

Abstract

The asymmetric synthesis of the oxindole alkaloid horsfiline is described. A palladium-catalyzed asymmetric allylic alkylation (AAA) is used to set the spiro(pyrrolidine-oxindole) stereogenic center.
(-)-Horsfiline was first isolated in 1991 from the leaves of the Horsfieldia superba plant by Bodo and co-workers.1 Biologically active alkaloids such as spirotryptostatin A and B, vincristine, and vinblastine have resulted in increased interest for oxindole natural products such as horsfiline. The unique spiro stereogenic center of horsfiline has challenged synthetic chemists to develop imaginative approaches towards its construction. As a result, there has been much synthetic effort towards horsfiline, resulting in several syntheses, three of which were asymmetric.2 In 1994, Borschberg confirmed the absolute configuration of horsfiline through the synthesis of both enantiomers via a diastereoselective oxidative rearrangement of a (L)-tryptophan derivative.2i In 1999, Fuji et al. used a chiral auxiliary for nitro-olefination of a substituted oxindole to set the stereochemistry at the quaternary chiral center.2h Palmisano and co-workers used an azomethine ylide cycloaddition reaction to form the pyrrolidine, and subsequently formed the oxindole by intramolecular cleavage of the chiral auxiliary.2f
We envisioned constructing the stereogenic quaternary carbon via a palladium-catalyzed asymmetric allylic alkylation (AAA) employing oxindole as the nucleophile. The use of 3-aryl oxindoles in palladium AAA has been previously reported,3 however oxindole nucleophiles with substituents other than aryl groups have not been employed in this chemistry. In our proposed synthesis of horsfiline, an ester or aldehyde substituent in the 3 position would provide a stabilized, compatible nucleophile for palladium allylic alkylation (Scheme 1). The carbonyl group also provides a handle for further manipulation. Oxidative cleavage of the allyl group followed by reductive amination would introduce the nitrogen of the pyrrolidine. Cyclization via reductive amination or SN2 substitution would complete the tricyclic system.
Scheme 1.
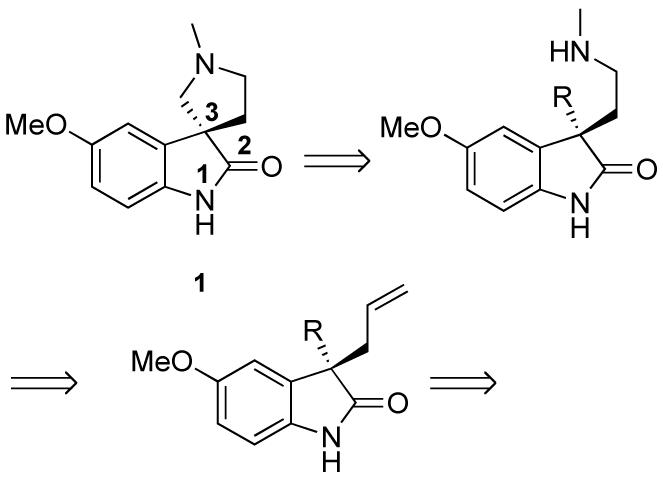
Retrosynthetic Analysis of Horsfiline Using Pd-AAA
Initially, an aldehyde was installed in the 3 position of the oxindole following a known procedure using sodium methoxide and ethyl formate.4 However, the product from the AAA reaction with allyl acetate was unstable to purification (Figure 1), resulting in deformylation.
Figure 1.
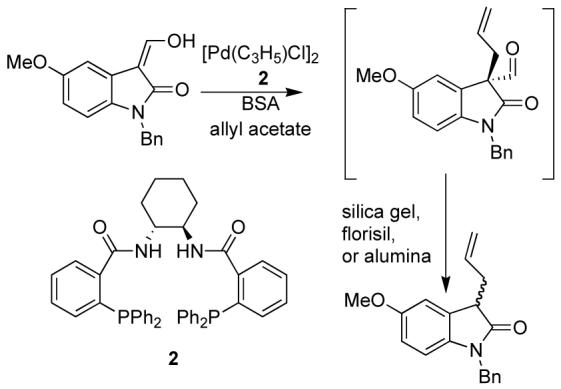
Deformylation of the initial Pd AAA Adduct
To avoid this decomposition, the aldehyde functionality was changed to an ester. A rhodium-catalyzed C-H insertion method developed by Padwa and co-workers was used to build the desired oxindole.5 Refluxing p-anisidine with 2,4-dimethoxybenzaldehyde in toluene followed by reduction of the imine with sodium borohydride in methanol resulted in quantitative yield of the desired amine 3 (Scheme 2). Acylation with the acid chloride derivative of ethyl diazoacetate in the presence of triethylamine, led to formation of amide 4 in 90% yield. Padwa discovered that Rh2(acac)4 led to β-lactam formation with diazoamides containing benzyl protecting groups, while the corresponding Rh2(CF3CONH2)4 catalyst led to oxindole formation. Therefore Rh C-H insertion of amide 4, followed by protection of the oxindole with TIPSOTf resulted in 79-86% yield of 5 over two steps varying with the catalyst load from 1 to 3 mol%. Protection of the oxindole was necessary to avoid hydrolysis.8
Scheme 2.
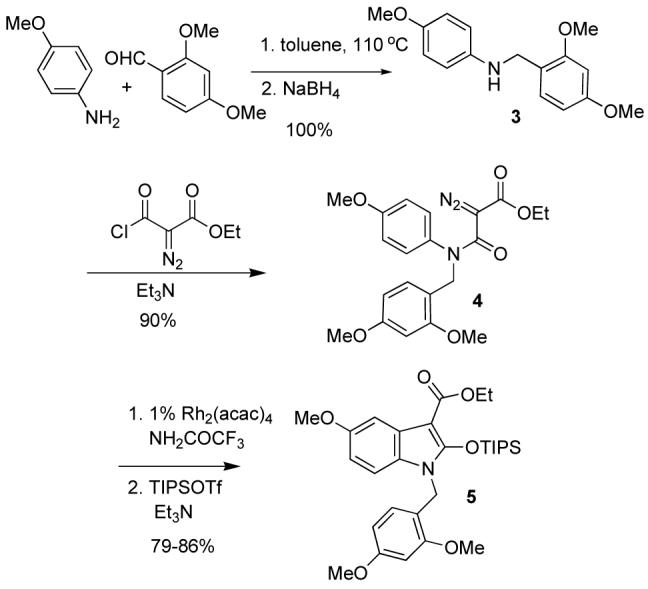
Synthesis of Oxindole Core
With the protected oxindole in hand, the next step was the asymmetric allylic alkylation. A fluoride source was added to generate the enolate anion nucleophile. Initially, CsF and ligand 2 were investigated with DME as the solvent, resulting in 77% ee. A significant improvement in ee occurred by switching the counterion from cesium to tetra-n-butylammonium. Various conditions were then screened for optimization (Table 1). Other Trost family ligands, such as 6 and 7, resulted in lower enantioselectivity.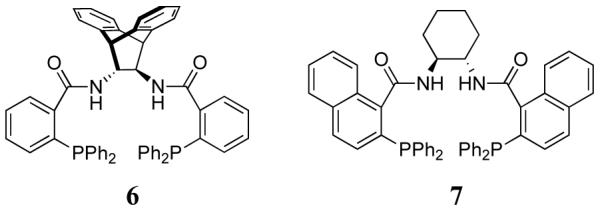
Table 1.
Pd AAA Reaction with oxindole 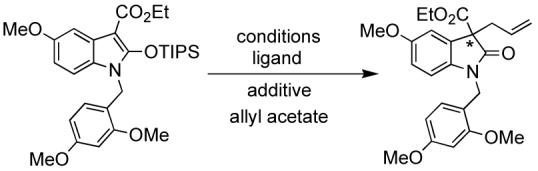
| Entry | Conditions | Ligand | Additive | ee(%)a |
|---|---|---|---|---|
| 1 | 2.5% [Pd(C2H5)Cl]2 DME, rt | 7.5% 2 | CsF | 77% |
| 2 | 2.5% [Pd(C2H5)Cl]2 DME, rt | 7.5% 6 | CsF | 21% |
| 3 | 2.5% [Pd(C2H5)Cl]2 DME, rt | 7.5% 7 | CsF | 62% |
| 4 | 2.5% [Pd(C2H5)Cl]2 DME, 0-4°C | 7.5% 2 | CsF | 74% |
| 5 | 2.5% [Pd(C2H5)Cl]2 DCM, rt | 7.5% 2 | CsF | 61% |
| 6 | 2.5% [Pd(C2H5)Cl]2DME, rt | 7.5% 2 | 15% TBAT | 78% |
| 7 | 2.5% [Pd(C2H5)Cl]2 toluene, rt | 7.5% 2 | 15% TBAT | 81% |
| 8 | 1% [Pd(C2H5)Cl]2 toluene, rt | 3% 2 | 15% TBAT | 84% |
| 9 | 1% [Pd(C2H5)Cl]2 toluene, rt | 3% 2 | 5% TBAT | 79% |
| 10 | 0.25% [Pd(C2H5)Cl]2 toluene, rt | 1% 2 | 15% TBAT | 84% |
3.0 eq of CsF was added.
Enantiomeric excess was determined by HPLC, AD column, 90% heptane/10% iPrOH.
Surprisingly, lowering the temperature resulted in a slightly lower ee (74%). Changing the fluoride source to TBAT and the solvent to toluene increased the ee to 84%. Moreover, the major enantiomer could be purified to 98% ee with 69% yield, by recrystallizing out the minor/major enantiomer pair using heptane or cyclohexane. Furthermore, the yield for this reaction was 96-100% even when the catalyst loading was dropped from 1% to 0.25% [Pd(C2H5)Cl]2.
Oxidative cleavage of the allyl group was accomplished by catalytic osmium tetraoxide and N-methylmorpholine N-oxide (NMO) followed by cleavage of the diol with lead tetraacetate in methylene chloride (Scheme 3). Sodium periodate cleavage required somewhat aqueous conditions, which led to the formation of the five membered lactone as a byproduct. The initial plan to close the pyrrolidine was to reduce the ester and aldehyde to the diol, followed by bis mesylation and SN2 substitution with methylamine. Another route involved reductive amination of the aldehyde, reduction of the ester to the alcohol followed by intramolecular SN2 substitution. However, all attempts to reduce the ester resulted in low yields of the alcohol with the major product resulting from loss of CO.
Scheme 3.
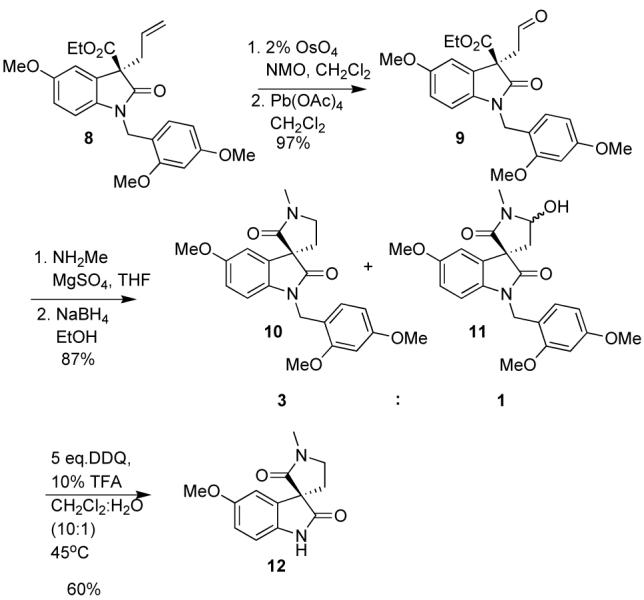
Ring Closure
Reductive amination of the aldehyde using NaBH3CN and the hydrochloride salt of methylamine resulted in the formation of the five membered lactam in low yields (25%). Switching to a solution of methylamine in THF and acetic acid with NaBH3CN led to a number of products, which was dependent on the amount and order in which the acetic acid was added to the reaction mixture. Reductive amination in a two step procedure, by first forming the imine in dry THF with MgSO4 and then reducing with NaBH4 in EtOH,6 provided lactam 10 in 65% yield. The byproduct of the reaction was the lactam alcohol 11, which presumably forms from cyclization of the hemiaminal onto the ester. Once lactam 10 was formed, only deprotection and a chemoselective reduction remained. The removal of the 2,4-dimethoxybenzyl group from the oxindole nitrogen was accomplished in 60% yield using DDQ in refluxing aqueous methylene chloride.
The chemoselective reduction of 12 proved to be a difficult challenge (Table 2). Initially, it was believed that 3 equiv. of DIBAL-H would first deprotonate the secondary amide protecting it from further reduction. However, it appeared that the oxindole was reduced preferentially under these conditions. Even by initially deprotonating with nBuLi, and then adding DIBAL-H, the desired product was not observed. Numerous reducing agents and conditions were applied to both lactams 10 and 12 in an attempt to successfully differentiate the two amides. Finally, it was found that the addition of 1 equiv. of nBuLi, followed by 2 equivalents of LAH in THF (from a freshly prepared and titrated solution), led to the desired product in 25-30% yield. Although the nBuLi was titrated before use7, low yields could have resulted from incomplete deprotonation, perhaps due to extraneous water. To avoid addition of excess base, a solution of trityllithium in DME was prepared. This solution was then added to the solution of amide 12 in DME until a slight pink color remained indicating the complete deprotonation of the secondary amide. Upon addition of 2 hydride equivalents of the LAH solution at 0 °C, horsfiline was formed in 45% yield.
Table 2.
Chemoselective Reduction 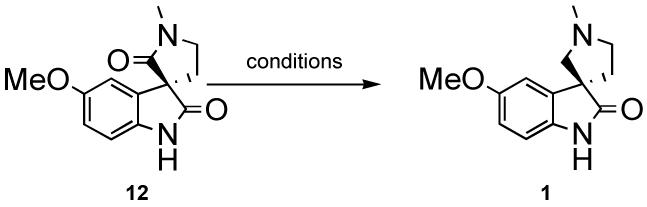
| Conditions | Yield |
|---|---|
| 3eq. DIBLAH, -78°C | NR |
| 3eq. DIBLAH, 0°C-->rt | reduced oxindole |
| 1. nBuLi(1eq) 2.DIBALH(2eq) -78°C-->0°C |
NR |
| 1. nBuLi(1eq) 2.DIBALH(2eq) 0°C-->rt |
NR |
| 1.nBuLi (-78°C) 2.DIBALH/nBuLi 0°C -->rt |
NR |
| BH3 in THF rt | NR |
| 1.nBuLi/TIPSOTf 2.2eq DIBALH |
only SM and silylated SM |
| 1.nBuLi(1eq -78°C-->0°C) 2.LAH soln (2H- eq.-78°C-->0°C) |
20% |
| 1. NaH (rt 30min) 2.LAH soln (2H- eq.-78°C-->0°C) |
<5% |
| 1. nBuLi(-78°C for 30 min) 2. LAHs soln (4H- eq, -78°C-->0°C) |
0% |
| 1. nBuLi(-78°C for 30 min) 2. LAHs soln (4H- eq, -78°C-->0°C) column(NH3/MeOH/EtOAc) |
32% |
|
1. Ph3CLi in DME 2. LAH soln (2H- eq DME,rt) |
48% |
It is worthy to note that horsfiline, along with other oxindole natural products, are prone to racemization via a retro-Mannich reaction that occurs in the presence of acid8 (Scheme 4). Since this synthesis avoids revealing the pyrrolidine until the last step, this problem is carefully avoided. Optical rotation verifies this and also determined that the (+)- horsfiline enantiomer had been synthesized.
Scheme 4.
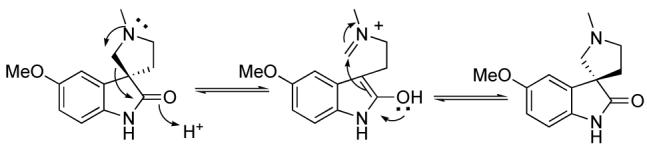
Racemization Mechanism
The enantiomer formed in the Pd AAA reaction can be rationalized by using the model previously described9 (Scheme 5). The nucleophile prefers to approach underneath a flap, and in an orientation that minimizes steric interactions with a nearby wall.
Scheme 5.
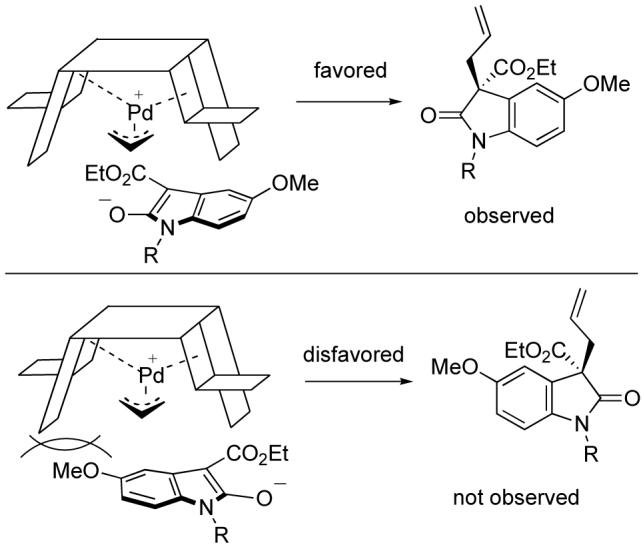
Rationalization of Stereochemistry
In conclusion, a concise total synthesis of horsfiline was achieved in 8 steps and 11.1% yield starting from p-anisidine and 2,4-dimethoxybenzaldehyde using an oxindole nucleophile in the palladium asymmetric allylic alkylation. A new class of prochiral nucleophiles, 3-carbalkoxyoxindoles, prove to be good substrates for asymmetric allylic alkylation. This synthesis circumvents the problems associated with racemization of the natural product by only exposing the pyrrolidine ring to a chemoselective reduction in the final step of the synthesis.
Supplementary Material
Acknowledgment
We thank the National Science Foundation and the National Institutes of Health (GM33049) for their generous support of our programs. Mass spectra were provided by the Mass Spectrometry Regional Center for the University of California-San Francisco, supported by the NIH Division of Research Resources. We thank Novartis for predoctoral support of M.B. We are indebted to Johnson-Matthey who generally provided palladium salts.
References
- 1.Jossang A, Jossang P, Hadi H, Sevenet T, Bodo B. J. Org. Chem. 1991;56:6527. [Google Scholar]
- 2 a.).Murphy JA, Tripoli R, Khan TA, Mali UW. Org. Lett. 2005;7:3287. doi: 10.1021/ol051095i. [DOI] [PubMed] [Google Scholar]; b.) Marti C, Carreira EM. Eur. J. Org. Chem. 2003;12:2209. [Google Scholar]; c.) Lizos DE, Murphy JA. Org. Biomol. Chem. 2003;1:117. doi: 10.1039/b208114h. [DOI] [PubMed] [Google Scholar]; d.) Selvakumar N, Azhagan AM, Srinivas D, Krishna GG. Tet. Lett. 2002;43:9175. [Google Scholar]; e.) Kumar U.K.Syam, Illa H, Junjappa H. Org. Lett. 2001;3:4193. doi: 10.1021/ol016824i. [DOI] [PubMed] [Google Scholar]; f). Cravotto G, Giovenzana G, Pilati T, Sisti M, Palmisano G. J. Org. Chem. 2001;66:8447. doi: 10.1021/jo015854w. [DOI] [PubMed] [Google Scholar]; g.) Fischer C, Meyers C, Carreira EM. Helv. Chim. Acta. 2000;83:1175. [Google Scholar]; h.) Lakshmaiah G, Kawabata T, Shang M, Fuji K. J. Org. Chem. 1999;64:1699. doi: 10.1021/jo981577q. [DOI] [PubMed] [Google Scholar]; i). Pellegrini C, Strässler C, Weber M, Borschberg H. Tetrahedron: Asymmetry. 1994;5:1979. [Google Scholar]; j). Bascop S, Sapi J, Laronze J, Levy J. Heterocycles. 1994;38:725. [Google Scholar]; k.) Jones K, Wilkinson J. Chem. Commun. 1992:1767. [Google Scholar]; l.) Chang MY, Pai C-L, Kung Y-H. Tet. Lett. 2005;46:8463. [Google Scholar]
- 3.Trost BM, Fredericksen MU. Angew. Chem. Int. Ed. 2005;44:308. doi: 10.1002/anie.200460335. [DOI] [PubMed] [Google Scholar]
- a.).Wenkert E, Bose AK, Reid TL. J. Am. Chem. Soc. 1953;75:5514. [Google Scholar]; b.) Wenkert E, Bhattacharyya NK, Reid TL, Stevens TE. J. Am. Chem. Soc. 1956;78:797. [Google Scholar]
- 5.Brown DS, Elliott MC, Moody CJ, Mowlem TJ, Marino JP, Padwa A. J. Org. Chem. 1994;59:2447. [Google Scholar]
- 6.Trost BM, Godleski SA, Genet JP. J. Am. Chem. Soc. 1978;100:3930. [Google Scholar]
- 7.Love BE, Jones EC. J. Org. Chem. 1999;64:3755. doi: 10.1021/jo982433e. [DOI] [PubMed] [Google Scholar]
- 8.Brown RT. In: Heterocyclic Compounds. Saxon JE, editor. Vol. 25. Wiley Interscience; New York: 1983. pp. 85–97. [Google Scholar]
- 9.Trost BM, Schroeder GM. J. Am. Chem. Soc. 1999;121:6759. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.