

SUMMARY
The risk of atherosclerosis is inversely associated with plasma levels of high-density lipoprotein cholesterol (HDL-C). However, HDL metabolism is incompletely understood, and there are few effective approaches to modulate HDL-C levels. Here we show that inhibition in the liver of the classical proprotein convertases (PCs), but not the atypical PCs S1P and PCSK9, decreases plasma HDL-C levels. This metabolic effect of hepatic PCs is critically dependent on expression of endothelial lipase (EL), an enzyme that directly hydrolyzes HDL phospholipids and promotes its catabolism. Hepatic PCs reduce EL function through direct inactivating cleavage of EL as well as through activating cleavage of angiopoietin-like protein 3 (ANGPTL3), an endogenous inhibitor of EL. Thus, inhibition of hepatic PCs results in increased EL activity, leading to reduced HDL-C as well as impaired reverse cholesterol transport. The hepatic PC-ANGPTL3-EL-HDL pathway is therefore a novel mechanism controlling HDL metabolism and cholesterol homeostasis.
INTRODUCTION
Proprotein convertases (PCs) belong to a family of subtilisin-related serine endopeptidases that is conserved from bacteria to mammals. This subfamily comprises nine members: furin, PCSK1, PCSK2, PCSK4, PACE4, PCSK5, S1P, PCSK7, and PCSK9. The physiological role of the individual PCs in mammals has recently been directly examined through genetic manipulation. The prototype furin and two closely related PCs, PCSK5 and PACE4, are essential for normal embryonic development; inactivation of any of genes that encode these proteins causes embryonic death in mice at various stages (Scamuffa et al., 2006). However, their postnatal physiological roles are still unknown.
Classically, the PCs are known to proteolytically activate peptides/proproteins by cleavage at paired basic amino acid sites. The recognition sequence is relatively nonspecific, with the general formula (R/K)-Xn-(R/K), where n = 0, 2, 4, or 6 (Seidah and Chretien, 1999). In this regard, PCSK9 and S1P are atypical PCs that cleave at nonbasic sites. Not surprisingly, the list of substrates of PCs has grown, and speculated physiological roles of PCs have broadened to include homeostasis as well as pathological states such as neurodegenerative disease, pathogen invasion, and cancer (Basak, 2005). However, most of the classical PC substrates have not yet been demonstrated in vivo.
Moreover, PCs are considered to be redundant, as early in vitro work showed that several PCs can cleave the same substrates and that they are often coexpressed in the same cell type (Seidah and Chretien, 1999). Consistent with this concept, upon hepatic inactivation of furin, the processing of the insulin receptor was normal, and only mild effects of reduced cleavage were observed for the substrates albumin, α5 integrin, lipoprotein receptor related protein, vitronectin, and α1-microglobulin/bikunin. None of the tested substrates displayed a complete block in processing (Roebroek et al., 2004). Thus, understanding the physiological role of PCs may require inhibition of all of the closely related PCs expressed in a particular cell type.
S1P and PCSK9 have been shown to modulate cholesterol metabolism. S1P, an endoplasmic reticulum-localized PC that cleaves membrane-bound sterol regulatory element-binding proteins (SREBPs) at an RSVL site, is required to release the active form of SREBPs to maintain cellular cholesterol homeostasis (Goldstein et al., 2002). PCSK9, after autocleavage at a VFAQ site, promotes degradation of low-density lipoprotein receptor (LDLR) through an unknown mechanism, a pathway supported by genetic data from humans (Abifadel et al., 2003; Cohen et al., 2005) as well as in vivo studies in mice (Benjannet et al., 2004; Maxwell and Breslow, 2004; Park et al., 2004). However, whether the classical PCs are also involved in lipid homeostasis is unknown.
Both loss-of-function and overexpression studies have established that the lipolytic enzyme endothelial lipase (EL) is a negative regulator of plasma high-density lipoprotein (HDL) levels (Ishida et al., 2003; Jaye et al., 1999; Jin et al., 2003; Ma et al., 2003). We recently reported that EL is cleaved by members of the PC family including furin, PACE4, and PCSK5A and that cleavage of EL resulted in its inactivation in vitro (Jin et al., 2005). Because these classical PCs are ubiquitously expressed, it was unclear whether their tissue-specific expression has any effects on EL cleavage in vivo and, as such, controls EL activity and HDL levels. Here, we provide evidence that hepatic PCs are able to modify EL activity both directly and indirectly and thus influence HDL metabolism and reverse cholesterol transport in mice.
RESULTS AND DISCUSSION
Inhibition of Hepatic PCs Decreases HDL Cholesterol Levels
The prosegment of human furin (profurin) was used to inhibit classical PCs in vivo for several reasons. Purified full-length profurin has been reported to be a potent inhibitor of furin and PCSK5, and to a lesser extent PCSK7 and PACE4, in a cell-free system (Zhong et al., 1999), and profurin is effective in inhibiting the processing of various cellular precursors including proNGF, proBDNF (Zhong et al., 1999), proVEGF-C (Nour et al., 2003), and proBACE1 (Benjannet et al., 2001) in vitro. In addition, profurin is an intracellular inhibitor of some classical PCs including furin, PCSK5, and PACE4 (see Figure S1 in the Supplemental Data available with this article online), but autocatalytic cleavage of PCSK9 and S1P was not altered by profurin overexpression (Figure S2). To confirm that profurin can inhibit murine PCs in liver, primary murine hepatocytes were isolated 3 days after in vivo infection with either adenovirus encoding human profurin and an internal ribosomal entry site-linked EGFP reporter gene (Ad-profurin) or control adenovirus containing no transgenic expression cassette (Ad-null). The transduction efficiency was ∼90% by fluorescence-activated cell sorting analysis of enhanced green fluorescent protein (EGFP) expression (data not shown). The expression of human profurin protein was confirmed by western blotting (Figure 1A) using an antibody specific for human profurin (Zhong et al., 1999). PC activity was measured in the conditioned medium. In the presence of profurin overexpression, secreted PC activity was approximately 50% of that of the control (Figure S3). All of the PC activity detected in the conditioned medium was inhibited by another PC-specific inhibitor, α1-PDX (Watanabe et al., 1995), indicating that overexpression of profurin only partially inhibits hepatic PC activity (data not shown). Plasma alanine aminotransferase levels were not altered after overexpression of profurin (data not shown), and morphologic assessment of mouse livers by hematoxylin and eosin staining did not reveal any gross changes, indicating that partial PC inhibition does not cause damage to hepatocytes (Figure S4).
Figure 1. Profurin and Plasma Lipid Levels after Inhibition of Hepatic Proprotein Convertases in Wild-Type Mice.

A single dose of Ad-null or Ad-profurin was injected in wild-type mice. (A) Western blotting of profurin protein expression in vivo after transduction. Similar amounts of proteins from liver lysates (10 μg/lane) were loaded.
(B and C) Blood was sampled at the indicated time points, and plasma lipid profile for high-density lipoprotein cholesterol (HDL-C) (B) and phospholipids (C) was determined as described in Experimental Procedures.
n ≥ 4 mice per group; *p < 0.05, **p < 0.01. Each experiment was repeated 12 times. In this and all other figures, error bars represent ± SD.
When profurin was overexpressed in mice, plasma HDL cholesterol (HDL-C) and phospholipids were significantly reduced at day 3 compared to those in the Ad-null-injected mice and returned to baseline by day 14 (Figures 1B and 1C). However, glucose and free fatty acids were not altered (data not shown). The plasma lipid profiles on day 3 after injection were further examined by fast protein liquid chromatography (FPLC) analysis, and the HDL-C levels were found to be reduced (Figure S5). Agarose gel electrophoresis was also used to separate lipoproteins based upon their charge. α-migrating lipoproteins (HDL) and β-migrating lipoproteins (LDL) were decreased in the Ad-profurin-treated mice, with no increase in pre-β-migrating lipoproteins (very low-density lipoprotein and pre-β HDL) (Figure S6). The protein composition in the HDL fractions was analyzed, and apoA-I, apoA-II, apoE, apoC-I, and apoC-III were all significantly reduced (Table S1). The reduced apoA-II and apoE proteins in HDL fractions were confirmed by immunoblotting (Figures S7A and S7B). Profurin expression had no effect on the secretion of apoA-I from primary murine hepatocytes (Figure S7C). In Ad-profurin-treated plasma, the mature form of apoA-I was barely detectable, but pro-apoA-I was comparable to that seen in control plasma (Figure S7D). No differences in hepatic expression of ABCA1 or SR-BI, both known to influence HDL levels (Acton et al., 1996; Christiansen-Weber et al., 2000; Rader, 2006), were observed in the mice overexpressing profurin (Figure S8).
EL-Dependent Effects of Inhibition of Hepatic PCs on HDL Metabolism
We hypothesized that hepatic PC-mediated cleavage and inactivation of EL influences HDL metabolism and that inhibition of hepatic PCs reduces HDL-C by increasing EL activity. To test whether EL is a substrate of hepatic PCs in vivo, the EL protein was analyzed in plasma from either Ad-null- or Ad-profurin-treated mice. To separate the full-length EL from albumin, two-dimensional electrophoresis was performed, and EL protein was detected by using a polyclonal antibody to mouse EL. As shown in Figure 2A, inhibition of hepatic PCs reduced generation of the 40 kDa cleavage product and increased the full-length 68 kDa form of EL in the Ad-profurin-treated plasma. Neither of these two forms of EL protein was detected in plasma from EL knockout mice (data not shown).
Figure 2. Inhibition of Hepatic Proprotein Convertases Affects HDL Levels through Endothelial Lipase in Mice.

(A) Effects of inhibition of hepatic proprotein convertases (PCs) on the cleavage of endothelial lipase (EL) in mice. Ten microliters of plasma was separated by two-dimensional electrophoresis, and EL protein was detected by western blotting. F: full-length protein (dotted box); N: cleaved N-terminal fragment (oval).
(B) Effects of inhibition of hepatic PCs on postheparin plasma phospholipase activity. n = 4; *p < 0.05.
(C) Effects of inhibition of hepatic PCs on HDL-phospholipid catabolism in wild-type (WT) mice. n = 4; p < 0.01.
(D) Effects of inhibition of hepatic PCs on HDL levels in EL knockout mice (top) and western blotting of profurin in liver lysates after transduction (bottom).
(E) Effects of inhibition of hepatic PCs on HDL-phospholipid catabolism in EL knockout mice. n = 4; p < 0.01.
(F) Effects of inhibition of hepatic PCs on HDL levels in HL knockout mice (top) and western blotting of profurin in liver lysates after transduction (bottom).
(G and H) Effects of PCSK5A on HDL levels in wild-type (G) and EL knockout mice (H).
n ≥ 4 mice per group; *p < 0.05.
To test whether inhibition of hepatic PCs increases plasma phospholipase activity, this parameter was measured in mouse postheparin plasma, where it was found to be increased 2.3-fold in Ad-profurin-treated plasma compared to control plasma (p < 0.01, Figure 2B). To examine whether inhibition of hepatic PCs increases HDL-phospholipid catabolism, HDL from wild-type mice was labeled with [14C]phosphatidylcholine and injected into mice that had been infected with Ad-null or Ad-profurin 3 days earlier. The clearance of HDL-PC was significantly increased in the Ad-profurin-treated mice (Figure 2C). The fractional catabolic rates were 1.67 ± 0.49 versus 4.32 ± 0.93 pool/hr (p < 0.01) for control- and profurin-treated mice, respectively. This rapid catabolism of HDL particles may explain the increased ratio of pro-apoA-I to mature apoA-I seen in ABCA1-deficient patients (Bojanovski et al., 1987). Thus, increased catabolism of HDL due to increased EL activity is likely the mechanism by which inhibition of hepatic PCs decreases HDL levels.
To prove that endogenous EL expression is required for the lipid phenotype observed after hepatic PC inhibition, Ad-profurin was injected into EL knockout mice. In contrast to wild-type mice, HDL levels were not changed after hepatic PC inhibition in these mice (Figure 2D). The fractional catabolic rates of HDL were not significantly different between control- and profurin-treated EL knockout mice (1.03 ± 0.16 versus 1.28 ± 0.34 pool/hr, respectively) (Figure 2E). To exclude the in vivo effects of another phospholipase in the same family as EL, hepatic lipase (HL) (Wong and Schotz, 2002), which is not cleaved by PCs (data not shown), a similar experiment was performed in HL knockout mice. HDL levels were markedly reduced and virtually undetectable at day 3 after Ad-profurin administration in these mice (Figure 2F). Therefore, regulation of HDL levels by hepatic PCs is dependent on the expression of endogenous EL, but not HL.
To test whether increased hepatic PC expression would lead to an elevation of HDL levels, PCSK5A, a representative classical PC, was overexpressed in wild-type mice. Overexpression of PCSK5A resulted in a significant increase of HDL-C levels, with a peak increase of ∼50% (Figure 2G). Both total cholesterol and phospholipid levels were also increased in parallel (data not shown). The change in HDL levels was similar to that in EL knockout mice as reported by Ishida et al. (2003) and Ma et al. (2003). Consistently, plasma EL cleavage products were increased after PCSK5A overexpression (Figure S9). To determine whether the effects of PCSK5A overexpression are dependent on EL, a similar experiment was performed in EL knockout mice. As expected, HDL-C levels were not different between the PCSK5A-expressing group and control group (Figure 2H), confirming that the ability of PCSK5A overexpression to raise HDL is dependent on endogenous EL.
Inhibition of Hepatic PCs Decreases the Cleavage of Angiopoietin-like Protein 3
Angiopoietin-like protein 3 (ANGPTL3), a liver-derived member of the vascular endothelial growth factor family, has been shown to be an inhibitor of LPL (Shimizugawa et al., 2002) and EL (Shimamura et al., 2007) in cell-free systems and has been suggested to play a potential role in regulating HDL levels (Koishi et al., 2002). To test whether ANGPTL3 inhibits EL in vitro, a plasmid expressing murine ANGPTL3 was transfected into HEK293 cells stably expressing EL. ANGPTL3 inhibited ∼60% of EL activity without affecting the amount of EL mass in the conditioned medium (Figure 3A) but did not inhibit HL activity (Figure S10). Interestingly, murine ANGPTL3 has been shown to be cleaved in vitro and in vivo at a classical PC recognition sequence, RAPR224 (Ono et al., 2003). The cleavage of murine ANGPTL3 was totally blocked in HEK293 cells stably expressing profurin (Figure 3B). The importance of the cleavage of ANGPTL3 for EL activity was further examined by comparing the inhibitory effects of the full-length ANGPTL3 on EL activity to the effects of the cleavage-product N terminus of ANGPTL3. As shown in Figure 3C, the N terminus of ANGPTL3 inhibited EL activity more effectively than the uncleavable ANGPTL3 did (p < 0.05). Thus, cleavage of ANGPTL3 is an activation process releasing the active form of ANGPTL3 (N terminus), which is more effective at inhibiting EL.
Figure 3. Effects of Inhibition of Hepatic PCs on ANGPTL3.
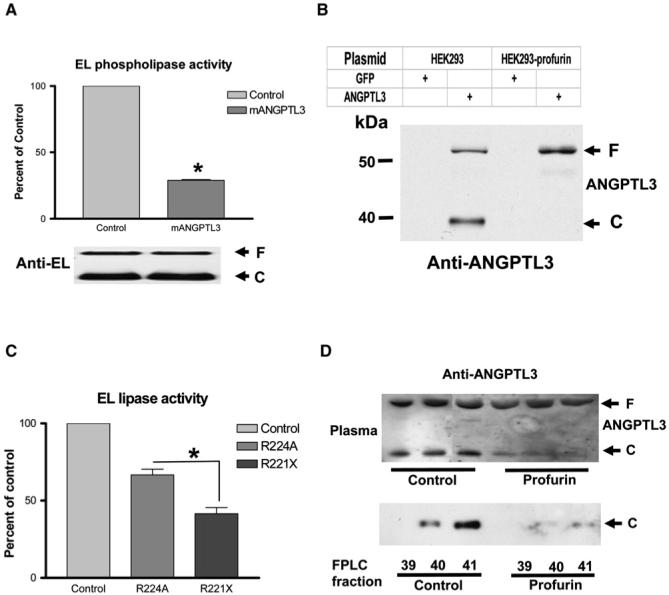
(A) ANGPTL3 inhibited EL activity in vitro.
(B) Profurin prevented the cleavage of ANGPTL3 in vitro.
(C) The N terminus of ANGPTL3 (R221X) was a more potent inhibitor of EL than uncleavable ANGPTL3 (R224A). Data are expressed as percent of the control. n = 3; *p < 0.05.
(D) Effects of inhibition of hepatic PCs on the cleavage of ANGPTL3 in mice. Upper panel: plasma (0.1 μl/lane); lower panel: FPLC fractions 39-41, which elute later than HDL. F: full-length protein; C: cleaved C-terminal fragment.
To test whether hepatic PC inhibition decreased the cleavage of ANGPTL3 in vivo, we examined endogenous ANGPTL3 in plasma by western blotting. Profurin dramatically reduced ANGPTL3 cleavage in mice (Figure 3D, upper panel). After mouse plasma was separated by FPLC, the C-terminal ANGPTL3 protein was detected in lipid poor fractions and was not associated with HDL fractions. Importantly, fewer cleavage products of ANGPTL3 were generated in mice treated with Ad-profurin compared to the control (Figure 3D, lower panel). When primary hepatocytes were isolated after infection, Ad-profurin-transduced hepatocytes produced significantly less of the cleavage products, with an increase in the full-length ANGPTL3 (data not shown). Thus, ANGPTL3 is an authentic hepatic PC substrate in vivo. Inhibition of PC cleavage reduced activation of ANGPTL3, leading to reduced inhibition of EL by ANGPTL3 (Figure 3C). This likely contributes to the decreased HDL levels seen in profurin-treated mice.
Inhibition of Hepatic PCs Decreases Reverse Cholesterol Transport
Because a major atheroprotective mechanism of HDL is the promotion of cellular efflux of cholesterol from macrophages, we addressed whether reduced HDL levels in the Ad-profurin-treated serum are associated with reduced macrophage cholesterol efflux and reverse cholesterol transport (RCT). The ability of serum from mice injected with either Ad-null or Ad-profurin to mediate cholesterol efflux was assessed in vitro (Yancey et al., 2004). The ABCA1-mediated efflux was abolished and the efflux via SR-BI was decreased by 93% using serum from Ad-profurin-treated mice (Figures 4A and 4B). To examine the effect of hepatic PC inhibition on macrophage RCT in vivo (Zhang et al., 2003), [3H]cholesterol-labeled, cholesterol loaded J774 cells were injected intraperitoneally into mice 3 days after the mice were transduced with either Ad-null or Ad-profurin. The Ad-profurin-treated mice had significantly lower plasma [3H]cholesterol levels at all time points studied: 64% (p < 0.01) lower at 2 hr, 71% (p < 0.01) lower at 6 hr, 72% (p < 0.01) lower at 24 hr, and 73% (p < 0.01) lower at 48 hr (Figure 4C). The level of tracer in the livers of Ad-profurin-treated mice was 37% lower (Figure 4D, p < 0.05). The [3H]bile acids in the Ad-profurin-treated mice were also significantly (41%, p < 0.05) lower compared to controls (Figure 4E). Consistently, the Adprofurin-treated mice excreted 59% less tracer in feces (p < 0.05) over 48 hr than control mice (Figure 4F). We observed similar results when EL was overexpressed in wild-type mice (data not shown).
Figure 4. Effects of Inhibition of Hepatic PCs on Reverse Cholesterol Transport.

(A) The measurement of ABCA1-specific efflux to mouse plasma in J774 cells. Data in (A) and (B) are expressed as percent of the control. n = 3; *p < 0.05.
(B) The measurement of SR-BI-specific efflux to mouse plasma in Fu5AH cells.
(C-F) Mice were injected intraperitoneally with ac-LDL-loaded J774 macrophages labeled with [3H]cholesterol. The percent of macrophage-derived [3H]cholesterol appearing in plasma (C), liver at 48 hr (D), bile at 48 hr (E), and feces over 48 hr (F) after macrophage injection is shown. Data are expressed as percent of the initial amount of [3H]cholesterol injected intraperitoneally. n = 6; *p < 0.05.
Our studies demonstrate that hepatic PCs play a physiological role in HDL metabolism in vivo and that both EL and ANGPTL3 are physiological substrates of hepatic PCs mediating the effects on HDL through two pathways. Hepatic PCs regulate EL function through cleavage of EL, a direct inactivation of EL lipolytic activity, and cleavage of ANGPTL3, an indirect process of activation of an inhibitor of EL. Thus, inhibition of hepatic PCs increases EL activity, resulting in a reduction of plasma HDL levels (Figure S11). The relative contribution of these two pathways is unknown, and further work will be required to determine the overall importance of each component of the system. The physiological significance of hepatic PCs is further supported by the fact that inhibition of hepatic PCs reduces RCT. In parallel to the known role of S1P and PCSK9 in regulating LDL metabolism through the LDLR, the PC-ANGPTL3-EL-HDL pathway is a critical mechanism for fine tuning HDL metabolism. This proteolytic cascade opens a new avenue in the proprotein convertase field and suggests several novel targets for raising HDL-C levels and promoting RCT. More experiments to determine which endogenous proprotein convertases are most relevant in regulation of HDL metabolism and to demonstrate that there is physiological regulation of this system are warranted. Insights gained from these studies could lead to new ways to therapeutically regulate HDL levels.
Our current studies indicate the physiological importance of EL, whose function is normally suppressed by hepatic PCs and ANPTL3. Importantly, this PC-ANGPTL3-EL system has a large capacity to modulate HDL levels. It is conceivable that this mechanism could be altered and could contribute to the changes of HDL metabolism under pathophysiological or disease conditions, a possibility that is currently under investigation. Furthermore, these studies provide an example of how the liver regulates HDL catabolism through an irreversible proteolytic cascade. This model may apply to other proteolytic events in lipid and lipoprotein metabolism as well.
EXPERIMENTAL PROCEDURES
Construction of Adenoviral Vectors
Recombinant, replication-defective adenoviruses encoding human profurin, murine pcsk5a, or murine angptl3 were generated by the Vector Core at the University of Pennsylvania. To better trace the expression of human profurin in vivo, an adenovirus encoding the human profurin cDNA and an internal ribosomal entry site-linked EGFP reporter gene, both driven by the CMV promoter, was generated. The control adenovirus, Ad-null, contained no transgenic expression cassette.
Western Blotting
SDS-PAGE and immunoblot analysis were carried out as previously described (Jin et al., 2005). The primary antibody used was a rabbit polyclonal antibody against murine EL (Jin et al., 2003). A rabbit polyclonal antibody to murine ANGPTL3 was generated according to the method previously described (Jin et al., 2003). FPLC fractions 39-41 were used for the detection of ANGPTL3. For two-dimensional gel electrophoresis, plasma samples were pipetted into the bottom of a protein isoelectric focusing (IEF) apparatus (GE Healthcare/Amersham) and were focused onto a freshly thawed Immobiline DryStrip (GE Healthcare/Amersham) after stepwise voltage increases to a total of 71,500 volt hours. After IEF, the proteins were subjected to second dimension separation according to their size.
Animal/Plasma Analysis
Female 8-week-old C57BL/6 mice obtained from Taconic and 8-week old EL knockout mice were used for the experiments. All mice were fed a chow diet. For blood sampling, mice were fasted for 4 hr and bled from the retro-orbital plexus. 1 × 1011 particles of adenovirus were administered via tail vein on day 0 of the study. Blood samples were taken at the days indicated after virus injection. Plasma lipids were measured using previously reported methods, and FPLC analysis was performed on pooled samples (Jin et al., 2003). The University of Pennsylvania Institutional Animal Care and Usage Committee approved the experiments, and all procedures were performed as recommended by the NIH Guide for the Care and Use of Laboratory Animals and the Laboratory Animal Welfare Act.
Lipase Activity Assays
EL-specific and HL-specific triglyceride lipase and postheparin plasma phospholipase activities were assayed as previously described (Jin et al., 2005).
PC Activity Assays
PC activity assay was performed using the fluorogenic peptide Pyr-RVRR-AMC (Bachem) as the substrate. One unit is defined as the amount of furin that will release 1 pmol of AMC from the fluorogenic peptide Pyr-RVRR-AMC in a total reaction volume of 100 μl in 1 min (1 pmol of AMC/min) at 37°C in 100 mM HEPES (pH 7.5 at 25°C) with 0.5% Triton X-100, 1 mM CaCl2, 1 mM 2-mercaptoethanol, and 100 μM Pyr-RVRR-AMC.
Histology
Animals were sacrificed and livers were fixed with 4% paraformaldehyde at 4°C. Paraffin sections (4 μm) were stained with hematoxylin and eosin and were examined and photographed with a Nikon Eclipse E-800 microscope.
Native Gel Electrophoresis
Lipoproteins were separated by agarose gel electrophoresis using Titan gels (Helena) according to the manufacturer’s standard protocol.
Proteomics
Proteins in the HDL fraction were extracted with methanol/chloroform and analyzed as previously described (Sewell et al., 2006). All data were normalized to volume. This experiment was repeated eight times.
Primary Hepatocyte Culture
C57BL/6J mice were injected with the indicated adenovirus, and 3 days later, primary hepatocytes were isolated as previously described (Park et al., 2004). The hepatocytes were allowed to attach on type I collagen-coated six-well plates for 4 hr in DMEM. The cells were then incubated with serum-free DMEM for another 24 hr.
HDL Turnover Experiments
Wild-type mice were infected with either Ad-null or Ad-profurin 3 days before the turnover study. Mouse HDL was isolated from wild-type mice and labeled with [14C]phosphatidylcholine, and kinetic studies were performed as previously described (Jin et al., 2003). The fractional catabolic rates of HDL phospholipid were calculated with the WinSAAM program by fitting a biexponential curve to the plasma counts normalized to the 1 min time point.
Plasmids and Cell Culture
Constructs expressing uncleavable ANGPTL3 (R224A) and the N terminus of ANGPTL3 (R221X), each with a C-terminal Myc-His tag, were generated. Conditioned media containing R224A or R221X were generated and quantitated using a modified ELISA with anti-Myc antibody. Equal amounts of R224A or R221X protein were used to incubate cells stably expressing EL for 24 hr.
Measurement of Cholesterol Efflux In Vitro and RCT In Vivo
SR-BI-mediated cholesterol efflux and ABCA1-mediated free cholesterol efflux was measured as previously described (Yancey et al., 2004). In vivo RCT was performed as previously described (Zhang et al., 2003).
Statistics
Data are presented as means ± SD. Multiple (>2) group study data were subjected to two-way analysis of variance (ANOVA) and the Newman-Keuls multiple comparison test. Two-tailed Student’s t tests were used for the mean of two groups, and statistical significance for all comparisons was assigned at p < 0.05. All calculations were performed using GraphPad Prism version 4.03 (GraphPad Software).
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Donovan, J. Liu, A. DiFloria, L. Morrell, R. Brown, A. Rodrigues, A. Faruqi, and E. Edouard for excellent technical assistance. N.G. Seidah provided expert advice and the anti-profurin antibody. This work was supported by NIH grant R01 HL-081861(W.J.), an American Heart Association Scientist Development Grant (W.J.), and NIH grant R01 HL-55323 (D.J.R.). W.J. designed and performed experiments and wrote the manuscript. X.W. performed the in vivo RCT studies. J.S.M. performed mouse kinetic studies and data analysis. T.Q. helped enable the PCSK5 studies and provided EL knockout mice. G.H.R. supervised the in vitro efflux assay and its data analysis. J.M.G. and D.J.R. conceptualized the study and participated in data interpretation. All authors made comments on the manuscript.
REFERENCES
- Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- Basak A. Inhibitors of proprotein convertases. J. Mol. Med. 2005;83:844–855. doi: 10.1007/s00109-005-0710-0. [DOI] [PubMed] [Google Scholar]
- Benjannet S, Elagoz A, Wickham L, Mamarbachi M, Munzer JS, Basak A, Lazure C, Cromlish JA, Sisodia S, Checler F, et al. Post-translational processing of beta-secretase (beta-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-beta production. J. Biol. Chem. 2001;276:10879–10887. doi: 10.1074/jbc.M009899200. [DOI] [PubMed] [Google Scholar]
- Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin MC, Hamelin J, Varret M, Allard D, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 2004;279:48865–48875. doi: 10.1074/jbc.M409699200. [DOI] [PubMed] [Google Scholar]
- Bojanovski D, Gregg RE, Zech LA, Meng MS, Bishop C, Ronan R, Brewer HB., Jr. In vivo metabolism of proapolipoprotein A-I in Tangier disease. J. Clin. Invest. 1987;80:1742–1747. doi: 10.1172/JCI113266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen-Weber TA, Voland JR, Wu Y, Ngo K, Roland BL, Nguyen S, Peterson PA, Fung-Leung WP. Functional loss of ABCA1 in mice causes severe placental malformation, aberrant lipid distribution, and kidney glomerulonephritis as well as high-density lipoprotein cholesterol deficiency. Am. J. Pathol. 2000;157:1017–1029. doi: 10.1016/S0002-9440(10)64614-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Rawson RB, Brown MS. Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch. Biochem. Biophys. 2002;397:139–148. doi: 10.1006/abbi.2001.2615. [DOI] [PubMed] [Google Scholar]
- Ishida T, Choi S, Kundu RK, Hirata K, Rubin EM, Cooper AD, Quertermous T. Endothelial lipase is a major determinant of HDL level. J. Clin. Invest. 2003;111:347–355. doi: 10.1172/JCI16306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaye M, Lynch KJ, Krawiec J, Marchadier D, Maugeais C, Doan K, South V, Amin D, Perrone M, Rader DJ. A novel endothelial-derived lipase that modulates HDL metabolism. Nat. Genet. 1999;21:424–428. doi: 10.1038/7766. [DOI] [PubMed] [Google Scholar]
- Jin W, Millar JS, Broedl U, Glick JM, Rader DJ. Inhibition of endothelial lipase causes increased HDL cholesterol levels in vivo. J. Clin. Invest. 2003;111:357–362. doi: 10.1172/JCI16146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Fuki IV, Seidah NG, Benjannet S, Glick JM, Rader DJ. Proprotein convertases [corrected] are responsible for proteolysis and inactivation of endothelial lipase. J. Biol. Chem. 2005;280:36551–36559. doi: 10.1074/jbc.M502264200. [DOI] [PubMed] [Google Scholar]
- Koishi R, Ando Y, Ono M, Shimamura M, Yasumo H, Fujiwara T, Horikoshi H, Furukawa H. Angptl3 regulates lipid metabolism in mice. Nat. Genet. 2002;30:151–157. doi: 10.1038/ng814. [DOI] [PubMed] [Google Scholar]
- Ma K, Cilingiroglu M, Otvos JD, Ballantyne CM, Marian AJ, Chan L. Endothelial lipase is a major genetic determinant for high-density lipoprotein concentration, structure, and metabolism. Proc. Natl. Acad. Sci. USA. 2003;100:2748–2753. doi: 10.1073/pnas.0438039100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. USA. 2004;101:7100–7105. doi: 10.1073/pnas.0402133101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nour N, Basak A, Chretien M, Seidah NG. Structure function analysis of the prosegment of the proprotein convertase PC5A. J. Biol. Chem. 2003;278:2886–2895. doi: 10.1074/jbc.M208009200. [DOI] [PubMed] [Google Scholar]
- Ono M, Shimizugawa T, Shimamura M, Yoshida K, Noji-Sakikawa C, Ando Y, Koishi R, Furukawa H. Protein region important for regulation of lipid metabolism in angiopoietin-like 3 (ANGPTL3): ANGPTL3 is cleaved and activated in vivo. J. Biol. Chem. 2003;278:41804–41809. doi: 10.1074/jbc.M302861200. [DOI] [PubMed] [Google Scholar]
- Park SW, Moon YA, Horton JD. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J. Biol. Chem. 2004;279:50630–50638. doi: 10.1074/jbc.M410077200. [DOI] [PubMed] [Google Scholar]
- Rader DJ. Molecular regulation of HDL metabolism and function: implications for novel therapies. J. Clin. Invest. 2006;116:3090–3100. doi: 10.1172/JCI30163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roebroek AJ, Taylor NA, Louagie E, Pauli I, Smeijers L, Snellinx A, Lauwers A, Van de Ven WJ, Hartmann D, Creemers JW. Limited redundancy of the proprotein convertase furin in mouse liver. J. Biol. Chem. 2004;279:53442–53450. doi: 10.1074/jbc.M407152200. [DOI] [PubMed] [Google Scholar]
- Scamuffa N, Calvo F, Chretien M, Seidah NG, Khatib AM. Proprotein convertases: lessons from knockouts. FASEB J. 2006;20:1954–1963. doi: 10.1096/fj.05-5491rev. [DOI] [PubMed] [Google Scholar]
- Seidah NG, Chretien M. Proprotein and prohormone convertases: a family of subtilases generating diverse bioactive polypeptides. Brain Res. 1999;848:45–62. doi: 10.1016/s0006-8993(99)01909-5. [DOI] [PubMed] [Google Scholar]
- Sewell DA, Yuan CX, Robertson E. Proteomic signatures in laryngeal squamous cell carcinoma. ORL J. Otorhinolaryngol. Relat. Spec. 2006;69:77–84. doi: 10.1159/000097406. [DOI] [PubMed] [Google Scholar]
- Shimamura M, Matsuda M, Yasumo H, Okazaki M, Fujimoto K, Kono K, Shimizugawa T, Ando Y, Koishi R, Kohama T, et al. Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler. Thromb. Vasc. Biol. 2007;27:366–372. doi: 10.1161/01.ATV.0000252827.51626.89. [DOI] [PubMed] [Google Scholar]
- Shimizugawa T, Ono M, Shimamura M, Yoshida K, Ando Y, Koishi R, Ueda K, Inaba T, Minekura H, Kohama T, Furukawa H. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J. Biol. Chem. 2002;277:33742–33748. doi: 10.1074/jbc.M203215200. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Hirano A, Stenglein S, Nelson J, Thomas G, Wong TC. Engineered serine protease inhibitor prevents furin-catalyzed activation of the fusion glycoprotein and production of infectious measles virus. J. Virol. 1995;69:3206–3210. doi: 10.1128/jvi.69.5.3206-3210.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong H, Schotz MC. The lipase gene family. J. Lipid Res. 2002;43:993–999. doi: 10.1194/jlr.r200007-jlr200. [DOI] [PubMed] [Google Scholar]
- Yancey PG, Asztalos BF, Stettler N, Piccoli D, Williams DL, Connelly MA, Rothblat GH. SR-BI- and ABCA1-mediated cholesterol efflux to serum from patients with Alagille syndrome. J. Lipid Res. 2004;45:1724–1732. doi: 10.1194/jlr.M400133-JLR200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- Zhong M, Munzer JS, Basak A, Benjannet S, Mowla SJ, Decroly E, Chretien M, Seidah NG. The prosegments of furin and PC7 as potent inhibitors of proprotein convertases. In vitro and ex vivo assessment of their efficacy and selectivity. J. Biol. Chem. 1999;274:33913–33920. doi: 10.1074/jbc.274.48.33913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.