

Abstract
The spirochete Borrelia burgdorferi, the etiologic agent of Lyme disease, causes severe subacute arthritis in susceptible inbred mouse strains, such as C3H/HeN, but only mild arthritis in resistant strains such as C57BL/6. The degree of Lyme arthritis severity is controlled in part by host genetics and several quantitative trait loci have been identified which contribute to this regulation. In addition, the anti-inflammatory cytokine IL-10 assumes an important role in the control of arthritis in C57BL/6 mice. However, the identification of genes and signaling pathways that dictate arthritis severity has remained elusive. In an attempt to elucidate such genes and pathways, the power of microarray analysis was combined with information gleaned from gene manipulation models. As a result of this approach, two novel gene profiles were identified: an IFN-inducible profile in arthritis-susceptible C3H and IL-10−/− mice, and an epidermal/differentiation profile in C57BL/6 mice. Application of this information to TLR2−/− mice, which also develop severe arthritis, indicated that they also upregulated IFN-responsive genes. These results provided new insight into the regulation of Lyme arthritis development and illustrated the utility of combining gene expression analyses with genetically manipulated mouse models in unraveling mechanisms underlying specific disease processes.
Keywords: Borrelia burgdorferi, gene regulation, arthritis, microarray analysis, mouse models
Introduction
Lyme disease in humans results from infection with the tick borne spirochete Borrelia burgdorferi (Burgdorfer et al., 1982). Symptoms involve a variety of tissues, following invasion by the pathogen. Approximately 60% of individuals not treated at the time of the tick bite will develop clinical arthritis, with a range of severities reported for this subacute inflammatory arthritis. A small percentage of patients with the early arthritis may progress to a chronic disease, which becomes refractory to antibiotic therapy (treatment resistant), and may be autoimmune mediated. The subacute, inflammatory, arthritis can be studied in mice, where it is characterized as a tendonitis dominated by PMN infiltration, synovial hyperproliferation, and edema. One rear ankle joint generally displays greater involvement, similar to the situation in humans where a single knee joint is likely to be involved (Fig. 1) (Barthold et al., 1990; Barthold et al., 1991; Nocton and Steere, 1995; Steere and Glickstein, 2004; Steere et al., 1987).
Fig. 1. Characteristics of human Lyme arthritis.

While both subacute and treatment resistant Lyme arthritis have been described in humans, they exhibit markedly different characteristics.
Several recent reviews have characterized the enzootic cycle of this organism, which relies on the mouse as the major reservoir in nature (Hayes and Piesman, 2003; Rosa et al., 2005). Inbred strains of mice have been used in many investigations of the mechanism of arthritis development, as C3H mice develop severe arthritis while C57BL/6 mice display mild to moderate arthritis (Barthold et al., 1990; Barthold et al., 1991; Ma et al., 1998). These studies have suggested that the inflammatory response to the bacteria invading the joint tissue drives the arthritic response. Importantly, two studies independently demonstrated that the overall inflammatory state of the joint is not directly dependent on the number of spirochetes in tissues (Brown and Reiner, 1998; Ma et al., 1998). These findings strongly implicate differential regulation of the host’s inflammatory response in determining the severity of disease in inbred mouse strains. Mice harboring scid or rag mutations develop disease similar to wild type background, indicating B and T lymphocytes are not required for disease nor do they modulate its severity (Barthold et al., 1992; Brown and Reiner, 1999b). Although not linked to the MHC, the severity of Lyme arthritis is genetically linked, with intercross populations between severely arthritic and mildly arthritic mice identifying quantitative trait loci on chromosomes 1, 4, 5, 11, and 12 (Brown and Reiner, 2000; Roper et al., 2001; Weis et al., 1999; Yang et al., 1992). While numerous studies with mice lacking particular cytokines have failed to unambiguously establish the requirement for any one particular cytokine in Lyme arthritis development, the anti-inflammatory molecule IL-10 is uniquely required for limiting the severity of arthritis in C57BL/6 mice (Brown et al., 1999; Wooten and Weis, 2001).
This review will summarize recent findings from our laboratory and others using novel genetic models and analyses to assess the pathways involved in Lyme arthritis development and resistance.
Innate inflammatory responses in host defense and arthritis development
Interestingly, in vitro experiments with cells from humans and mice respond to products of B. burgdorferi, with the interaction of bacterial lipoproteins with TLR2 in conjunction with CD14 and TLR1 providing the major inflammatory stimulus (Aliprantis et al., 1999; Benhnia et al., 2005; Hirschfeld et al., 1999; Lien et al., 1999; Norgard et al., 1996; Sellati et al., 1999; Wooten et al., 1998). These in vitro findings led to the prediction that mice lacking TLR2 would be protected from severe arthritis. In fact, TLR2−/− mice developed more severe arthritis than wild type mice, on both the B6 and C3H backgrounds, implying that arthritis is driven by TLR2-independent pathways. TLR2−/− animals were severely compromised in host defense, harboring many fold more spirochetes in tissues than wild type mice (Wooten et al., 2002). The effect of CD14 ablation on Lyme arthritis was less dramatic than TLR2, and these studies led to the conclusion that CD14 accelerates and directs the resolution of disease (Benhnia et al., 2005) Mice deficient in the major adapter for TLR signaling, MyD88 are severely compromised in host defense, harboring up to 100-fold more B. burgdorferi than wild type controls (Behera et al., 2006; Bolz et al., 2004; Liu et al., 2004). MyD88−/− mice also develop more severe arthritis than wild type and heterozygous littermates.
The observation that TLR2−/− mice had greatly increased numbers of spirochetes in their ankle tissues suggested that heavy spirochete burden was responsible for the increased severity of arthritis. This result was followed by an experiment designed to assess the relative contribution of acquired and innate host defenses to bacterial control. TLR2−/− mice were crossed with scid mice to develop double mutant mice lacking B and T lymphocytes and non-functional TLR2. These mice harbored even greater numbers of spirochetes than TLR2−/− mice, but displayed suppression of the aggravated arthritis of mice singly deficient in TLR2 (Fig. 2). This finding directly demonstrated that the increased arthritis severity in TLR2−/− mice was not linked to increased bacterial number and, instead, was likely dependent on the recruitment of lymphocytes to the joint tissue (Wang et al., 2005). An additional histopathological feature of the arthritis in TLR2−/− and MyD88−/− mice was an increase in mononuclear cell infiltration, greater than that characteristic of the severe arthritis seen in wild type C3H mice (Bolz et al., 2004; Wang et al., 2005). This led to the hypothesis that the increase in mononuclear cells was due to lymphocytes. This was tested by immunohistochemical staining of cells in the joint tissue and by flow cytometry of cells released from arthritic joints. Both types of analysis indicated a selective increase in T cell infiltration into joint tissue of TLR2 deficient mice, on both the C57BL/6 and C3H backgrounds (Wang et al., 2007), and data not shown (Table 1).
Fig. 2. TLR2−/− mice with the scid mutation (TLR2−/−/scid) harbor increased levels of spirochetes with reduced arthritis severity.
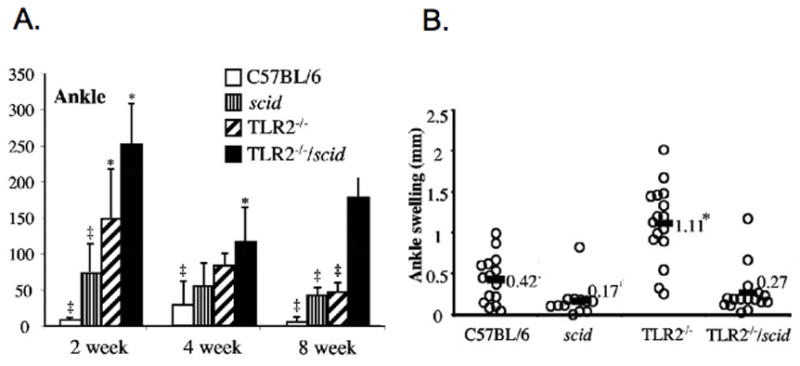
A. Enumeration of B. burgdorferi numbers within the rear ankle joints of wildtype and immunodeficient mice was conducted at 2,4, and 8 weeks post-infection by quantitative PCR. Values are expressed as number of B. burgdorferi genomes/1000 mouse genomes. Statistical significance was assessed by one-way analysis of variance followed by Tamhane post hoc multiple comparison. P < 0.05 was considered significant. * indicates values for immunodeficient mice that were significantly greater versus wildtype mice. ‡ indicates values for wildtype and singly immunodeficient mice that were significantly less than those for TLR2−/−/scid mice. B. Assessment of rear ankle swelling in B. burgdorferi-infected mice. Values obtained at 4 wks post-infection were subtracted from those obtained prior to infection (Yang et al., 1992). Statistical significance was assessed by one-way analysis of variance followed by Tamhane post hoc multiple comparison. P < 0.05 was considered significant. * indicates significantly greater ankle swelling in TLR2−/− mice versus all other mice (Wang et al., 2005). Copyright 2005 American Society for Microbiology, used with permission.
Table 1.
Enhanced T cell infiltration in joints of B. burgdorferi infected TLR2−/− mice
| Cell typea | Mouse strain | Infection Status
|
|
|---|---|---|---|
| Uninfected | B. burgdorferi | ||
| T cells | C3H/HeN | 2.12 × 104 | 7.1 × 104 b |
| TLR2−/− C3H | 1.0 × 104 | 1.8 × 105 b | |
|
| |||
| B cells | C3H/HeN | 5.3 × 104 | 1.4 × 105 |
| TLR2−/− C3H | 1.2 × 104 | 1.6 × 105 | |
Cells were released by collagnease digestion of rear ankle tissues and stained with anti-CD3 and anti-CD19
Values indicate signficant differences (p< 0.05) between wild type and TLR2−/−C3H mice {Wang, 2007 #473}. Copyright 2007 Elsevier, used with permission.
The selective increase in T cells led to an assessment of chemokine induction in ankle tissue from B burgdorferi infected TLR2−/− mice. Chemokines important in the recruitment of macrophages and T cells were increased in joint tissue from TLR2−/− mice, on both the B6 and C3H backgrounds, relative to wild type mice (Wang et al., 2007). The upregulation of the T cell chemokines CXCL9 and CXCL10 was striking, particularly in light of the increased numbers of T cells in joint tissues from these mice. Together, these findings paint a picture of increased T cell recruitment stemming from heightened production of T cell chemokines in TLR2−/− mice. The reduced PMN recruitment in the TLR2−/− scid mice suggests that the T cells were contributing to the recruitment of other inflammatory cells that was missing in TLR2 deficiency, and in fact the PMN recruitment to the joints of infected TLR2−/− mice was similar to that seen in wild-type mice. Induction of the B cell recruiting chemokine CXCL13 was reduced. Importantly, there is not evidence for a direct pathological effect of the enhanced T cells in the joints of TLR2−/− mice, rather, their presence may reflect the dysregulated production of chemokines and could be an indicator of uncontrolled production of other inflammatory products (Wang et al., 2007).
The finding of severe arthritis in TLR2 deficient mice indicated that the in vitro evidence for involvement of TLR2 in inflammatory signaling was not a predictor of a pathway required for arthritis development. Therefore, we felt that further studies on the response of selected inflammatory cells to B. burgdorferi in vitro was unlikely to identify pathways important in arthritis development. This conclusion prompted us to expand the search for TLR2-independent pathways involved in Lyme arthritis development and to incorporate unbiased approaches for identification of such pathways.
Expression profiling during arthritis development
A novel experimental approach was undertaken, using gene expression analysis to globally assess pathways activated at various time points following infection of C3H mice by B. burgdorferi. This process avoided the bias of previous in vitro experiments as the whole joint tissue was assessed, not isolated cell types, with the hypothesis that pathways identified in this manner would be robust. The specificity of the response was demonstrated by comparing gene expression profiles with the mildly arthritic infected C57BL/6 mouse (Crandall et al., 2006). This control is highly relevant and unique for an arthritis model as C57BL/6 mice harbor as many B. burgdorferi in their tissues as C3H mice as early as 1 week of infection, but ultimately develop only mild disease (Ma et al., 1998). Thus, we could compare differential response to an identical infection challenge in mouse strains destined to develop severe versus mild disease (Fig. 3).
Fig. 3. Expression profiling of ankle joint tissue from mice developing differing severities of arthritis.
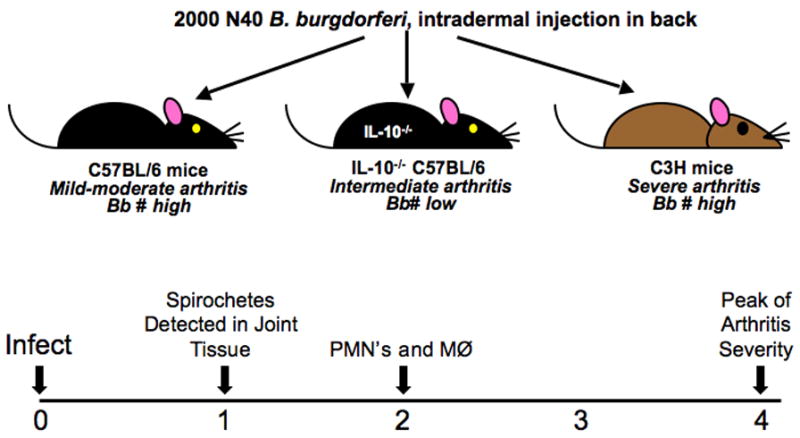
Affymetrix microarray analysis was conducted on cDNA produced from RNA extracted from the rear ankle joints of uninfected, and B. burgdorferi-infected C57BL/6, C57BL/6 IL-10−/−, and C3H mice at 1, 2, and 4 weeks post-infection. Data were reported as the fold change induction or repression relative to the values obtained for uninfected mice. Fold change values of ≥ 2 were considered to be significant (Crandall et al., 2006). C57BL/6 exhibit mild to moderate arthritis, C3H mice develop severe arthritis, and IL-10−/− mice display an arthritis phenotype intermediate to that seen in C3H and C57BL/6 mice. 1 week post-infection represents the earliest timepoint at which spirochetes can be detected in joint tissue, PMNs and macrophage traffic to the joint by 2 weeks, and 4 weeks represents the peak of arthritis severity.
Expression profiles at 1 week
Both C3H and C57BL/6 mice displayed robust induction of transcripts at 1 week of infection, however, only two genes were upregulated greater than 2-fold in both mouse strains. The gene induction profile of C3H mice was IFN inducible whereas an epidermal differentiation profile was identified in C57BL/6 mice (Fig. 4). Upregulation of these profiles were unique to joint tissue, as neither profile was induced in spleens or ears obtained from infected mice (Crandall et al., 2006), and data not shown. This suggests a totally distinct early response occurs within B. burgdorferi-infected joints of mice developing severe arthritis (C3H) compared with mice destined to develop mild disease (C57BL/6). To distinguish arthritis-associated gene induction profiles from genetically determined differences in response to infection in the two mouse strains, a third mouse was studied. IL-10−/− C57BL/6 mice develop more severe arthritis than wild type C57BL/6 mice, thus we thought that genes that were induced in both C3H and IL-10−/− C57BL/6 mice would be selectively linked to the development of severe arthritis. In fact, there was a switch from the C57BL/6 profile to one that shared more upregulated transcripts with C3H mice in the IL-10 deficient C57BL/6 mice (35 vs 3) at 1 wk of infection (Table 2) (Crandall et al., 2006).
Fig. 4. Distinct gene induction profiles were obtained in joint tissue of C3H and C57BL/6 mice at 1 week post- infection.
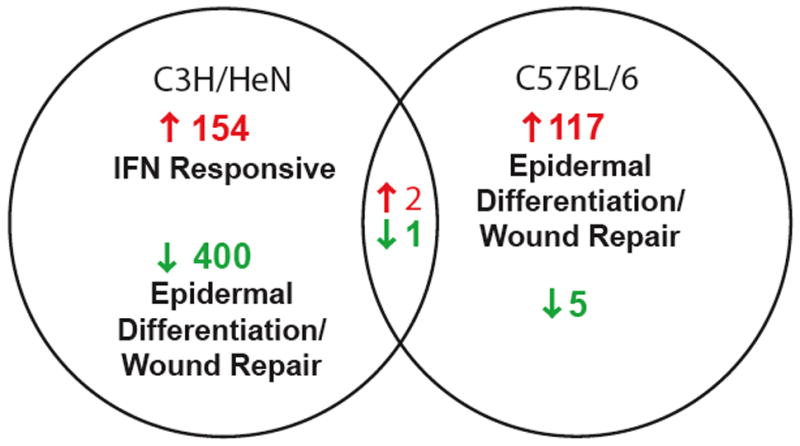
154 of 156 of the most highly induced genes were unique to C3H mice at 1 week post-B. burgdorferi infection and the majority were annotated as IFN-inducible. In contrast, 117 of 119 induced genes were unique to C57BL/6 mice with the majority of these genes annotated as being associated with epidermal differentiation or wound repair. The downregulated genes in C3H mice included many of the genes upregulated in C57BL/6 mice. 6 genes were significantly repressed in C57BL/6 mice. Only two genes exhibited a shared upregulation between C3H and C57BL/6 mice, and 1 gene was commonly downregulated in the two strains (Crandall et al., 2006). Copyright 2006 The American Association of Immunologists, Inc., used with permission.
Table 2.
Selected genes exhibiting significant fold changes in infected ankle joints
| 1 weeka | ||||
|---|---|---|---|---|
| Gene Title | Gene Symbol | C57BL/6 | C3H/HeN | IL10−/− |
| IFN-inducible
| ||||
| interferon gamma induced GTPase | Igtp | NC | 128 | NC |
| interferon inducible GTPase 2 | Iigp2 | NC | 123 | 2.4 |
| interferon inducible GTPase 1 | Iigp1 | NC | 113 | 2.5 |
| T-cell specific GTPase | Tgtp | NC | 42 | 2.5 |
| guanylate nucleotide binding protein 1 | Gbp1 | NC | 31 | NC |
| guanylate nucleotide binding protein 2 | Gbp2 | NC | 12 | 2.8 |
| 2′-5′ oligoadenylate synthetase-like 2 | Oasl2 | NC | 4.4 | 4.2 |
|
| ||||
| Epidermal/Wound Repair
| ||||
| filaggrin | Flg | 47 | −12 | −23 |
| keratin complex 2, basic, gene 1 | Krt2-1 | 38 | −124 | −41 |
| hornerin | Hrnr | 36 | −8.6 | −18 |
| keratin complex 2, basic, gene 17 | Krt2-17 | 24 | −86 | −6.8 |
| loricrin | Lor | 8.5 | −18 | −2.7 |
| elongation of very long chain fatty acids | Elovl4 | 14 | −4.5 | NC |
| keratin complex 2, basic, gene 6a | Krt2-6a | 3.7 | −2.2 | NC |
Gene expression in ankle joint tissue at 4 weeks post infection was compared to uninfected ankle tissue. Numbers indicate fold change. Changes less than 2-fold are designated “NC” (Crandall et al., 2006). Copyright 2006 The American Association of Immunologists, Inc., used with permission.
Identification of the IFN-inducible profile exhibited by C3H mice and the epidermal differentiation/wound repair upregulated in C57BL/6 mice was unexpected. However, key clues concerning spirochetal dissemination to the joints and resultant degree of arthritis development in these different mouse strains were revealed as a consequence of these microarray analyses. First, the presence of the IFN-responsive profile in arthritis-susceptible C3H mice and its absence in arthritis-resistant C57BL/6 mice suggests that the resident cells of the C3H joint may be mounting a potent pro-inflammatory charge in an attempt to eradicate spirochetes arriving in the joint. In support of this idea, many of the epidermal differentiation and wound repair transcripts increased in C57BL/6 mice were significantly decreased in C3H and IL-10 deficient C57BL/6 mice (Crandall et al., 2006), indicating that regulation of these genes is tethered to the degree of inflammation present in the joint and not governed by strain-specific disparities between C3H and C57BL/6 mice. When taken together, these data posit the hypothesis that upregulation of IFN-responsive transcripts by 1 week post-infection provides the impetus for inflammatory cellular trafficking to the joint, thus setting the stage for arthritis development. A corollary to this hypothesis is that triggering of the epidermal profile, and by extension, suppression of the IFN response, reduces the pro-inflammatory status of the joint, contributing to the prevention of Lyme arthritis (Fig. 5).
Fig. 5. The gene induction profile obtained at 1 week post-infection determines the inflammatory events that occur at 2 and 4 weeks post-infection.
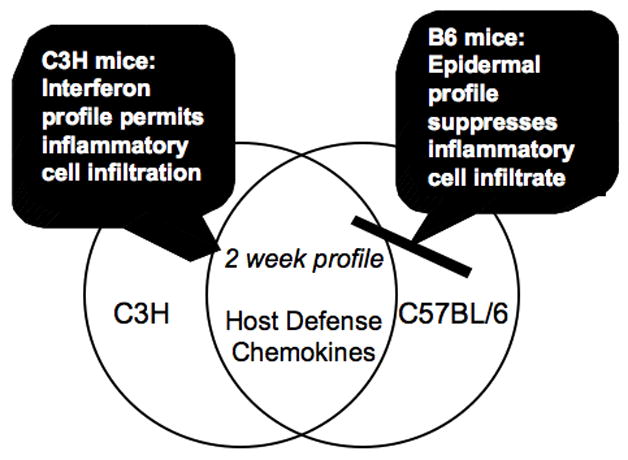
By 2 weeks post-infection, chemokines and host defense-associated genes are induced in both C3H and C57BL/6 mice, providing further evidence that the gene profiles triggered by 1 week post-infection govern the inflammatory status of the joint, which in turn modulates arthritis severity (Crandall et al., 2006).
Expression profiles at 2 weeks: peak of spirochetal burden and maximal induction of host defense genes within ankle joints
Affymetrix microarray analysis was also utilized to identify transcripts within rear ankle joints that were significantly altered as a consequence of B. burgdorferi infection at 2 and 4 weeks post-infection relative to the expression amplitude seen within uninfected joints. By 2 weeks post-infection, the expression profiles exhibited by all three examined mouse strains were dominated by chemokines and genes associated with host defense, as a common core of >200 genes were induced in all three strains (Fig. 5). One characteristic signature of Lyme arthritis is the influx of neutrophils into the joint (Barthold et al., 1990; Barthold et al., 1993), a process represented by the elevation of several PMN-recruiting chemokines. In addition, mononuclear, T, and B cell chemokines were also upregulated in joint tissue. Numerous genes associated with host defense were also induced, including markers for neutrophils, macrophages, complement components, antigen presenting cells, and antigen processing machinery (Crandall et al., 2006). The number of B. burgdorferi within ankle tissues peaks at 2 weeks post-infection (Ma et al., 1998), thus the host defense response at this time point likely reflects the host’s efforts to clear the pathogen. The common induction of chemokines and genes affiliated with host defense in all three mouse strains by 2 weeks post-infection further reinforces the idea that commitment to a severely versus a mildly arthritic pathway occurs very early following B. burgdorferi infection (Fig. 5).
Induction of the epidermal differentiation profile is maintained in C57BL/6 mice at 2 weeks post-infection. In contrast, this profile is suppressed in C3H and IL-10 deficient mice. While the IFN-responsive profile has returned to baseline in C3H mice at this timepoint, it is elevated in IL-10 deficient mice, suggesting that these mice share the IFN-responsive profile with C3H mice but are delayed in its induction (Fig. 6) (Crandall et al., 2006). These results reinforce the idea that upregulation of the IFN profile is associated with the resultant infiltration of pro-inflammatory cells into the joint and the development of severe arthritis, and not due to strain-specific differences in gene expression patterns. Intriguingly, transcripts classically associated with the NF-kB pathway were not significantly induced in ankle joints of C3H or C57BL/6 mice at any timepoint. Expression levels of these cytokines were also low in spleen and ears isolated from B. burgdorferi-infected mice (Crandall et al., 2006), and data not shown. The failure of arthritis-susceptible C3H mice to upregulate genes located downstream of NF-kB, such as the pro-inflammatory cytokines IL-6, IL-12, and TNFα, is consistent with the observation that TLR2 deficient mice still develop severe arthritis. These mice also exhibit a significant defect in host defense, as they harbor large numbers of B. burgdorferi within their joints. This observation suggests that while the NF-kB pathway is dispensable for inflammation, it assumes a critical role in controlling the bacterial burden within the joint (Wooten et al., 2002). However, several key NF-kB dependent cytokines were elevated in IL-10 deficient mice, indicating that the expression of these genes in wildtype C57BL/6 and C3H mice is tightly controlled by IL-10 (Brown et al., 1999; Crandall et al., 2006; Lazarus et al., 2006).
Fig. 6. The phenotypes observed by microarray analysis correlate with the inflammatory status of the joint and not with the mouse strain.
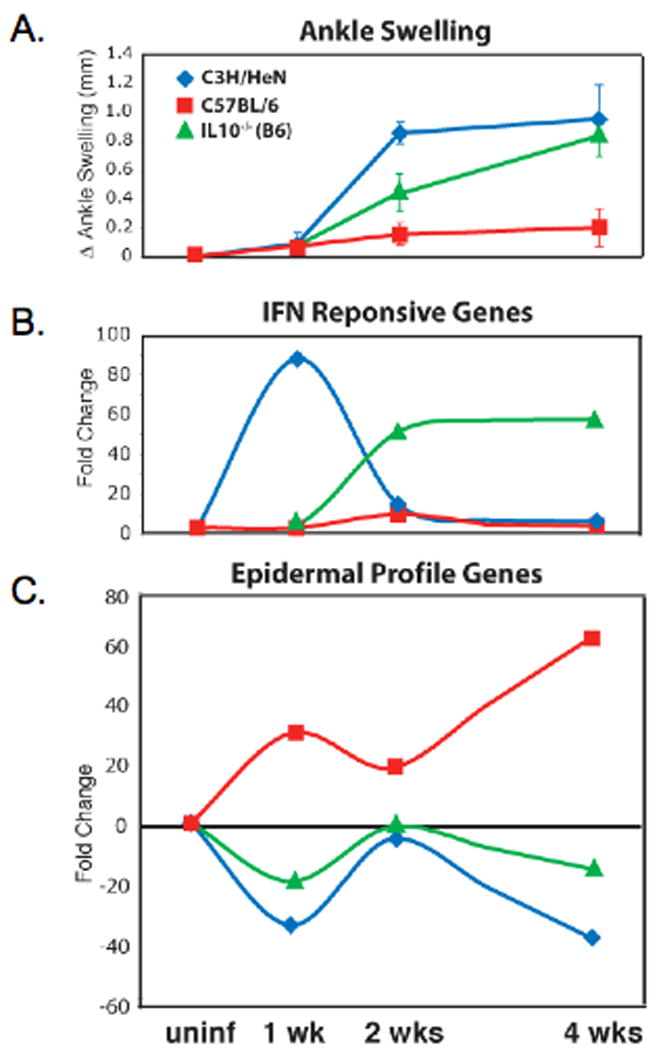
A. Ankle swelling, an indicator of arthritis severity was measured in C3H, C57BL/6, and IL-10−/− mice at 1, 2, and 4 weeks post-B. burgdorferi infection. C3H mice develop severe arthritis, C57BL/6 mice get mild arthritis, and IL-10−/− mice exhibit an intermediate arthritis phenotype. B. The IFN profile induced in C3H mice at 1 week post-infection returns to baseline by 2 weeks after infection, whereas IL-10−/− mice are delayed in induction of this profile, exhibiting upregulation of IFN-responsive genes at 2 weeks post-infection. C. C57BL/6 mice maintain induction of the epidermal profile at 2 weeks post-infection, while this profile remains suppressed in C3H and IL-10−/− mice (Crandall et al., 2006). Copyright 2006 The American Association of Immunologists, Inc., used with permission.
Expression at 4 weeks of infection: similarities of severely arthritic C3H mice with other models of arthritis
By 4 weeks post-infection, microarray analysis revealed the presence of induced genes in C3H mice that are associated with chondrocyte foci formation and new bone development, both of which are hallmarks of the reactive and reparative processes that occur within the microenvironment of the severely arthritic joint. While many of these genes were selectively upregulated in C3H mice, some such as cathespin C were also elevated in the C57BL/6 and IL-10 deficient mice. Several matrix metalloproteinase (Mmp) and tissue-inhibitor of matrix metalloproteinase (Timp) genes were also induced in C3H and IL-10 deficient mice (Table 3). Two such genes, Mmp3 and Timp1, have previously been documented by Behera and colleagues as being elevated both following B. burgdorferi infection of primary human chondrocytes and within synovial fluid samples obtained from Lyme arthritis patients (Behera et al., 2005). Induction of Mmp3 by infected C3H mice was also noted by this group, and confirmed by our microarray analysis (Behera et al., 2005; Crandall et al., 2006). In addition, Gebbia and colleagues previously demonstrated that B. burgdorferi-stimulated monocytes produce MMPs (Gebbia et al., 2004).
Table 3.
Genes induced at 4 wks post-infection in B. burgdorferi-infected ankles that are also upregulated in rodent models of rheumatoid arthritis
| RA Modela |
|||||||
|---|---|---|---|---|---|---|---|
| Gene Title | Gene Symbol | IL-1Ra | HTLV-1 | SCW | CIA | PGIA | AIA |
| Chemokines/Chemokine Receptors
| |||||||
| Chemokine (C-C motif) ligand 2 | Ccl2 | + | + | − | − | + | + |
| Chemokine (C-C motif) ligand 7 | Ccl7 | + | + | − | − | + | − |
| Chemokine (C-C motif) ligand 8 | Ccl8 | + | + | + | − | + | − |
| Chemokine (C-C motif) ligand 9 | Ccl9 | + | + | − | − | + | − |
| Chemokine (C-X-C motif) ligand 1 | Cxcl1 | + | + | − | − | + | − |
| Chemokine (C-X-C motif) ligand 12 | Cxcl12 | + | + | + | + | − | − |
| Chemokine (C-X-C motif) ligand 13 | Cxcl13 | + | + | + | + | + | − |
| Chemokine (C-X-C motif) ligand 14 | Cxcl14 | + | + | + | − | + | − |
| Chemokine (C-X-C motif) ligand 16 | Cxcl16 | + | + | + | + | − | − |
| Chemokine (C-C motif) receptor 2 | Ccr2 | + | + | + | − | + | + |
| Chemokine (C-C motif) receptor 5 | Ccr5 | + | + | + | − | + | + |
|
| |||||||
| MMPs and TIMPs
| |||||||
| matrix metalloproteinase 3 | Mmp3 | + | + | + | + | + | + |
| matrix metalloproteinase 13 | Mmp13 | − | − | − | + | + | +/− |
| tissue inhibitor of metalloproteinase 1 | Timp1 | + | + | + | − | + | + |
|
| |||||||
| Chondrocyte Related
| |||||||
| bone morphogenetic protein 1 | Bmp1 | + | + | + | + | − | − |
| cathepsin C | Ctsc | + | + | + | − | − | + |
| cathepsin K | Ctsk | + | + | + | + | + | +/− |
| procollagen, type III, alpha 1 | Col3a1 | + | + | + | − | + | − |
| procollagen, type V, alpha 1 | Col5a1 | + | + | + | − | + | − |
| procollagen, type V, alpha 2 | Col5a2 | + | + | + | − | + | − |
+ = gene reported to be upregulated in the examined arthritic rodent model by microarray analysis or RT-PCR, or demonstrated to contribute to arthritis develoment by other published experiments. − = gene induction not observed by microarray analysis. +/− = gene induction observed by microarray analysis, but not RT-PCR.
The following references were used to compile this table: B. burgdorferi:(Crandall et al., 2006); IL-1Ra, HTLV-1, and SCW: (Newman and Weiner, 2005), (Fujikado et al., 2006), (Rioja et al., 2005); CIA: (Ibrahim et al., 2002), (Zheng et al., 2005), (Nanki et al., 2005), (De Klerck et al., 2005); PGIA: (Adarichev et al., 2005a); AIA: (Shahrara et al., 2003), (Szekanecz et al., 2000), (Schurigt et al., 2005)
While numerous chemokine transcripts were upregulated by all three examined mouse strains, most exhibited higher fold change values in the C3H or IL-10 deficient mice (Crandall et al., 2006), a result that is consistent with previous reports documenting the induction of neutrophil-recruiting chemokines within arthritic joints of B. burgdorferi-infected mice (Brown et al., 2003; Brown et al., 2004). A notable exception to this, the B cell recruiting chemokine Cxcl13 was more markedly elevated within C57BL/6 ankle joints (Table 3). This is an intriguing observation that warrants further study, since Cxcl13 is a putative diagnostic marker for neuroborreliosis (Narayan et al., 2005; Rupprecht et al., 2005), and increased Cxcl13 expression has been noted in lymphocytoma skin lesions from European Lyme disease patients (Mullegger et al., 2007).
Although the development of subacute Lyme arthritis in mice does not involve MHC alleles (Brown and Reiner, 2000; Yang et al., 1992), B or T cells (Barthold et al., 1992), all of which have been implicated in various animal models of rheumatoid arthritis (RA) (Steere and Glickstein, 2004), the histopathological manifestations of Lyme arthritis shares some key features with RA and certain HLA-DRB1 alleles possessed by individuals with treatment-resistant Lyme arthritis also occur in RA patients (Kalish et al., 1993; Steere et al., 1988; Steere et al., 1990; Steere et al., 2003). For these reasons, some lessons learned from animal models of RA can be applied to Lyme disease, and vice versa. To assess whether any of the genes induced at 4 weeks post-B. burgdorferi infection are shared with rodent models of RA, the L2L microarray analysis tool was utilized. This website developed at the University of Washington is a user-friendly tool that allows researchers to upload their microarray data and determine how the results compare with those obtained from previously published microarray experiments whose data have been painstakingly compiled into one location (Newman and Weiner, 2005). Use of the L2L microarray analysis tool allowed the identification of several chemokines, such as Ccl8 and Cxcl13, that were also upregulated in the streptococcal cell wall (Rioja et al., 2005), IL-1ra deficient mouse, the HTLV-1 tax transgenic (Fujikado et al., 2006), and proteoglycan-induced (PGIA) (Adarichev et al., 2005b) rodent models of RA. The chemokine receptors Ccr2 and Ccr5 were induced in all of these systems (Adarichev et al., 2005b; Fujikado et al., 2006; Rioja et al., 2005), as well as in the rat adjuvant-induced arthritis (AIA) rodent models (Shahrara et al., 2003). Shared MMP and chondrocyte related genes were also noted. For example, increased Mmp3 and bone morphogenetic protein 1 (Bmp1) expression was shared with some of the above-mentioned RA models (Fujikado et al., 2006; Rioja et al., 2005), as well as with the well-characterized collagen-induced arthritis (CIA) model (Ibrahim et al., 2002) (Table 3). When these observations are taken together, they suggest that although Lyme arthritis and RA rodent models exhibit different trigger mechanisms they share common effector molecules.
Implication of the absence of classic NF-kB dependent gene transcripts in arthritis-susceptible C3H mice
Triggering of NF-kB dependent genes has a documented impact on host defense to B. burgdorferi, as IL-10 deficient mice, which induce numerous pro-inflammatory cytokines following infection, exhibit a reduced spirochetal burden in their ankle joints relative to wild-type mice (Brown et al., 1999; Crandall et al., 2006; Lazarus et al., 2006). However, the lack of NF-kB-dependent gene induction in severely arthritic C3H mice suggests regulation of these events and Lyme arthritis development is discrete in wild-type mice. In further support of this idea, TLR2−/− mice on both the C3H and C57BL/6 backgrounds fail to produce pro-inflammatory cytokines and develop severe arthritis when challenged with B. burgdorferi (Wang et al., 2007; Wooten et al., 2002). As mentioned earlier in this review, infected arthritic TLR2−/− ankle joints exhibit an increase in mononuclear cell infiltrate dominated by T cells. Induction of the T lymphocyte-recruiting and IFN-inducible chemokines Cxcl9 and Cxcl10 was also noted (Table 4). Intriguingly, both wild-type C3H and TLR2−/− synovial cells co-cultured with either B. burgdorferi-stimulated T cells or T cell supernatants exhibited an upregulation of several chemokine transcripts, including Cxcl9 and Cxcl10 (Wang et al., 2007). These observations prompted the question: Do TLR2−/− mice display other features of the IFN profile? Examination of other IFN-responsive genes found to be highly induced by our microarray analysis, including the interferon gamma inducible GTPase (Igtp) gene (Table 4) indicated that they were also significantly upregulated in TLR2−/− mice on the C3H background (Wang et al., 2007). Taken together, the data obtained from studies conducted with TLR2−/− mice indicate that induction of the IFN-responsive profile occurs independently of TLR2 signaling, suggesting that regulation of this profile is mediated by one or more genes located downstream of TLR2. The identification of these genes awaits further investigation, but potential candidates include IL-10 or a factor(s) produced by a cell type whose recruitment is dependent on TLR2 signaling.
Table 4.
Interferon-inducible transcripts in ankle tissue from B. burgdorferi -infected mouse ankles
| Interferon-inducible transcripta |
||||
|---|---|---|---|---|
| Infected with Mouse strainc | Cxcl9 | Cxcl10 | Igtp | |
| BSKb | (pooled) | 64 ± 54 | 9 ± 7 | 13.0 ± 7.0 |
| B. burgdorferi | C57BL/6 | 312 ± 136 | 46 ± 25 | 25.1 ± 20.1 |
| C3H/HeN | 475 ± 210 | 56 ± 11 | 60.1 ± 21. 1 | |
| TLR2−/−B6 | 4079 ± 1388d | 704 ± 232d | 174.0 ± 49.3d | |
| TLR2−/−C3H | 4259 ± 2183d | 1321 ± 959d | 225.3 ± 219 | |
RNA was isolated from ankles at 2-week post infection, and RT-PCR was performed. Transcript copy numbers were normalized to 10,000 β-actin.
Transcript copy numbers in mock infected mice (BSK) were similar for all mouse strains, and were pooled to define the basal level for IFN-inducible transcripts.
Each infected group consisted of 5 animals. The BSK group contained 8 mice (2 mice per genotype).
Significantly greater levels were detected in TLR2−/−(B6 and C3H) than in wild type joint tissues (p < 0.05), using One-way ANOVA with Bonferroni as the post test {Wang, 2007 #473}. Copyright 2007 Elsevier, used with permission.
What IFN is responsible for the IFN profile?
The microarray data do not provide a clear-cut indicator of which IFN is governing expression of the IFN-responsive profile as the majority of these genes can be induced by either Type I (IFNα and IFNβ in the mouse) or Type II (IFNγ) IFN. Additionally, elevated transcripts for the IFNα subtypes, IFNβ, and IFNγ genes were not detected in the ankle joints of C3H mice (Crandall et al., 2006). Although IL-10 can inhibit Type I IFN production, its role as a negative regulator of IFNγ is better understood (Grutz, 2005; Moore et al., 2001). However, there are data that argue against involvement of Type II IFN in the induction of this profile. First, a previous study has demonstrated that IFNγ −/− mice on the C3H background still develop Lyme arthritis (Brown and Reiner, 1999a). Secondly, IFNγ is located downstream of TLR2 and therefore would be predicted to be absent in TLR2−/− mice, which still develop severe arthritis. For these reasons and because there are no currently identified signaling pathways linking TLR2 and IFNα/β (Noppert et al., 2007; Uematsu and Akira, 2007), induction of the IFN-responsive profile in TLR2 −/− mice is most consistent with dependence on Type I IFN.
Based on this assumption, the following working model is proposed in an attempt to understand regulation of the IFN-inducible profile and arthritis development in TLR2−/− mice: B. burgdorferi lipoprotein signaling through TLR2 in wild-type animals leads to NF-kB-mediated signaling events that result in the production of pro-inflammatory and anti-inflammatory cytokines. An anti-inflammatory cytokine such as IL-10 inhibits the feedback amplification of Type I IFN, which has been produced as a consequence of signaling through an unknown pathogen-associated molecular pattern (PAMP) receptor. In TLR2−/− mice, pro- and anti-inflammatory cytokines are not produced and feedback inhibition of Type I IFN does not occur. This results in unchecked amplification of the Type I IFN response and the development of severe arthritis (Fig. 7). Several recent studies support a direct association between Type I IFN production and inflammatory pathologies, including the development of arthritis. For example, a number of researchers have established a link between Type I IFN production and systemic lupus erythematosus (SLE) (Blanco et al., 2001; Crow et al., 2003; Mathian et al., 2005; Santiago-Raber et al., 2003). In addition, some patients receiving Type I IFN as a therapeutic intervention for multiple sclerosis or Hepatitis C virus infection have developed arthritis as a result of this treatment regimen (Strueby et al., 2005; Wilson et al., 2002). Future experiments will examine the involvement of other PAMP-mediated signaling events in the induction and control of IFN-responsive genes, and attempt to identify key regulators of this profile.
Fig. 7. In the absence of TLR2 the IFNβ response is unchecked, resulting in exaggerated arthritis.
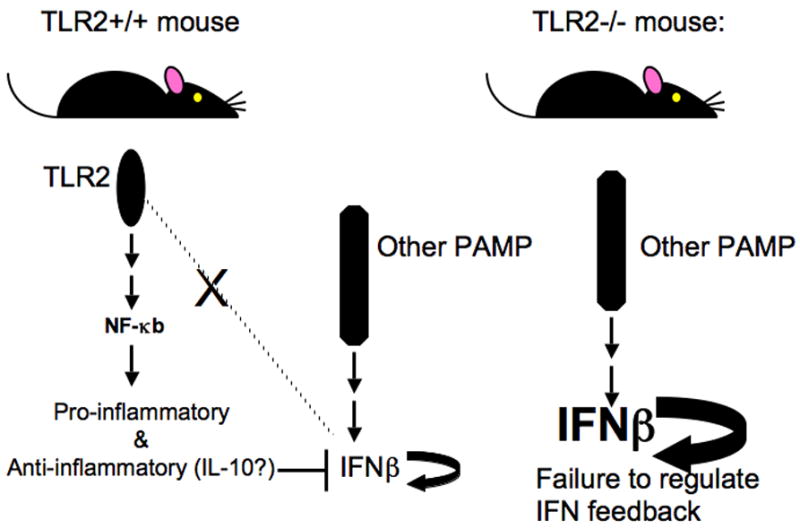
In wild-type animals, B. burgdorferi lipoproteins signal through TLR2, resulting in downstream activation of NF-kB and production of pro-inflammatory and anti-inflammatory cytokines. The presence of anti-inflammatory cytokines (i.e. IL-10) blocks the feedback amplification of Type I IFN, which is present due to PAMP receptor-mediated signaling events. In TLR2−/− mice, the lack of TLR2 signaling prevents cytokine production, resulting in uninhibited amplification of the Type I IFN response and the development of severe arthritis.
Conclusions
The use of Affymetrix microarray analysis resulted in the identification of two previously unidentified and unexpected gene profiles at 1 week post-B. burgdorferi infection: the IFN-inducible profile in the ankle joints of C3H and an epidermal profile in C57BL/6 mice infected with B. burgdorferi. These data provide valuable insight into the regulation of Lyme arthritis development, by providing the provocative idea that induction of IFN-responsive genes in C3H mice sets the stage for the onset of severe arthritis. Differential expression of NF-kB-dependent genes classically associated with inflammation was not seen in these two mouse strains, consistent with the idea that TLR2−/− mice still develop arthritis. Since TLR2−/− mice also contain a high number of bacteria within their joints, the microarray data suggest that the NF-kB pathway is involved in host defense, not inflammation. However, induction of NF-kB-dependent genes was observed in C57BL/6 IL-10−/− mice, indicating that expression of these genes in wild-type mice is tightly controlled by IL-10. By 2 weeks post-infection, part of the IFN-inducible profile was elevated in IL-10−/− mice and the epidermal profile was suppressed. The common phenotypes seen in both the C3H and IL-10−/− mice indicate that development of Lyme arthritis transcends strain background and is instead, linked to the inflammatory state of the joint. Chemokines and host defense-related genes were upregulated in all three strains of mice by 2 weeks post-infection, reinforcing the idea that commitment to a pro-versus anti-arthritic pathway occurs prior to this timepoint. Numerous chemokines, Mmps, and chondrocyte-related genes were induced at 4 weeks post-infection, many of which have also been reported to be elevated in rodent models of RA. The identification of effector molecules that are shared between Lyme arthritis and RA illustrates the broad applicability of our data to other arthritis models. These microarray analyses resulted in the discovery of novel pathways regulating arthritis development in C3H, C57BL/6, and IL-10−/− mice, and prompted us to question whether the IFN-responsive profile is also induced in B. burgdorferi-infected TLR2−/− mice. Upregulation of IFN-inducible genes was observed in TLR2−/− mice, providing new insights into the development of Lyme arthritis in these mice. Our results demonstrate the power of gene expression profiling in revealing novel genes and processes that can dramatically influence our interpretation of disease processes, and illustrate the utility of microarray analysis, when used in combination with gene manipulation models, to mechanistically dissect out portions of regulatory pathways governing disease development.
Acknowledgments
This work was supported by Public Health Services grants AI-32223 and AR-43521 to J.J.W, an Arthritis Foundation fellowship to J.C.M, financial support from the Associated Regional University Pathologists, and by grant 5P30CA42014 to the University of Utah. J.C.M was also supported by the Training Program in Microbial Pathogenesis 5T32-AI055434. H.C. was supported by the National Institute of Diabetes and Digestive and Kidney Diseases training grant DK07115. We would like to thank John Weis, Kirstin Roundy, and the members of the Weis laboratory for insightful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adarichev VA, et al. Gene expression profiling in murine autoimmune arthritis during the initiation and progression of joint inflammation. Arthritis Res Ther. 2005a;7:R196–207. doi: 10.1186/ar1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adarichev VA, et al. Gene expression profiling in murine autoimmune arthritis during the initiation and progression of joint inflammation. Arthritis Res Ther. 2005b;7:R196–R207. doi: 10.1186/ar1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliprantis AO, et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- Barthold SW, et al. Lyme borreliosis in selected strains and ages of laboratory mice. Journal of Infectious Diseases. 1990;162:133–138. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- Barthold SW, et al. Chronic Lyme borreliosis in the laboratory mouse. Am J Pathol. 1993;143:959–971. [PMC free article] [PubMed] [Google Scholar]
- Barthold SW, et al. Kinetics of Borrelia burgdorferi dissemination and evolution of disease after intradermal inoculation of mice. American Journal of Pathology. 1991;139:263–273. [PMC free article] [PubMed] [Google Scholar]
- Barthold SW, et al. Lyme borreliosis in genetically resistant and susceptible mice with severe combined immunodeficiency. Am J Trop Med Hyg. 1992;47:605–613. doi: 10.4269/ajtmh.1992.47.605. [DOI] [PubMed] [Google Scholar]
- Behera AK, et al. Induction of host matrix metalloproteinases by Borrelia burgdorferi differs in human and murine lyme arthritis. Infect Immun. 2005;73:126–134. doi: 10.1128/IAI.73.1.126-134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera AK, et al. Identification of a TLR-independent pathway for Borrelia burgdorferi-induced expression of matrix metalloproteinases and inflammatory mediators through binding to integrin alpha 3 beta 1. J Immunol. 2006;177:657–664. doi: 10.4049/jimmunol.177.1.657. [DOI] [PubMed] [Google Scholar]
- Benhnia MR, et al. Signaling through CD14 attenuates the inflammatory response to Borrelia burgdorferi, the agent of Lyme disease. J Immunol. 2005;174:1539–1548. doi: 10.4049/jimmunol.174.3.1539. [DOI] [PubMed] [Google Scholar]
- Blanco P, et al. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- Bolz DD, et al. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J Immunol. 2004;173:2003–2010. doi: 10.4049/jimmunol.173.3.2003. [DOI] [PubMed] [Google Scholar]
- Brown CR, et al. Susceptibility to experimental Lyme arthritis correlates with KC and monocyte chemoattractant protein-1 production in joints and requires neutrophil recruitment via CXCR2. J Immunol. 2003;171:893–901. doi: 10.4049/jimmunol.171.2.893. [DOI] [PubMed] [Google Scholar]
- Brown CR, et al. Treatment of mice with the neutrophil-depleting antibody RB6-8C5 results in early development of experimental lyme arthritis via the recruitment of Gr-1- polymorphonuclear leukocyte-like cells. Infect Immun. 2004;72:4956–4965. doi: 10.1128/IAI.72.9.4956-4965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Reiner SL. Clearance of Borrelia burgdorferi may not be required for resistance to experimental lyme arthritis. Infect Immun. 1998;66:2065–2071. doi: 10.1128/iai.66.5.2065-2071.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Reiner SL. Experimental lyme arthritis in the absence of interleukin-4 or gamma interferon. Infect Immun. 1999a;67:3329–3333. doi: 10.1128/iai.67.7.3329-3333.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Reiner SL. Genetic control of experimental lyme arthritis in the absence of specific immunity. Infect Immun. 1999b;67:1967–1973. doi: 10.1128/iai.67.4.1967-1973.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Reiner SL. Genes outside the major histocompatibility complex control resistance and susceptibility to experimental Lyme arthritis. Med Microbiol Immunol (Berl) 2000;189:85–90. doi: 10.1007/s004300000044. [DOI] [PubMed] [Google Scholar]
- Brown JP, et al. Dual Role of Interleukin-10 in Murine Lyme Disease: Regulation of Arthritis Severity and Host Defense. Infect Immun. 1999;67:5142–5150. doi: 10.1128/iai.67.10.5142-5150.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorfer W, et al. Lyme disease-a tick-borne spirochetosis? Science. 1982;216:1317–1319. doi: 10.1126/science.7043737. [DOI] [PubMed] [Google Scholar]
- Crandall H, et al. Gene expression profiling reveals unique pathways associated with differential severity of Lyme arthritis. J Immunol. 2006;177:7930–7942. doi: 10.4049/jimmunol.177.11.7930. [DOI] [PubMed] [Google Scholar]
- Crow MK, et al. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. 2003;36:481–490. doi: 10.1080/08916930310001625952. [DOI] [PubMed] [Google Scholar]
- De Klerck B, et al. Pro-inflammatory properties of stromal cell-derived factor-1 (Cxcl12) in collagen-induced arthritis. Arthritis Res Ther. 2005;7:R1208–R1220. doi: 10.1186/ar1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikado N, et al. Identification of arthritis-related gene clusters by microarray analysis of two independent mouse models for rheumatoid arthritis. Arthritis Res Ther. 2006;8:R100–R112. doi: 10.1186/ar1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebbia JA, et al. Selective induction of matrix metalloproteinases by Borrelia burgdorferi via toll-like receptor 2 in monocytes. J Infect Dis. 2004;189:113–119. doi: 10.1086/380414. [DOI] [PubMed] [Google Scholar]
- Grutz G. New insights into the molecular mechanism of interleukin-10-mediated immunosuppression. J Leukoc Biol. 2005;77:3–15. doi: 10.1189/jlb.0904484. [DOI] [PubMed] [Google Scholar]
- Hayes EB, Piesman J. How can we prevent Lyme disease? N Engl J Med. 2003;348:2424–2430. doi: 10.1056/NEJMra021397. [DOI] [PubMed] [Google Scholar]
- Hirschfeld M, et al. Cutting edge: inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by toll-like receptor 2. J Immunol. 1999;163:2382–2386. [PubMed] [Google Scholar]
- Ibrahim SM, et al. Gene-expression profile of collagen-induced arthritis. J Autoimmun. 2002;18:159–167. doi: 10.1006/jaut.2001.0580. [DOI] [PubMed] [Google Scholar]
- Kalish RA, et al. Association of treatment-resistant chronic Lyme arthritis with HLA-DR4 and antibody reactivity to OspA and OspB of Borrelia burgdorferi. Infect Immun. 1993;61:2774–2779. doi: 10.1128/iai.61.7.2774-2779.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus JJ, et al. IL-10 deficiency promotes increased Borrelia burgdorferi clearance predominantly through enhanced innate immune responses. J Immunol. 2006;177:7076–7085. doi: 10.4049/jimmunol.177.10.7076. [DOI] [PubMed] [Google Scholar]
- Lien E, et al. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem. 1999;274:33419–33425. doi: 10.1074/jbc.274.47.33419. [DOI] [PubMed] [Google Scholar]
- Liu N, et al. Myeloid differentiation antigen 88 deficiency impairs pathogen clearance but does not alter inflammation in Borrelia burgdorferi-infected mice. Infect Immun. 2004;72:3195–3203. doi: 10.1128/IAI.72.6.3195-3203.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, et al. Distinct characteristics of resistance to Borrelia burgdorferi-induced arthritis in C57BL/6N mice. Infection & Immunity. 1998;66:161–168. doi: 10.1128/iai.66.1.161-168.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathian A, et al. IFN-alpha induces early lethal lupus in preautoimmune (New Zealand Black × New Zealand White) F1 but not in BALB/c mice. J Immunol. 2005;174:2499–2506. doi: 10.4049/jimmunol.174.5.2499. [DOI] [PubMed] [Google Scholar]
- Moore KW, et al. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Mullegger RR, et al. Chemokine signatures in the skin disorders of Lyme borreliosis in Europe: predominance of CXCL9 and CXCL10 in erythema migrans and acrodermatitis and CXCL13 in lymphocytoma. Infect Immun. 2007;75:4621–4628. doi: 10.1128/IAI.00263-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanki T, et al. Pathogenic role of the Cxcl16-Cxcr6 pathway in rheumatoid arthritis. Arthritis Rheu. 2005;52:3004–3014. doi: 10.1002/art.21301. [DOI] [PubMed] [Google Scholar]
- Narayan K, et al. The nervous system as ectopic germinal center: CXCL13 and IgG in lyme neuroborreliosis. Ann Neurol. 2005;57:813–823. doi: 10.1002/ana.20486. [DOI] [PubMed] [Google Scholar]
- Newman JC, Weiner AM. L2L: a simple tool for discovering the hidden significance in microarray expression data. Genome Biol. 2005;6:R81.81–R81.18. doi: 10.1186/gb-2005-6-9-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocton JJ, Steere AC. Lyme disease. Advances in Internal Medicine. 1995;40:69–117. [PubMed] [Google Scholar]
- Noppert SJ, et al. The role of type I interferons in TLR responses. Immunol Cell Biol. 2007;85:446–457. doi: 10.1038/sj.icb.7100099. [DOI] [PubMed] [Google Scholar]
- Norgard MV, et al. Activation of human monocytic cells by Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides proceeds via a pathway distinct from that of lipopolysaccharide but involves the transcriptional activator NF-kappa B. Infect Immun. 1996;64:3845–3852. doi: 10.1128/iai.64.9.3845-3852.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioja I, et al. Gene expression profiles in the rat streptococcal cell wall-induced arthritis model identified using microarray analysis. Arthritis Res Ther. 2005;7:R101–R117. doi: 10.1186/ar1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RJ, et al. Genetic control of susceptibility to experimental Lyme arthritis is polygenic and exhibits consistent linkage to multiple loci on chromosome 5 in four independent mouse crosses. Genes Immun. 2001;2:388–397. doi: 10.1038/sj.gene.6363801. [DOI] [PubMed] [Google Scholar]
- Rosa PA, et al. The burgeoning molecular genetics of the Lyme disease spirochaete. Nat Rev Microbiol. 2005;3:129–143. doi: 10.1038/nrmicro1086. [DOI] [PubMed] [Google Scholar]
- Rupprecht TA, et al. The chemokine CXCL13 (BLC): a putative diagnostic marker for neuroborreliosis. Neurology. 2005;65:448–450. doi: 10.1212/01.wnl.0000171349.06645.79. [DOI] [PubMed] [Google Scholar]
- Santiago-Raber ML, et al. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003;197:777–788. doi: 10.1084/jem.20021996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurigt U, et al. Local expression of matrix metalloproteinases, cathepsins, and their inhibitors during the development of murine antigen-induced arthritis. Arthritis Res Ther. 2005;7 doi: 10.1186/ar1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellati TJ, et al. Activation of human monocytic cells by Borrelia burgdorferi and Treponema pallidum is facilitated by CD14 and correlates with surface exposure of spirochetal lipoproteins. J Immunol. 1999;163:2049–2056. [PubMed] [Google Scholar]
- Shahrara S, et al. Chemokine receptor expression and in vivo signaling pathways in the joints of rats with adjuvant-induced arthritis. Arthritis Rheum. 2003;48:3568–3583. doi: 10.1002/art.11344. [DOI] [PubMed] [Google Scholar]
- Steere AC, et al. Spirochetal antigens and lymphoid cell surface markers in Lyme synovitis. Arthritis Rheum. 1988;31:487–495. doi: 10.1002/art.1780310405. [DOI] [PubMed] [Google Scholar]
- Steere AC, et al. Association of chronic Lyme arthritis with HLA-DR4 and HLA-DR2 alleles. N Engl J Med. 1990;323:219–223. doi: 10.1056/NEJM199007263230402. [DOI] [PubMed] [Google Scholar]
- Steere AC, et al. Binding of outer surface protein A and human lymphocyte function-associated antigen 1 peptides to HLA-DR molecules associated with antibiotic treatment-resistant Lyme arthritis. Arthritis Rheum. 2003;48:534–540. doi: 10.1002/art.10772. [DOI] [PubMed] [Google Scholar]
- Steere AC, Glickstein L. Elucidation of Lyme arthritis. Nat Rev Immunol. 2004;4:143–152. doi: 10.1038/nri1267. [DOI] [PubMed] [Google Scholar]
- Steere AC, et al. The clinical evolution of Lyme arthritis. Ann Intern Med. 1987;107:725–731. doi: 10.7326/0003-4819-107-5-725. [DOI] [PubMed] [Google Scholar]
- Strueby L, et al. Arthritis and bursitis in multiple sclerosis patients treated with interferon-beta. Scand J Rheumatol. 2005;34:485–488. doi: 10.1080/03009740510026805. [DOI] [PubMed] [Google Scholar]
- Szekanecz Z, et al. Temporal expression of inflammatory cytokines and chemokines in rat adjuvant-induced arthritis. Arthritis Rheu. 2000;43:1266–1277. doi: 10.1002/1529-0131(200006)43:6<1266::AID-ANR9>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Uematsu S, Akira S. Toll-like receptors and type I interferons. J Biol Chem. 2007;282:15319–15323. doi: 10.1074/jbc.R700009200. [DOI] [PubMed] [Google Scholar]
- Wang X, et al. Relative contributions of innate and acquired host responses to bacterial control and arthritis development in Lyme disease. Infect Immun. 2005;73:657–660. doi: 10.1128/IAI.73.1.657-660.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. T cell infiltration is asoociated with increased Lyme arthritis in TLR2−/− mice. FEMS Immunol Med Microbiol. 2007 doi: 10.1111/j.1574-695X.2007.00356.x. In Press. [DOI] [PubMed] [Google Scholar]
- Weis JJ, et al. Identification of quantitative trait loci governing arthritis severity and humoral responses in the murine model of Lyme disease. J Immunol. 1999;162:948–956. [PubMed] [Google Scholar]
- Wilson LE, et al. Autoimmune disease complicating antiviral therapy for hepatitis C virus infection. Semin Arthritis Rheum. 2002;32:163–173. doi: 10.1053/sarh.2002.37277. [DOI] [PubMed] [Google Scholar]
- Wooten RM, et al. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. Journal of immunology. 2002;168(1):348–355. doi: 10.4049/jimmunol.168.1.348. [DOI] [PubMed] [Google Scholar]
- Wooten RM, et al. The role of CD14 in signaling mediated by outer membrane lipoproteins of Borrelia burgdorferi. Journal of immunology (Baltimore, Md.: 1950) 1998;160(11):5485–5492. [PubMed] [Google Scholar]
- Wooten RM, Weis JJ. Host-pathogen interactions promoting inflammatory Lyme arthritis: use of mouse models for dissection of disease processes. Cur Opin Microbiol. 2001;4:274–279. doi: 10.1016/s1369-5274(00)00202-2. [DOI] [PubMed] [Google Scholar]
- Yang L, et al. Evidence for B-lymphocyte mitogen activity in Borrelia burgdorferi-infected mice. Infect Immun. 1992;60:3033–3041. doi: 10.1128/iai.60.8.3033-3041.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, et al. Cxcl13 neutralization reduces the severity of collagen-induced arthritis. Arthritis Rheu. 2005;52:620–626. doi: 10.1002/art.20768. [DOI] [PubMed] [Google Scholar]