

Abstract
Upon maturation of the human immunodeficiency virus type 1 (HIV-1) virion, proteolytic cleavage of the Gag precursor protein by the viral protease is followed by morphological changes of the capsid protein p24, which will ultimately transform the virus core from an immature spherical to a mature conical structure. Virion infectivity is critically dependent on the optimal semistability of the capsid cone structure. We have reported earlier that glycineamide (G-NH2), when added to the culture medium of infected cells, inhibits HIV-1 replication and that HIV-1 particles with aberrant core structures were formed. Here we show that it is not G-NH2 itself but a metabolite thereof, α-hydroxy-glycineamide (α-HGA), that is responsible for the antiviral activity. We show that α-HGA inhibits the replication of clinical HIV-1 isolates with acquired resistance to reverse transcriptase and protease inhibitors but has no effect on the replication of any of 10 different RNA and DNA viruses. α-HGA affected the ability of the HIV-1 capsid protein to assemble into tubular or core structures in vitro and in vivo, probably by binding to the hinge region between the N- and C-terminal domains of the HIV-1 capsid protein as indicated by matrix-assisted laser desorption ionization-mass spectrometry results. As an antiviral compound, α-HGA has an unusually simple structure, a pronounced antiviral specificity, and a novel mechanism of antiviral action. As such, it might prove to be a lead compound for a new class of anti-HIV substances.
There are more than 20 approved antiretroviral drugs for the treatment of human immunodeficiency virus (HIV)-infected patients (6). The therapy is often individualized and involves a combination of different classes of inhibitors of the two viral enzymes reverse transcriptase and protease. Other classes of drugs recently introduced in combination therapy include fusion inhibitors, entry inhibitors, and an integrase strand transfer inhibitor. The introduction of antiretroviral therapy has significantly improved the prognosis of HIV type 1 (HIV-1)-infected patients with sustained reduction in viral load leading to reduced morbidity and mortality rates (14). However, the widespread use of anti-HIV drugs is accompanied by the appearance of drug resistance, and it has become more common for people who contract HIV to be infected with a multiclass drug-resistant strain (12, 27). In a recent study from 39 separate HIV treatment centers in 10 U.S. cities, ca. 8% of the virus strains isolated from the patients prior to initiating antiviral treatment had one or more drug-resistant mutations of HIV (26). In some areas where antiretroviral therapy is widely used (such as in San Francisco), the prevalence of patients infected with drug-resistant HIV strains is even higher (4, 8). This is also the case in some parts of Europe (28), where the transmission of drug-resistant HIV has shown a steady increase since 1994. In the United Kingdom, for example, more than 20% of people newly infected with HIV are affected by this problem (15). These findings underscore the need for new classes of drugs with a different mechanism of antiviral action than the currently available drugs (9, 18). A possible target for antiviral therapy is the viral capsid (CA/p24) protein (9, 18).
We have earlier shown that the addition of glycyl-prolyl-glycine amide (GPG-NH2) or glycineamide (G-NH2) to the culture medium of infected cells abrogated HIV-1 replication and proper capsid formation (2, 3, 10, 20, 21). We have now defined the active antiviral metabolite of GPG-NH2/G-NH2 (data not shown) to be α-hydroxy-glycineamide (α-HGA). We show here that α-HGA specifically affects HIV-1 replication and that its antiviral activity is retained against clinical HIV-1 isolates which have acquired resistance to other antiretroviral drugs. We also show that α-HGA affects HIV-1 capsid assembly probably by binding to the hinge region between the N- and C-terminal domains of the p24 capsid protein.
MATERIALS AND METHODS
Cells, media, and reagents.
Peripheral blood mononuclear cells (PBMC) and H9 and ACH-2 cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum and antibiotics. HeLa-tat cells were cultured in Dulbecco modified Eagle medium supplemented with 10% serum and antibiotics. HeLa-tat cells were transfected in six-well culture plates by using the nonliposomal transfection reagent FuGENE6 (Roche) as recommended by the manufacturer. G-NH2 and α-HGA (custom ordered from Pharmatory Oy, Oulu, Finland) were kindly provided by Tripep AB, Stockholm, Sweden.
Cell toxicity assay.
PBMC (600,000 cells/ml) and HeLa-tat, H9, L1210, FM3A, Molt4/C8, and CEM cells (200,000 cells/ml) were cultured with 25, 100, 250, 500, or 1,000 μM α-HGA for 14 days. Viable and dead cells were counted in a Bürcher chamber after staining with trypan blue solution (0.4%; Sigma, St. Louis, MO).
For testing possible effect on cell proliferation, PBMC (600,000 cells/ml) were cultured for 96 h in RPMI 1640 medium supplemented with 10% inactivated fetal calf serum, antibiotics, and phytohemagglutinin (PHA; 2.5 μg/ml). The cells were treated with α-HGA at different concentrations (10, 50, 100, 250, 500, and 2,000 μM), and [3H]thymidine (1 μCi/100,000 cells; Amersham Pharmacia Biotech, Freiburg, Germany) was added to the cultures 8 h before termination. Lysed cells were harvested on a nitrocellulose filter, and the radioactivity was recorded (in counts per minute) on a β-scintillation counter (Direct Beta Counter; Packard Instrument Company, Meriden, CT). Untreated cells and cells treated with the immunosuppressive agent cyclosporine (1 μg/ml; Sandimmun, Novartis, Täby, Sweden) served as controls.
Anti-HIV activity of α-HGA against clinical HIV-1 isolates.
Stocks of clinical HIV-1 isolates were prepared by collecting the supernatants from infected cultures of PBMC. The 50% tissue culture infective dose (TCID50) was determined by infecting PBMC with serial, 10-fold dilutions of the virus. PBMC (600,000 cells/ml) were infected with clinical isolates at 100 TCID50 and cultured in medium containing various concentrations (1, 4, 16, 64, and 256 μM) of α-HGA. Infected cultures without test compound served as controls, and each isolate was plated in duplicate. The medium was changed at days 5 to 7 postinfection, and the supernatants were monitored for their p24 antigen contents until the termination of incubation at day 14. To determine coreceptor usage, each isolate was cultured in MT2 cells (500,000 cells/ml). Syncytium formation indicates CXCR4 usage, whereas no cytopathic effect indicates CCR5 usage.
PCR and sequencing of drug-resistant clinical HIV-1 isolates.
To characterize the mutations in all clinical isolates, nested reverse transcription-PCR was used to amplify the reverse transcriptase and protease genes. Sequencing was performed with an ABI Prism 310 genetic analyzer. Drug resistance-related mutations were defined according to European guidelines (25). Clade affiliation for each clinical isolate was determined based on the reverse transcriptase and protease genes, using the Stanford University HIV Drug Resistance Database.
Antiviral activity of α-HGA against different RNA and DNA viruses.
The antiviral activity of α-HGA was further investigated against several different viruses with different sizes and genomes. These included viruses with negative-sense single-stranded RNAs (ssRNAs; vesicular stomatitis virus, respiratory syncytial virus, and parainfluenza virus), positive-sense ssRNAs (coxsackievirus B4 and Sindbis virus), double-stranded DNAs (herpes simplex virus types 1 and 2, vaccinia virus, cytomegalovirus, and varicella-zoster virus), and positive-sense double-stranded RNA (reovirus 1) and a virus with an ambisense ssRNA genome (Punta Toro virus). The antiviral assays were based on inhibition of virus-induced cytopathicity in HEL (for herpes simplex virus type 1 [KOS], herpes simplex virus type 2 [G], vaccinia virus, cytomegalovirus, varicella-zoster virus, and vesicular stomatitis virus), Vero (for parainfluenza virus 3, reovirus 1, Sindbis virus, coxsackievirus B4, and Punta Toro virus), or HeLa (for vesicular stomatitis virus, coxsackievirus B4, and respiratory syncytial virus) cell cultures. Confluent cell cultures in microtiter 96-well plates were inoculated with 100 TCID50 of virus. After a 1-h virus adsorption period, residual virus was removed, and the cell cultures were incubated in the presence of various concentrations (fivefold dilutions) of the test compound. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with α-HGA.
Enzyme-linked immunosorbent assay.
p24 enzyme-linked immunosorbent assay of infected PBMC was performed essentially as described elsewhere (11). Briefly, supernatants were added to microwell plates (MWPs) coated with anti-24 antibody and incubated at 37°C for 1 h. The MWPs were washed three times, and biotinylated anti-p24 antibody (1:1,500) was added. One hour after incubation, the MWPs were washed and incubated with horseradish peroxidase-conjugated streptavidin (1:2,000) for 30 min. Finally, the MWPs were washed and detected by adding the substrate o-phenylenediamine dihydrochloride (Sigma). Recombinant p24 at fixed concentrations was used as a standard. The plates were read in a Multiscan MS spectrometer (Labsystems).
TEM.
Transmission electron microscopy (TEM) analysis was performed with H9 cells infected with the HIV-1 SF-2, HeLa-tat cells transfected with the expression plasmid pC37M that expresses a matrix (MA/p17) and capsid (CA/p24) protein, and purified p24 that were assembled in vitro into tubular structures.
Enumeration of various types of virions produced from infected H9 cells was performed.
Cells were infected with SF-2 at 100 TCID50 by incubation for 2 h at 37°C. The cells were pelleted, washed, and resuspended in complete RPMI medium with or without α-HGA. Cells were cultured for 11 days, and growth medium was changed 7 days postinfection. The cells were fixed freshly upon embedding in Epon resin, essentially as described before (10). Sections were made approximately 60 nm thick to allow accommodation of the volume of the core structure parallel to the section plane. Duplicate samples were used, and a minimal beam dose technique was used throughout. Evaluation of morphology was done with a series of electron micrographs to depict different categories of virus morphology. Numerical analysis of 439 untreated and 401 α-HGA-treated virus particles was done to categorize various forms of the virus core morphology.
For in vivo (intracellular) p24 assembly, HeLa-tat cells (105) were seeded in single-culture plates 1 day before and transfected with the pC37M expression plasmid using the nonliposomal FuGENE6 transfection reagent (Roche). At 48 to 72 h posttransfection, the cells were fixed with freshly prepared 2.5% glutaraldehyde and prepared for TEM essentially as described before (1).
In addition, TEM analysis of in vitro-assembled tubular structures (see below) was performed essentially as described above. Briefly, in vitro-assembled p24 tubular structures were negatively stained with 2% ammonium molybdate at pH 8.0 to study possible polymerization.
In vitro HIV-1 p24 assembly.
A turbidity assay was also used to study possible in vitro polymerization of p24. Polymerization of p24 was then monitored spectrophotometrically, as the rate of p24 tube formation increases sample turbidity. The assay was performed at room temperature using a BioSpec-1601E spectrometer (Shimadzu), and the absorbance was set to a 350-nm wavelength. Purified p24 (200 μM) was mixed with various concentrations of α-HGA (5, 50, and 100 μM) and 50 mM sodium phosphate buffer (pH 8.0). Tubular p24 assembly was then induced by the addition of 2.0 M NaCl solution, and absorbance measurements were made every 10 s for up to 60 min. The assembly rate was then set by plotting the absorbance versus time.
Virus purification.
ACH-2 cells chronically infected with the HIV-LAI or HeLa-tat cells were transfected with the infectious HIV-1 clone pNL4-3 were cultured in medium containing various concentrations of α-HGA (10, 20, 50, and 100 μM), and then ACH-2 cells were stimulated with PHA (2.5 μg/ml) for virus production. Untreated cells were used as controls. At 48 h poststimulation, cells were harvested, and virus particles in the culture supernatants were purified as follows. Equal amounts of culture supernatants, untreated or treated with α-HGA, were clarified from cell debris by centrifugation, filtered, and mixed (4:1) with Viraffinity (CPG). The viral particles were then purified as recommended by the manufacturer, resuspended in sodium dodecyl sulfate (SDS) sample buffer, and subjected to SDS-polyacrylamide gel electrophoresis.
Immunoblotting.
Denatured whole-cell lysates and Viraffinity-purified viruses of α-HGA-treated and untreated HeLa-tat cells transfected with the infectious HIV-1 clone pNL4-3 or stimulated ACH-2 cell cultures were resolved by SDS-polyacrylamide gel electrophoresis in a 10 to 15% gradient gel, transferred electrophoretically to a nitrocellulose membrane, and detected with HIV-1-positive human sera.
MALDI-MS.
Recombinant proteins of the C-terminal p24 domain of pNL4-3 (amino acids 151 to 231), wild-type p24, and three other single amino acid substitution p24 mutants (D51N, D51E, and D51Q) and a p24 carrying a double mutation (7) at the C-terminal p24 dimer interface (W184A+M185A) were incubated without or with 100 μM α-HGA at 37°C for 24 h. After incubation, enzymatic cleavage was performed on each sample by using trypsin. The obtained peptide mixtures were then analyzed by matrix-assisted laser desorption ionization-mass spectrometry (MALDI-MS) scanning the mass range between 600 and 3,000 Da, covering a large section of the theoretical masses of p24 peptide fragments.
All tandem MS (MS/MS) data from the MALDI-time of flight (TOF)/TOF instrument was acquired by using the default 1-kV MS/MS method. MS/MS data acquisition from the ABI MALDI target plate was performed in a two-step process. First, MS spectra (peptide mass fingerprint) were recorded from each sample position, and each spectrum was generated by accumulating the data from 2,000 laser shots, while at the same time an internal calibration of the mass spectrum was performed using the autodigested fragments of trypsin, i.e., m/z 842.50 and m/z 2,211.01. Second, MS/MS was performed in a manual precursor selection by analyzing the acquired MS spectra. During MS/MS data acquisition, 2,500 shots (20 subspectra accumulated from 125 laser shots each) were accumulated for each spectrum, 30 ppm was used as the error tolerance in MS mode, and 0.1 Da was used for MS/MS fragments. The experiments were repeated three times, resulting in identical signal patterns.
RESULTS
Anti-HIV activities of α-HGA on clinical HIV-1 isolates.
The antiviral activity of α-HGA was tested on 15 clinical HIV-1 isolates representing clades A, B, C, and CRF01-AE of both CCR5 and CXCR4 coreceptor usage (Table 1). All isolates had acquired resistance against other antiretroviral drugs, as determined by sequencing. Fourteen isolates had multiple mutations (average, 6.6 mutations) with resistance to nucleoside analogue reverse transcriptase inhibitors (NRTIs; 11 isolates) and/or non-NRTIs (1 isolate), as well as protease inhibitors (8 isolates; see Table 1), and one isolate had a single NRTI mutation (M230S). The replication in PBMC of all of these clinical HIV-1 isolates was inhibited by α-HGA. The 50% effective concentration (EC50) values ranged from 4.2 to 34 μM, with a geometric mean value of 14 μM. From repeated experiments of the same isolate, the variation seen here between different isolates was most probably due to test to test variations and not to actual differences in sensitivity to the drug. Two representative inhibition curves for clinical isolates HI99-58 and HI98-100 are shown in Fig. 1A and B. For comparison, the susceptibility to α-HGA of the laboratory strain HIV-1IIIB is also shown (Fig. 1C). The EC50 values for laboratory strains SF2, pNL4-3, and HIV-1IIIB tested in continuous cell lines were repeatedly between 1 and 8 μM (data not shown). For the clinical isolates tested in PBMC, higher variations in EC50 values were observed when the same isolate was tested on different occasions, probably due to inherent intertest variations in the assay (data not shown).
TABLE 1.
Sensitivity to α-HGA of clinical HIV-1 isolates
| Isolate | Resistance mutation(s)a | Clade | CPE in MT2b | EC50 (μM α-HGA)c |
|---|---|---|---|---|
| HI90-79 | RT: M230V; P: M36I, L63P, A71T, I93L | B | Neg | 24 |
| HI90-317 | RT: M41L, T215Y; P: M36I, L63P, A71T, I93L | B | ND | 20 |
| HI93-49 | RT:L74V, V106I; P: L63P | B | Neg | 15 |
| HI93-124 | RT: N67D, T69D, K70R, T215F, K219Q; P: L63P | B | Pos | 33 |
| HI94-18 | RT: M230S; P: no mutations | B | Neg | 16 |
| HI95-10 | RT: V179I; P: M36I, L63P | RT: A; P: CRF01-AE | Neg | 14 |
| HI96-88 | RT: M41L, E44A, D67N, T69D, L210W, T215Y; P: L63P | B | Neg | 24 |
| HI96-90 | RT: M41L, E44A, V118L, M184V, L210W, T215Y; P: L10I, L63P | B | Neg | 8.6 |
| HI97-59 | RT: M184V, L210W, T215Y; P: L10I, L63T | B | Neg | 4.2 |
| HI98-9 | RT: M41L, M184V, T215Y; P: L10I, L24I, L63P, V77I, V82T/S | B | Neg | 34 |
| HI98-80 | RT: M41L, D67N, M184V, T215F, K219Q; P: M36I | A | Neg | 5.5 |
| HI98-82 | RT: M184V; P: L10I, I47M, L63P, I93L | B | Neg | 33 |
| HI98-96 | RT: D67N, T69N, K70R, A98G, M184V, T215F, K219Q; P: L63P, G73T, D60E, L90M | B | Neg | 4.6 |
| HI98-100 | RT: M41L, A98G, M184V, L210W, T215Y; P: L10F, K20R, M36I, M46I, L63P, V82A | C | Neg | 16 |
| HI99-58 | RT: D67N, K70R, A98S, Y181C, K219Q; P: M36I, L63P, I93L | B | Neg | 7.6 |
The mutations depicted are those giving resistance to reverse transcriptase (RT) and protease (P); those indicated in boldface are primary.
A positive (Pos) cytopathic effect (CPE) in MT2 cells indicates syncytium formation and CXCR4 usage, whereas negative (Neg) CPE indicates CCR5 usage. ND, not done.
The geometric mean EC50 (μM α-HGA) for all of these isolates is 14 μM.
FIG. 1.

Virus inhibition curves. Susceptibility to α-HGA of clinical isolates with different patterns of resistance-related mutations was determined in PBMC as described in Materials and Methods. Two typical examples of inhibition curves for clinical isolate HI99-58 (A) and HI98-100 (B) are shown. (C) For comparison, the susceptibility to α-HGA of the laboratory strain HIV-1IIIB in H9 cells is shown. The data are the average of duplicate samples.
Effect of α-HGA on cell viability and proliferation.
At concentrations up to 1,000 μM, α-HGA had no effect on cell viability on PBMC or any of the cell lines tested. α-HGA had no mitogenic activity against human PBMC at concentrations up to 2,000 μM. In contrast, 2 μg of PHA/ml markedly stimulated deoxythymidine incorporation into PBMC DNA. No effect on PHA-induced stimulation of DNA synthesis (cell proliferation) was observed when α-HGA was added to PBMC at concentrations up to 400 μM. However, at 2,000 μM, the PHA-induced stimulation was markedly inhibited.
Effect of α-HGA on replication of other viruses.
The antiviral activity of α-HGA against several different viruses, i.e., vesicular stomatitis virus, respiratory syncytial virus, parainfluenza virus, coxsackievirus B4, Sindbis virus, herpes simplex viruses types 1 and 2, vaccinia virus, cytomegalovirus, varicella-zoster virus, reovirus 1, and Punta Toro virus was also tested. The EC50s of α-HGA to all of these viruses exceeded 2,000 μM.
Effect of α-HGA on HIV core assembly.
Untreated and α-HGA-treated H9 cells were infected with the HIV-1 SF-2, and progeny virus was analyzed by TEM. α-HGA treatment at a concentration of 10 μM resulted in significant changes in virion morphology, as summarized in Fig. 2A. Numerical analysis of 439 virus particles produced in untreated cells and 401 virus particles from α-HGA-treated cells was performed, focusing on the virus core morphology. Representative TEM images are shown in Fig. 2B. TEM images of the virus particles were then categorized by the presence of three different core structures: aberrant, immature, and mature dense core structures (Fig. 2C). Although a small percentage of virus with aberrant core was present in the TEM images of untreated SF-2, ca. 62% of α-HGA-treated cultures showed virus with distorted cores. These effects on HIV morphology could not be explained by an effect of α-HGA on p25 to p24 processing since no band with a molecular weight of 25,000 appeared in α-HGA-treated pNL4-3 transfected HeLa-tat cells (Fig. 3) or stimulated ACH-2 cells (data not shown).
FIG. 2.

Effect of α-HGA (at 10 μM) on virion morphology. (A) Enumeration of virus core structures with respective morphology (numbered 1 to 8). A total of 439 virus particles obtained from untreated and 401 virus particles obtained from α-HGA-treated HIV-1 SF-2-infected H9 cells were analyzed by TEM. (B) Typical examples of TEM images representing the various core morphologies depicted in panel A obtained in the absence or presence of α-HGA. Scale bar, 100 nm. (C) The bars depict the relative number of immature virus particles, the virus particles with a mature and normal appearance, and those with aberrant core morphology.
FIG. 3.

Western blot analysis of lysates from HeLa-tat cells transfected with the infectious HIV-1clone pNL4-3 (A) and Viraffinity-purified virus particles (B) from cells treated with α-HGA showing no effect on processing of p25 to p24. The membrane was initially probed with a mixture of rabbit anti-p24 and anti-calnexin antibodies (lower panel in Fig. 3A). The membrane was then stripped off and reprobed with HIV+ sera (upper panel in Fig. 3A) obtained from two individuals. The image in Fig. 3B was obtained by using the same pool of HIV+ sera. The positions of molecular mass markers are indicated to the right (in kilodaltons). The positions of major viral proteins (and the cellular calnexin in Fig. 3A) are indicated to the left.
To study possible effects of α-HGA on in vitro p24 assembly, purified HIV-1 p24 was incubated with or without different concentrations of α-HGA, after which 2.0 M NaCl was added to induce p24 tubular formation. The absence of α-HGA resulted in an increased turbidity that was monitored spectrophotometrically at 350 nm (Fig. 4A). p24 tubular formation decreased in an α-HGA dose-dependent manner. At 100 μM α-HGA, corresponding to a p24/α-HGA molar ratio of 1/3.3, tube formation appeared to be completely inhibited. Glycine at 100 μM had no inhibitory effect on HIV-1 p24 assembly. In parallel, p24 assembled under similar conditions were analyzed by TEM. In the absence of α-HGA, long cylinders of the p24 were observed (Fig. 4B). In contrast, no tubular structures could be detected when p24 was incubated in the presence of 100 μM α-HGA (Fig. 4C). At 50 μM α-HGA, fewer and shorter tubular structures were observed (data not shown).
FIG. 4.
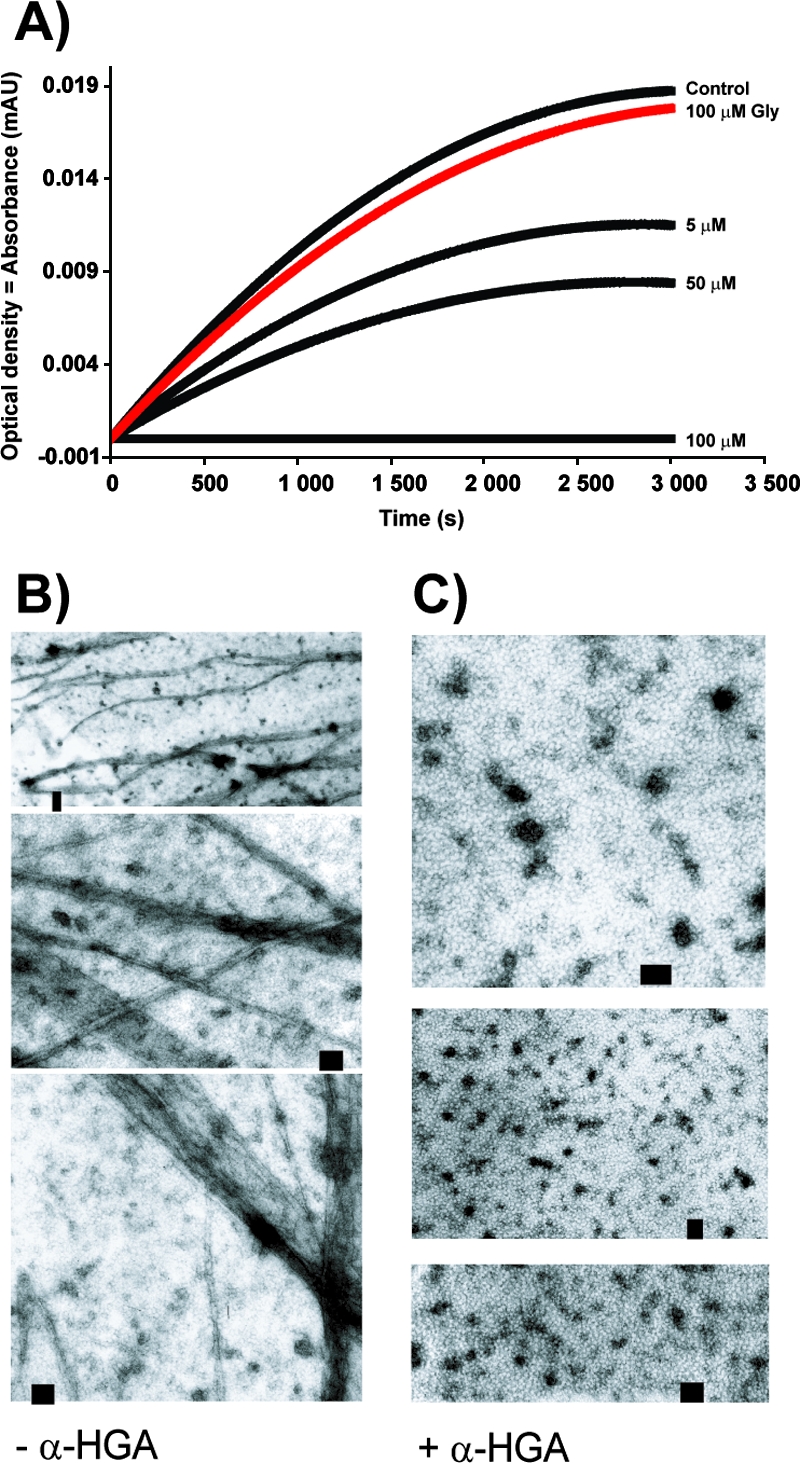
Inhibitory effect of α-HGA on in vitro assembly of the HIV-1 p24. (A) Turbidity assay showing the increase in light absorbance after the addition of 2.0 M NaCl to recombinantly produced p24 (30 μM) reflecting the assembly of the p24 into tubular structures with or without the addition of α-HGA. The α-HGA concentrations 5, 50, and 100 μM correspond to p24/α-HGA molar ratios of 6:1, 1:1.7, and 1:3.3, respectively. (B and C) TEM of negatively stained p24 (200 μM) with (C) or without (B) 100 μM α-HGA.
The effect on intracellular p24 tubular formation was also assessed in HeLa-tat cells transfected with the p17-p24 expression plasmid and analyzed by electron microscopy. In the absence of α-HGA, intracellular clusters of long tubular structures could be observed (Fig. 5A). Addition of 100 μM α-HGA to the transfected cell cultures abolished the intracellular assembly of these tubular structures (Fig. 5B).
FIG. 5.

Effect of α-HGA on in vivo p24 assembly. Intracellular (in vivo) p24 assembly in HeLa-tat cells transfected with the pC37M plasmid expressing the p17-p24 fusion protein without (A) or with (B) the addition of 100 μM α-HGA to the culture medium. The figure in the middle represents a higher magnification of Fig. 5A. Scale bar, 100 nm.
MALDI-MS.
A series of experiments were designed to investigate whether α-HGA can interact with HIV-1 p24. Recombinant p24 proteins were incubated with or without α-HGA and then enzymatically cleaved with trypsin. If the interactions occurred are strong, a mass shift of one or more of the generated peptide fragments obtained after incubation with α-HGA compared to the untreated control could reveal the positions where α-HGA interacts with the protein.
With the wild-type p24, five characteristic long peptide fragments were readily detected in all MALDI-MS experiments: PIVQNIQGQMVHQAISPR (m/z 2,016.12), LHPVHAGPIAPGQMR (m/z 1,580.87), ETINEEAAEWDR (m/z 1,462.66), MYSPTSILDIR (m/z 1,295.72), and QGPKEPFR (m/z 958.53) (Fig. 6A).
FIG. 6.

MALDI-MS analysis of mutant and wild-type p24 in the presence or absence of α-HGA. (A) Mass spectrum of HIV-1 p24 incubated at 37°C for 24 h and then cleaved with trypsin. (B) Mass spectra of HIV-1 p24 incubated with or without 100 mM α-HGA at 37°C for 24 h and then treated with trypsin. The signal of one of the identified fragment (MYSPTSILDIR; 1,295.72 Da) and another signal (1,367.70 Da) that appeared in its vicinity are highlighted. (C and D) MS/MS spectra of 1,295.72 and 1,367.70, respectively, showing the y- and b-ion series identified, suggesting that these signals originate from the same peptide fragment.
A new signal appeared in the vicinity of fragment MYSPTSILDIR (m/z 1,295.72), with the mass value increased 72 Da in all samples upon incubation of p24 with α-HGA compared to untreated p24 (Fig. 6B). Similar results were also observed when three p24 mutants (D51N, D51E, and D51Q) were analyzed (data not shown), which was not surprising since these mutations altered D51, an amino acid localized on a tryptic peptide of 40 residues (4,233.05 Da) that fell out of the detection range of our MS instrument. The MYSPTSILDIR peptide corresponds to the 10 amino acid residues (143 to 153)—as confirmed by MS/MS experiments using a MALDI-TOF/TOF instrument —in the hinge region connecting the N- and the C-terminal domains of p24. Analysis of the new signal of 1,367.70 Da revealed the same peptide sequence (Fig. 6C and D), supporting the assumption that a possible interaction between MYSPTSILDIR and α-HGA occurred.
The mass signal of 1,367.70 could not be detected when similar experiments were performed with the C-terminal domain (amino acids 151 to 231) or p24 containing mutations in the C-terminal dimer interface, W184A+M185A (data not shown).
DISCUSSION
α-HGA is a small (90 Da) antiretroviral molecule that was discovered as a metabolite of GPG-NH2/G-NH2 when incubated in bovine or porcine serum but not in human serum (data not shown). The EC50 of α-HGA for the laboratory strain HIV-1 SF-2 was ca. 5 μM and ranged between 4 and 34 μM for clinical HIV-1 isolates resistant to other antiretroviral drugs, indicating that the compound had no cross-resistance to anti-HIV drugs of other classes.
Characteristics attributed earlier to GPG-NH2, such as not interfering with CD4 or coreceptor binding, penetration, viral reverse transcription, mRNA synthesis, or protein translation (20-22), should apply to α-HGA. Consistent with previous results obtained with GPG-NH2 and G-NH2 (2, 10), we found that virus particles generated in the presence of α-HGA exhibit aberrant HIV-1 core structures with varying morphology. Detailed analysis of the core morphology of 439 control and 401 α-HGA treated HIV-1 particles showed that the number of virions with aberrant core morphologies were markedly different between the two groups, suggesting that α-HGA disrupts proper core assembly, possibly by binding to the HIV-1 p24. In support of this, α-HGA inhibited both in vitro and in vivo (intracellular) p24 tubular formation.
Previously, we have shown that, apart from affecting the morphogenesis of HIV-1 mature conical cores, the addition of GPG-NH2 to infected cells influenced the processing of the HIV-1 glycoprotein gp160 (21). In a recent study we have found that also this effect is due to GPG-NH2 being converted to α-HGA but is independent of its effect on p24 assembly (data not shown).
α-HGA had no antiviral activity against a variety of DNA and RNA viruses with different size, genome, and morphology, emphasizing the specificity of the α-HGA-HIV-1 p24 interaction. Other compounds that inhibit or interfere with HIV-1 p24 maturation or assembly have recently been reported (13, 19, 23). PA-457 (13) is a compound that binds to the proteolytic cleavage site of the p24 precursor (p25/CA-SP1) and thereby affects its maturation to p24. However, α-HGA did not affect p25 to p24 processing. Tang et al. reported on the binding of compound N-(3-chloro-4-methylphenyl)-N′-2-(5-[dimethylamino-methyl]-2-furyl)-methylsulfanyl-ethyl urea (CAP-1) to the N-terminal domain of p24 (23). CAP-1, which has a molecular mass of approximately four times that of α-HGA, has an EC50 for HIV-1 replication of ca. 75 μM (23). Recently, a 12-mer peptide was shown to interfere with p24 dimerization but not with HIV-1 replication in cell culture (19, 24).
Although inhibition of both in vitro and intracellular p24 tubular assembly suggested that α-HGA could bind to HIV-1 p24 and thereby interfere with its assembly, the exact binding site of α-HGA has yet to be determined. In attempts to determine this, we investigated the α-HGA-p24 interactions by both nuclear magnetic resonance (NMR) titration and MALDI-MS analysis. NMR titrations experiment of α-HGA with NTD-p24 and p24 with mutations at the p24 dimer interfaces (p24W184A+M185A), however, failed to prove any interactions between α-HGA and p24 (data not shown). The two mutated residues in the p24 used in these experiments were introduced to inhibit interactions necessary for p24 C-terminal dimer formation (7, 19, 29). In MALDI-MS, however, α-HGA repeatedly was shown to interact with the wild-type p24 and the three other p24 mutants (D51N, D51E, and D51Q) with intact p24 dimer interfaces. Analysis by MALDI-MS also indicated that α-HGA may bind to the hinge region of the p24 molecule. Based upon the increased mass of the newly observed signal (+72 Da compared to MYSPTSILDIR) in mass spectra after incubation of the proteins with α-HGA (Mw = 90), a condensation reaction may be assumed due to the nucleophilic amino group on α-HGA. Although the chemical character of α-HGA may allow such reaction, it is unclear on which residue it takes place. Interestingly, as was the case with the NMR titrations, p24 with mutations at the p24 dimer interfaces (p24W184A+M185A) was not found to bind α-HGA in the MALDI-MS experiments, indicating that binding of α-HGA to p24 may require intact p24 dimer interfaces and possible allosteric changes of p24 upon dimerization. However, it is apparent that crystallization of α-HGA with the C-terminal p24 domain may be needed in order define the exact binding site for α-HGA on the p24 molecule.
In conclusion, we have shown that the small compound α-HGA at μM concentrations specifically can interfere with the multiple semistable noncovalent protein interactions in HIV-1 capsid assembly and thereby inhibit virus replication in cell culture. The results from the present study, along with future characterization of α-HGA-p24 complexes might prove useful for further modification of α-HGA, improving its efficacy in inhibiting HIV-1 replication and its possible clinical utility.
Acknowledgments
We are indebted to Jack J. Skalicky and Wesley I. Sundquist at the University of Utah for help with the NMR titration assay. We thank Pia O‥sterwall, Sung Oun Stenberg, Yuba S. Tellez, and Ulrika Noborg for skillful technical assistance. We thank the original donors of the following reagents that were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: the CD4+ cell lines H9 (16) and ACH-2 (5) and the CD4− cell line HeLa-tat-III (17).
This study was supported by grants from the European Commission (HPAW-2002-90005), Swedish Research Council (grant K2000-06X-09501-10B), Swedish International development Cooperation Agency, SIDA (grant HIV-2006-050), and Tripep AB.
A.V. is a shareholder in Tripep AB, and M.L. is an employee of Tripep AB.
Footnotes
Published ahead of print on 21 July 2008.
REFERENCES
- 1.Abdurahman, S., M. Youssefi, S. Hoglund, and A. Vahlne. 2007. Characterization of the invariable residue 51 mutations of human immunodeficiency virus type 1 capsid protein on in vitro CA assembly and infectivity. Retrovirology 4:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson, E., P. Horal, A. Jejcic, S. Hoglund, J. Balzarini, A. Vahlne, and B. Svennerholm. 2005. Glycine-amide is an active metabolite of the antiretroviral tripeptide glycyl-prolyl-glycine-amide. Antimicrob. Agents Chemother. 49:40-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersson, E., P. Horal, A. Vahlne, and B. Svennerholm. 2004. No cross-resistance or selection of HIV-1 resistant mutants in vitro to the antiretroviral tripeptide glycyl-prolyl-glycine-amide. Antivir. Res. 61:119-124. [DOI] [PubMed] [Google Scholar]
- 4.Boden, D., A. Hurley, L. Zhang, Y. Cao, Y. Guo, E. Jones, J. Tsay, J. Ip, C. Farthing, K. Limoli, N. Parkin, and M. Markowitz. 1999. HIV-1 drug resistance in newly infected individuals. JAMA 282:1135-1141. [DOI] [PubMed] [Google Scholar]
- 5.Clouse, K. A., D. Powell, I. Washington, G. Poli, K. Strebel, W. Farrar, P. Barstad, J. Kovacs, A. S. Fauci, and T. M. Folks. 1989. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T-cell clone. J. Immunol. 142:431-438. [PubMed] [Google Scholar]
- 6.De Clercq, E. 2007. The design of drugs for HIV and HCV. Nat. Rev. Drug Discov. 6:1001-1018. [DOI] [PubMed] [Google Scholar]
- 7.Gamble, T. R., S. Yoo, F. F. Vajdos, U. K. von Schwedler, D. K. Worthylake, H. Wang, J. P. McCutcheon, W. I. Sundquist, and C. P. Hill. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278:849-853. [DOI] [PubMed] [Google Scholar]
- 8.Grant, R. M., F. M. Hecht, M. Warmerdam, L. Liu, T. Liegler, C. J. Petropoulos, N. S. Hellmann, M. Chesney, M. P. Busch, and J. O. Kahn. 2002. Time trends in primary HIV-1 drug resistance among recently infected persons. JAMA 288:181-188. [DOI] [PubMed] [Google Scholar]
- 9.Groeschen, H. M. 2007. Novel HIV treatment approved. Am. J. Health Syst. Pharm. 64:1886. [DOI] [PubMed] [Google Scholar]
- 10.Hoglund, S., J. Su, S. S. Reneby, A. Vegvari, S. Hjerten, I. M. Sintorn, H. Foster, Y. P. Wu, I. Nystrom, and A. Vahlne. 2002. Tripeptide interference with human immunodeficiency virus type 1 morphogenesis. Antimicrob. Agents Chemother. 46:3597-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horal, P., W. W. Hall, B. Svennerholm, J. Lycke, S. Jeansson, L. Rymo, M. H. Kaplan, and A. Vahlne. 1991. Identification of type-specific linear epitopes in the glycoproteins gp46 and gp21 of human T-cell leukemia viruses type I and type II using synthetic peptides. Proc. Natl. Acad. Sci. USA 88:5754-5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Humphreys, E. H., L. B. Hernandez, and G. W. Rutherford. 2007. Antiretroviral regimens for patients with HIV who fail first-line antiretroviral therapy. Cochrane Database Syst. Rev.:CD006517. [DOI] [PubMed]
- 13.Li, F., R. Goila-Gaur, K. Salzwedel, N. R. Kilgore, M. Reddick, C. Matallana, A. Castillo, D. Zoumplis, D. E. Martin, J. M. Orenstein, G. P. Allaway, E. O. Freed, and C. T. Wild. 2003. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. USA 100:13555-13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore, R. D., J. C. Keruly, K. A. Gebo, and G. M. Lucas. 2005. An improvement in virologic response to highly active antiretroviral therapy in clinical practice from 1996 through 2002. J. Acquir. Immune Defic. Syndr. 39:195-198. [PubMed] [Google Scholar]
- 15.Pillay, D. 2004. Current patterns in the epidemiology of primary HIV drug resistance in North America and Europe. Antivir. Ther. 9:695-702. [PubMed] [Google Scholar]
- 16.Popovic, M., M. G. Sarngadharan, E. Read, and R. C. Gallo. 1984. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-1II) from patients with AIDS and pre-AIDS. Science 224:497-500. [DOI] [PubMed] [Google Scholar]
- 17.Rosen, C. A., W. A. Haseltine, J. Lenz, R. Ruprecht, and M. W. Cloyd. 1985. Tissue selectivity of murine leukemia virus infection is determined by long terminal repeat sequences. J. Virol. 55:862-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shafer, R. W., and J. M. Schapiro. 2005. Drug resistance and antiretroviral drug development. J. Antimicrob. Chemother. 55:817-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sticht, J., M. Humbert, S. Findlow, J. Bodem, B. Muller, U. Dietrich, J. Werner, and H. G. Krausslich. 2005. A peptide inhibitor of HIV-1 assembly in vitro. Nat. Struct. Mol. Biol. 12:671-677. [DOI] [PubMed] [Google Scholar]
- 20.Su, J., E. Andersson, P. Horal, M. H. Naghavi, A. Palm, Y. P. Wu, K. Eriksson, M. Jansson, H. Wigzell, B. Svennerholm, and A. Vahlne. 2001. The nontoxic tripeptide glycyl-prolyl-glycine amide inhibits the replication of human immunodeficiency virus type 1. J. Hum. Virol. 4:1-7. [PubMed] [Google Scholar]
- 21.Su, J., M. H. Naghavi, A. Jejcic, P. Horal, Y. Furuta, Y. P. Wu, S. L. Li, W. W. Hall, L. Goobar-Larsson, B. Svennerholm, and A. Vahlne. 2001. The tripeptide glycyl-prolyl-glycine amide does not affect the early steps of the human immunodeficiency virus type 1 replication. J. Hum. Virol. 4:8-15. [PubMed] [Google Scholar]
- 22.Su, J., A. Palm, Y. Wu, S. Sandin, S. Hoglund, and A. Vahlne. 2000. Deletion of the GPG motif in the HIV type 1 V3 loop does not abrogate infection in all cells. AIDS Res. Hum. Retrovir. 16:37-48. [DOI] [PubMed] [Google Scholar]
- 23.Tang, C., E. Loeliger, I. Kinde, S. Kyere, K. Mayo, E. Barklis, Y. Sun, M. Huang, and M. F. Summers. 2003. Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 327:1013-1020. [DOI] [PubMed] [Google Scholar]
- 24.Ternois, F., J. Sticht, S. Duquerroy, H. G. Krausslich, and F. A. Rey. 2005. The HIV-1 capsid protein C-terminal domain in complex with a virus assembly inhibitor. Nat. Struct. Mol. Biol. 12:678-682. [DOI] [PubMed] [Google Scholar]
- 25.Vandamme, A. M., F. Houyez, D. Banhegyi, B. Clotet, G. De Schrijver, K. A. De Smet, W. W. Hall, R. Harrigan, N. Hellmann, K. Hertogs, C. Holtzer, B. Larder, D. Pillay, E. Race, J. C. Schmit, R. Schuurman, E. Schulse, A. Sonnerborg, and V. Miller. 2001. Laboratory guidelines for the practical use of HIV drug resistance tests in patient follow-up. Antivir. Ther. 6:21-39. [PubMed] [Google Scholar]
- 26.Weinstock, H. S., I. Zaidi, W. Heneine, D. Bennett, J. G. Garcia-Lerma, J. M. Douglas, Jr., M. LaLota, G. Dickinson, S. Schwarcz, L. Torian, D. Wendell, S. Paul, G. A. Goza, J. Ruiz, B. Boyett, and J. E. Kaplan. 2004. The epidemiology of antiretroviral drug resistance among drug-naive HIV-1-infected persons in 10 US cities. J. Infect. Dis. 189:2174-2180. [DOI] [PubMed] [Google Scholar]
- 27.Vella, S., and L. Palmisano. 2005. The global status of resistance to antiretroviral drugs. Clin. Infect. Dis. 41(Suppl. 4):S239-S2246. [DOI] [PubMed] [Google Scholar]
- 28.Wensing, A. M., D. A. van de Vijver, G. Angarano, B. Asjo, C. Balotta, E. Boeri, R. Camacho, M. L. Chaix, D. Costagliola, A. De Luca, I. Derdelinckx, Z. Grossman, O. Hamouda, A. Hatzakis, R. Hemmer, A. Hoepelman, A. Horban, K. Korn, C. Kucherer, T. Leitner, C. Loveday, E. MacRae, I. Maljkovic, C. de Mendoza, L. Meyer, C. Nielsen, E. L. Op de Coul, V. Ormaasen, D. Paraskevis, L. Perrin, E. Puchhammer-Stockl, L. Ruiz, M. Salminen, J. C. Schmit, F. Schneider, R. Schuurman, V. Soriano, G. Stanczak, M. Stanojevic, A. M. Vandamme, K. Van Laethem, M. Violin, K. Wilbe, S. Yerly, M. Zazzi, and C. A. Boucher. 2005. Prevalence of drug-resistant HIV-1 variants in untreated individuals in Europe: implications for clinical management. J. Infect. Dis. 192:958-966. [DOI] [PubMed] [Google Scholar]
- 29.von Schwedler, U. K., K. M. Stray, J. E. Garrus, and W. I. Sundquist. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77:5439-5450. [DOI] [PMC free article] [PubMed] [Google Scholar]