47 ± 17 mmHg (both P < 0.0001). Therapy engendered no significant change in pulmonary capillary wedge pressure (Table III).
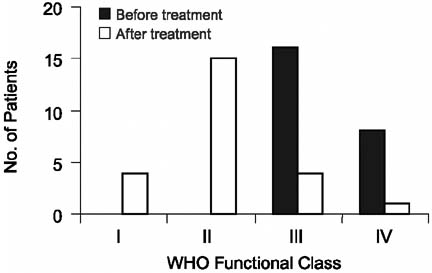
Fig. 1 World Health Organization (WHO) functional class in the patients before and after endothelin-1 receptor antagonist therapy. The mean functional class changed from class III to class II after therapy. Fourteen patients improved by 1 class, 7 patients improved by 2 classes, 1 patient improved by 3 classes, 1 patient experienced no change, and 1 patient's condition worsened by 1 class.

Fig. 2 Improvement in exercise tolerance, as measured by 6-minute walk distance. After treatment with endothelin-1 antagonists, distance improved from a mean of 226 ± 159 meters to 351 ± 113 meters (n = 14; P < 0.004). When the 3 patients who had been assigned a walk distance of 0 feet before therapy were omitted from the analysis, the improvement in distance was significant (n = 11; P < 0.019).
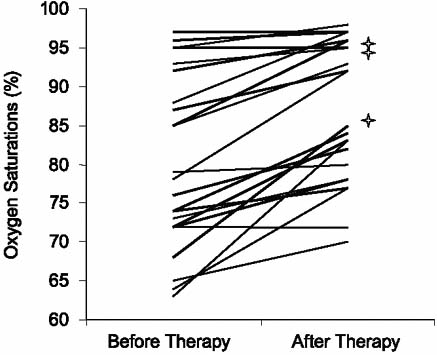
Fig. 3 Improvement in oxygen saturations after endothelin-1 receptor antagonist therapy. The asterisks denote the 3 patients who took sitaxsentan.
Table III. Response to Endothelin-1 Receptor Antagonists

Discussion
Although patients with Eisenmenger syndrome have a better life expectancy than patients with idiopathic pulmonary hypertension (despite the similar pathophysiology),2 progressive complications of exercise intolerance and heart failure occur. Transplantation may not convey clear survival benefits, because patients with Eisenmenger syndrome often survive to the 4th or 5th decade of life without transplantation. An effective and well-tolerated oral therapy would prove more convenient and tolerable than intravenous prostacyclin treatment for patients with Eisenmenger syndrome, who are likely to require long-term therapy. Epoprostenol has been found to have a role in the treatment of patients with pulmonary hypertension that is associated with CHD.13 However, epoprostenol use is complicated by infections, pump malfunctions, and the inconvenience of continuous intravenous delivery.13 The present study suggests that ET-1 receptor blockers can function as a preferable alternative.
We have shown objective evidence of improved exercise tolerance by 6MWT distances, improved oxygen saturations, and improved pulmonary artery pressures even in patients with complex cyanotic CHD and Eisenmenger syndrome. Not all patients with Eisenmenger syndrome require treatment. Our criteria for treating patients were WHO FC III or IV symptoms with objective evidence of exercise intolerance or the development of sequelae related to secondary erythrocytosis. Our patients had lower baseline oxygen saturations in comparison with the patients who were enrolled in BREATHE-5, in whom no change in oxygen saturation was found. In our study, the substantial increase in oximetry at rest with ET-1 antagonists may be related to reduced right-to-left shunting and augmented pulmonary blood flow that was caused by a reduction in pulmonary vascular resistance. Due to the relatively slow progression of pulmonary vascular disease in Eisenmenger syndrome, early therapy is unlikely to be beneficial and may increase left-to-right shunting and systemic ventricular load.
The improvement that we describe was similar to that described in previous trials of bosentan and sitaxsentan in subjects who had idiopathic pulmonary artery hypertension.15,16,20,21 There resulted significant improvements in 6MWT distance, WHO FC, pulmonary artery pressure, cardiac output, and pulmonary vascular resistance. Although some preliminary evidence indicates sustained efficacy with 12 months of therapy, the long-term effects of bosentan and sitaxsentan warrant further evaluation.22 Previous studies have also documented a significant improvement in resting oxygen saturations (up to 3%) and a decrease in mean pulmonary artery pressure.23,24
Of note, therapy was not equally beneficial. Of the 24 patients, 2 were listed for or underwent heart–lung transplantation, and 1 additional patient died due to progressive disease while on therapy. Eight patients required add-on therapy for symptom recurrence a mean of 18 ± 11 months after starting an ET-1 receptor antagonist; add-on therapy included sildenafil (6 patients) and treprostinil (2 patients). Adverse reactions secondary to ET-1 receptor antagonists are well known and include hepatotoxicity, anemia, pruritus, and edema.21 In our study, the side effects of therapy were minor; 2 patients had changes in their liver-function tests that necessitated the temporary disruption of therapy, although both resumed treatment without incident. There were no significant changes in hematocrit levels after the start of therapy. A possibility exists that a vasodilator could disturb the fine balance between pulmonic and systemic circulations; however, this did not manifest itself in worsening functional class, an increase in pulmonary capillary wedge pressure, or a decline in oxygen saturations.
Limitations
Our study has several limitations, including its retrospective nature, a small group of heterogeneous patients, and the lack of a placebo group. Our study considered surrogate markers of disease severity, including 6MWTs, oxygen saturations, and WHO FC. Post-therapy hemodynamics were evaluated not via cardiac catheterization but by echocardiography, as in the BREATHE-5 trial. We did not evaluate quality of life formally by use of any survey or questionnaire, and we did not look at hospital admissions or improvement in survival. However, our study is unique as the longest series of patients with Eisenmenger syndrome who were being treated with endothelin-receptor blockers, and it adds to the medical literature regarding the long-term outcome and safety profile of these drugs in patients with complex cyanotic CHD.
Conclusion
Eisenmenger syndrome encompasses a unique group of patients for whom there is no established therapy other than combined heart–lung transplantation. Our study shows that endothelin receptor blockers improve exercise tolerance, oxygenation, and functional status in a heterogeneous group of adults with Eisenmenger syndrome, without adverse effects.
Footnotes
Address for reprints: Wendy M. Book, MD, Adult Congenital Cardiac Program, 1365 Clifton Road NE, Suite A2445, Atlanta, GA 30322
E-mail: wendy_book@emoryhealthcare.org
References
- 1.Vongpatanasin W, Brickner ME, Hillis LD, Lange RA. The Eisenmenger syndrome in adults. Ann Intern Med 1998;128 (9):745–55. [DOI] [PubMed]
- 2.Granton JT, Rabinovitch M. Pulmonary arterial hypertension in congenital heart disease. Cardiol Clin 2002;20(3):441–57, vii. [DOI] [PubMed]
- 3.Curran WD, Akindipe O, Staples ED, Baz MA. Lung transplantation for primary pulmonary hypertension and Eisenmenger's syndrome. J Cardiovasc Nurs 2005;20(2):124–32. [DOI] [PubMed]
- 4.Bowyer JJ, Busst CM, Denison DM, Shinebourne EA. Effect of long term oxygen treatment at home in children with pulmonary vascular disease. Br Heart J 1986;55(4):385–90. [DOI] [PMC free article] [PubMed]
- 5.Sandoval J, Aguirre JS, Pulido T, Martinez-Guerra ML, Santos E, Alvarado P, et al. Nocturnal oxygen therapy in patients with the Eisenmenger syndrome. Am J Respir Crit Care Med 2001;164(9):1682–7. [DOI] [PubMed]
- 6.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004;351(14): 1425–36. [DOI] [PubMed]
- 7.Mehta S. Drug therapy for pulmonary arterial hypertension: what's on the menu today? Chest 2003;124(6):2045–9. [DOI] [PubMed]
- 8.Lacassie HJ, Germain AM, Valdes G, Fernandez MS, Allamand F, Lopez H. Management of Eisenmenger syndrome in pregnancy with sildenafil and L-arginine. Obstet Gynecol 2004;103(5 Pt 2):1118–20. [DOI] [PubMed]
- 9.Michelakis ED, Tymchak W, Noga M, Webster L, Wu XC, Lien D, et al. Long-term treatment with oral sildenafil is safe and improves functional capacity and hemodynamics in patients with pulmonary arterial hypertension. Circulation 2003;108(17):2066–9. [DOI] [PubMed]
- 10.Benigni A, Remuzzi G. Endothelin antagonists. Lancet 1999; 353(9147):133–8. [DOI] [PubMed]
- 11.Schiffrin EL. State-of-the-Art lecture. Role of endothelin-1 in hypertension. Hypertension 1999;34(4 Pt 2):876–81. [DOI] [PubMed]
- 12.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988;332 (6163):411–5. [DOI] [PubMed]
- 13.Rosenzweig EB, Kerstein D, Barst RJ. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation 1999;99(14):1858–65. [DOI] [PubMed]
- 14.Luscher TF, Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation 2000;102(19):2434–40. [DOI] [PubMed]
- 15.Barst RJ, Langleben D, Frost A, Horn EM, Oudiz R, Shapiro S, et al.; STRIDE-1 Study Group. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med 2004;169(4):441–7. [DOI] [PubMed]
- 16.Barst RJ, Rich S, Widlitz A, Horn EM, McLaughlin V, McFarlin J. Clinical efficacy of sitaxsentan, an endothelin-A receptor antagonist, in patients with pulmonary arterial hypertension: open-label pilot study. Chest 2002;121(6):1860–8. [DOI] [PubMed]
- 17.Galie N, Beghetti M, Gatzoulis MA, Granton J, Berger RM, Lauer A, et al.; BREATHE-5 Investigators. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation 2006;114(1):48–54. [DOI] [PubMed]
- 18.Schulze-Neick I, Gilbert N, Ewert R, Witt C, Gruenig E, Enke B, et al. Adult patients with congenital heart disease and pulmonary arterial hypertension: first open prospective multicenter study of bosentan therapy. Am Heart J 2005;150 (4):716. [DOI] [PubMed]
- 19.Christensen DD, McConnell ME, Book WM, Mahle WT. Initial experience with bosentan therapy in patients with the Eisenmenger syndrome. Am J Cardiol 2004;94(2):261–3. [DOI] [PubMed]
- 20.Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet 2001; 358(9288):1119–23. [DOI] [PubMed]
- 21.Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension [published erratum appears in N Engl J Med 2002;346 (16):1258]. N Engl J Med 2002;346(12):896–903. [DOI] [PubMed]
- 22.Sitbon O, Badesch DB, Channick RN, Frost A, Robbins IM, Simonneau G, et al. Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary arterial hypertension: a 1-year follow-up study. Chest 2003;124(1):247–54. [DOI] [PubMed]
- 23.Apostolopoulou SC, Manginas A, Cokkinos DV, Rammos S. Effect of the oral endothelin antagonist bosentan on the clinical, exercise, and haemodynamic status of patients with pulmonary arterial hypertension related to congenital heart disease. Heart 2005;91(11):1447–52. [DOI] [PMC free article] [PubMed]
- 24.Gatzoulis MA, Rogers P, Li W, Harries C, Cramer D, Ward S, et al. Safety and tolerability of bosentan in adults with Eisenmenger physiology. Int J Cardiol 2005;98(1):147–51. [DOI] [PubMed]