

Abstract
The complete DNA sequence of Candida albicans DIT2, encoding cytochrome P450 family 56 (CYP56), was obtained, and heterologous expression was achieved in Escherichia coli, where CYP56 was targeted to the membrane fraction. In reconstituted assays with the purified enzyme, CYP56 was shown to catalyze the conversion of N-formyl tyrosine into N,N′-bisformyl dityrosine, a reaction that was dependent on cytochrome P450 reductase, NADPH, and oxygen, yielding a turnover of 21.6 min−1 and a ks of 26 μM. The Hill number was calculated as 1.6, indicating that two molecules of the substrate could bind to the protein. Azole antifungals could bind to the heme of CYP56 as a sixth ligand with high affinity. Both chromosomal alleles of CYP56 were disrupted using the SAT1 flipper technique, and CYP56 was found to be nonessential for cell viability under the culture conditions investigated. Susceptibility to azole drugs that bind to cytochromes P450 was tested, and the mutant showed unaltered susceptibility. However, the mutant showed increased susceptibility to the echinocandin drug caspofungin, suggesting an alteration in 1,3-glucan synthase and/or cell wall structure mediated by the presence of dityrosine. Phenotypically, the wild-type and mutant strains were morphologically similar when cultured in rich yeast extract-peptone-dextrose medium. However in minimal medium, the cyp56Δ mutant strain exhibited hyphal growth, in contrast to the wild-type strain, which grew solely in the yeast form. Furthermore, CYP56 was essential for chlamydospore formation.
Cytochromes P450 (CYPs), a superfamily of heme-thiolate monooxygenase enzymes, are found in almost all eukaryotes, where an ancestral form, CYP51, is required for sterol 14 demethylation. CYPs catalyze a diverse range of reactions, which play important roles in biosynthetic pathways, the bioremediation of external pollutants, and the metabolism of xenobiotics. The Saccharomyces cerevisiae genome project revealed the presence of three cytochrome P450 genes: CYP51, involved in sterol 14 demethylation; CYP61, which is required for sterol 22 desaturation (6, 11); and CYP56, known to be involved in meiotic spore wall biogenesis (4). The genomes of several other yeasts and fungi have been published; some fungi possess more than 100 putative CYP genes (12). Analysis of the Candida albicans genome, however, revealed the presence of 10 CYP genes (7), 1 of which is a possible homologue of S. cerevisiae CYP56 (also known as DIT2). CYP56, along with DIT1, is involved in the biosynthesis of N,N′-bisformyl dityrosine, a component of the outer layer of the yeast spore wall (3). CYP56 is one of a number of homologues of meiotic genes from S. cerevisiae that have prompted interest in elucidating putative meiotic processes in C. albicans (9, 18, 19). Some time ago, the presence of dityrosine in vegetatively growing C. albicans was reported (27). However, to date, the role of this polymer in normal C. albicans cell wall metabolism and structure has not been investigated. Therefore, the study of the biological function of dityrosine in C. albicans and the potential for CYP56 as a new target for antifungal drugs was of therapeutic interest.
The cell wall is an essential and dynamic structure required for normal cell function, including the control of the cellular permeability barrier, osmotic balance, cell shape, and morphogenesis. The processes of cell wall biosynthesis have been identified as excellent targets for the development of new antifungal agents (e.g., the echinocandins, which target 1,3-glucan synthase [26]). However, cytochromes P450, specifically CYP51 (sterol 14α-demethylase in the ergosterol pathway), are the targets of the most commonly used antifungal drugs, the azoles (5, 28). This work reports the identification and characterization of a CYP56 homologue implicated in the biosynthesis of a cell wall component of the pathogenic yeast C. albicans. The enzyme was studied here for the first time, and through gene deletion, the importance of this gene in the viability, antifungal drug susceptibility, and cellular biology of C. albicans was investigated.
MATERIALS AND METHODS
Strains and media.
The C. albicans strains and the primers used for gene deletion in this study are listed in Tables 1 and 2, respectively. For routine experiments, C. albicans cells were grown in yeast extract-peptone-dextrose (YEPD) and yeast minimal medium (YM) at 30°C. The Escherichia coli strain used for expression studies was BL21(DE3).
TABLE 1.
C. albicans strains used in this study
| Strain | Parent | Genotype | Reference |
|---|---|---|---|
| SC5314 | Wild-type strain | 11 | |
| YNM8 | SC5314 | cyp56Δ::SAT1/CYP56 | This study |
| YNM9 | YNM8 | cyp56Δ::FRT/CYP56 | This study |
| YNM10 | YNM9 | cyp56Δ::FRT/cyp56Δ::SAT1 | This study |
| YNM11 | YNM10 | cyp56Δ::FRT/cyp56Δ::FRT | This study |
| YNM12 | YNM11 | cyp56Δ::FRT/cyp56Δ::FRT RP10-CYP56 | This study |
TABLE 2.
Primers used in this study for the deletion of CYP56 in C. albicans
| Primer | Sequencea | Reference |
|---|---|---|
| DITFOR | 5′-TTGGCCAAGGAATATTTCTG-3′ | This study |
| DITREV | 5′-TGTGCTTTGATGTACCTGTG-3′ | This study |
| CYP56F | 5′-CATCGCCATATGGCTCAACTATTGAAATAT-3′ | This study |
| CYP56R | 5′-ACTGCTGCATGCCTAGTGATGGTGATGGTGATG GTGATGGTGATGGGAAACTGTAAGCTTTTC-3′ | This study |
| DIT2F | 5′-ATGCGAGCTCAGTTTGTTACCAATATGCAC-3′ | This study |
| DIT2R | 5′-ATCCGGATCCGCCATACTATGCCATTAAGG-3′ | This study |
| CYP56KF | 5′-GGCCGGTACCAGTTTGTTACCAATATGCAC-3′ | This study |
| CYP56XR | 5′-GGCCCTCGAGAATGGTGGGAAAACTATCTC-3′ | This study |
| DITSIIF | 5′-GCCGCCGCGGTTCTAGGTGAGTATAAAGTG-3′ | This study |
| DITSIR | 5′-GGCCGAGCTCGCCATACTATGCCATTAAGG-3′ | This study |
| SAT1 | 5′-GCCCGACGTCGCACTCGAGCGTCAAAACTAG-3′ | 12 |
| SAT2 | 5′-CTAGTGATTTCTGCAGGACCACCTTTG-3′ | 12 |
| MAL1 | 5′-CATGCAAGCCAGGATCCAATAATGATTGG-3′ | 12 |
| MAL2 | 5′-GTTCACTCATTGTCGACGATTATTAGTTAAACC-3′ | 12 |
| FLP1 | 5′-TTCCGTTATGTGTAATCATCC-3′ | 12 |
| FLP2 | 5′-CGCTGAGTTTCGATATTGTC-3′ | 12 |
| FLP3 | 5′-GTATATGTGCCTACTAACGC-3′ | 12 |
| RT300 | 5′-GTACTTGACATGCCATACTATGC-3′ | This study |
| RT768 | 5′-AGTTTCGATAAAGAAAACAAGA-3′ | This study |
| TEM1R | 5′-TGGTTATTCTGATCCTGTTG-3′ | This study |
| ACT10 | 5′-GCGGAATTCAGAGTCGACATT-3′ | This study |
Underlined sequences represent the following restriction sites: SacI for DIT2F, BamHI for DIT2R, KpnI for CYP56KF, XhoI for CYP56XR, SacII for DITSIIF, and SalI for DITSIR.
Isolation of the CYP56 gene sequence.
In the assembly of the C. albicans SC5314 genome sequence (http://www.candidagenome.org) that was examined (assembly 19), the sequence of the CYP56 gene was incomplete at the 5′ end, probably due to an unfinished gap. In order to complete the C. albicans CYP56 gene sequence, primers DITFOR and DITREV were designed to amplify the CYP56 open reading frame (ORF) and flanking sequences based on the nucleotide sequence of the homologous Candida dubliniensis CYP56 gene, obtained from the C. dubliniensis genome sequence (http://www.sanger.ac.uk/sequencing/Candida/dubliniensis/) (Table 2) (7, 24). The C. albicans CYP56 gene was amplified by PCR using genomic DNA from C. albicans strain SC5314 as a template to obtain the complete CYP56 sequence. Positive clones were verified for integrity by DNA sequencing.
Protein studies.
For heterologous expression of C. albicans CYP56 in E. coli, forward primer CYP56F (Table 2), containing an NdeI site, was designed. A triplet encoding alanine replaced the second triplet encoding phenylalanine in the protein sequence in order to improve the expression (2). The reverse primer, CYP56R (Table 2), incorporated an SphI restriction site as well as triplets encoding 10 histidine residues to facilitate the purification of the heterologously expressed CYP56 protein using Ni2+-nitrilotriacetic acid (NTA) affinity chromatography. The C. albicans CYP56 gene was isolated by PCR and cloned into a pSP19g10L expression vector for E. coli (14). The construct, pSP19g10L-CYP56, was transformed into E. coli BL21(DE3) cells for heterologous expression.
Expression of CYP56 protein.
Transformants containing pSP19g10L-CYP56 were grown in 5 ml of Luria-Bertani (LB) broth containing 0.1 mg·ml−1 sodium ampicillin overnight at 37°C and 225 rpm and were subsequently used to inoculate 500-ml volumes of Terrific Broth containing 0.1 mg·ml−1 carbenicillin. The cultures were then shaken at 200 rpm and 37°C for 5 to 6 h until an optical density of 0.6 was obtained, after which 5-aminolevulinic acid and isopropyl-β-d-thiogalactopyranoside were added to 1 mM final concentrations in order to assist maximal hemoprotein production and induce protein expression, respectively, by shaking overnight at 160 rpm and 28°C. CYP56 was purified by Ni2+-NTA agarose affinity chromatography, and protein purity was assessed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis. The cytochrome P450 content was determined spectrophotometrically using the method of Omura and Sato (22).
Spectral binding studies of CYP56.
Cytochrome P450 substrate binding spectra were determined according to the method of Jefcoate (10) by using Ni2+-NTA agarose-purified CYP56 at 2 μM in 0.1 M potassium phosphate (pH 7.4)-20% (vol/vol) glycerol. The spectral difference in absorbance between 390 and 412 nm (ΔA390-412) was determined at several incremental N-formyl tyrosine concentrations (26, 51, 77, 103, 128, 153, 178, 203, 228, and 253 μM). The maximal difference in absorbance (ΔAmax) was determined by nonlinear regression (Levenberg-Marquardt algorithm) of ΔA390-412 against the substrate concentration using the Hill equation: ΔA = ΔAmax/(1 + k/[S]n) where [S] is the substrate concentration. The ΔAmax values derived were used to construct a Hill plot {log[ΔA/(ΔAmax − ΔA)] versus log[substrate]} to determine the ks and Hill number (n). ks in this study is defined as the dissociation constant for the enzyme-substrate complex and is equivalent to k in the Hill equation (23). Curve fitting was performed using the ProfFit program (version 5.0.1; Quantumsoft, Zurich, Switzerland).
Binding of azole antifungal agents to CYP56 was performed as previously described by Lamb et al. (15, 16). The binding of fluconazole, itraconazole, and voriconazole to CYP56 was studied by progressively titrating the azole antifungal drugs (dissolved in dimethyl sulfoxide) against a 2 μM solution of Ni2+-NTA agarose-purified CYP56 while monitoring the absorption spectrum between 350 and 500 nm. Fluconazole concentrations used were 52, 103, 129, 154, 179, 205, 230, 255, 279, and 305 nM. Itraconazole concentrations used were 23, 45, 67, 89, 111, 133, 154, 176, and 197 nM. Voriconazole concentrations used were 91, 137, 183, 228, 274, 319, 365, 411, 456, 502, 547, 593, 639, and 684 nM. The enzyme-azole dissociation constant (kd) was determined by nonlinear regression (Levenberg-Marquardt algorithm) of ΔA430-412 against the azole concentration using the Hill equation: ΔA = ΔAmax/(1 + k/[azole]n) where k is equivalent to the kd for azole binding (23).
Reconstitution of dityrosine synthase (CYP56) activity in vitro.
Dityrosine synthase activity was determined using an in vitro reconstitution assay as previously described (9) with 10 μM N-formyl tyrosine as the substrate. The reaction was terminated by the addition of 2.5 ml of chloroform-methanol (1:1) to the 1-ml reaction volume with vortexing for 1 min at room temperature. The upper phase was isolated by centrifugation, followed by aspiration. The lower phase was washed with 1 ml of 10% (wt/vol) ammonium hydroxide and the upper phase isolated. The two upper-phase fractions were pooled and centrifuged at 16,000 × g for 10 min to remove debris prior to concentration of the supernatant using a SpeediVac (Heto Maxi Dry plus) desiccator under a vacuum at room temperature. The isolated assay products were separated by thin-layer chromatography (TLC) with silica gel 60F (Merck, Germany) using a 1-propanol-ammonium hydroxide (70:30) solvent system. Control samples containing dityrosine and N,N′-bisformyl dityrosine, generated by use of horseradish peroxidase, were used for comparison (1). The separated components were visualized under UV light and recovered from the TLC plate by extraction with 10% (wt/vol) ammonium hydroxide from the excised silica matrix. The separated assay products were identified by high-performance liquid chromatography (HPLC)-mass spectrometry (MS) in the multiple-reaction-monitoring (MRM) mode. The LC-MS system used consisted of a Dionex HPLC system using a PepMap C18 column (0.3 mm by 15 cm) run isocratically at 4 μl·min−1 in 0.1% (vol/vol) methanoic acid-50% (vol/vol) methanol. The eluent from the C18 column was analyzed using an LCQ-XP DECA ion trap MS (Thermo Finnigan, United Kingdom) in positive-ion mode with a spray voltage of 3.5 kV, sheath and auxiliary gas flows of 30% and 10% (derived from arbitrary units), respectively, a capillary temperature of 200°C, and a capillary voltage of 10 V.
LC-MS analysis of the control samples confirmed that the molecular mass ions of dityrosine and N,N′-bisformyl dityrosine were 361 and 417, respectively. Tandem MS (MS-MS) was employed to determine the fragmentation patterns of the two molecular mass ions. The dityrosine 361 molecular mass ion fragmented to produce two abundant mass ions of m/z 315 (100% relative abundance) and 344 (53% relative abundance). The N,N′-bisformyl dityrosine 417 molecular mass ion fragmented to produce four abundant mass ions of m/z 343 (100% relative abundance), 370.9 (22% relative abundance), 389 (47% relative abundance), and 398.8 (22% relative abundance). The MS was set to analyze the MRM transitions of the column eluent compared to those of N,N′-bisformyl dityrosine and dityrosine synthesized as standards. Selected-reaction-monitoring (SRM) transitions of 361 to 315 m/z and 417 to 343 m/z were used to detect the presence of dityrosine and N,N′-bisformyl dityrosine, respectively, in the C18 column eluent.
Deletion of the CYP56 gene in C. albicans.
The C. albicans CYP56 gene in strain SC5314 was deleted using the SAT1 flipper described by Reuss et al. (24). A deletion construct was created by PCR by amplifying the flanking 5′ and 3′ regions of the CYP56 gene with primer pairs CYP56KF-CYP56XR and DIT2IIF-DIT2SIR, respectively (Table 2). These 5′ and 3′ fragments were cloned into the KpnI/XhoI and SacII/SacI sites, respectively, flanking the SAT1 flipper in plasmid pSFS2A to yield pNM10. The entire deletion construct was released from plasmid pNM10 on a KpnI-SacI fragment and used to transform C. albicans SC5314 as described previously (24). Integration of the cassette at the correct locus was confirmed by PCR with primers CYP56KF (which binds to chromosomal DNA) and FLP1 (which binds to cassette sequences). In order to delete the second copy of CYP56, the marker was excised and recycled for a second round of transformation as previously described (24). This resulted in the generation of strain YNM11, which contained a deletion between nucleotides +85 and +1320 (where the first A of the ATG start codon is +1) in both copies of the CYP56 gene. This was confirmed by Southern blot analysis. The wild-type CYP56 gene and its upstream and downstream regulatory regions were reintroduced by PCR amplification of regions −770 to +1787 of the CYP56 gene using primers RT300 and RT768. This product was ligated into the SpeI and XbaI sites of the integrating vector CIp20 to yield pNM12. Plasmid CIp20 is a derivative of the RP10 integrating vector CIp10, which contains the SAT1 resistance marker from pSFS2A in place of the C. albicans URA3 marker (24). Targeted integration of pNM12, containing the complete CYP56 ORF, into the CYP56 mutant was achieved by linearization of the plasmid within the RP10 region by digestion with NcoI and transformation into YNM11 by electroporation, yielding the CYP56-complemented strain YNM12. The correct genomic integration at the RP10 locus was confirmed by PCR using a primer that annealed within the cassette (ACT10) and a primer that annealed with flanking chromosomal sequences (TEM1R).
Susceptibility testing.
MICs were determined in triplicate by a modified broth microdilution method (20). For amphotericin B, the MIC was defined as the lowest concentration producing 100% inhibition of growth compared with that of the growth control well; for all other antifungal agents, the MIC was defined as the lowest concentration producing 80% inhibition. The antifungal agents used were amphotericin B (Bristol-Myers Squibb), voriconazole, fluconazole (Pfizer, United Kingdom), itraconazole (Janssen-Kyowa Co., Ltd., United Kingdom), and caspofungin (Merck & Co., Inc.).
Cell wall integrity assays.
The phenotype of C. albicans cells grown in the presence of chemicals that induce stress responses was evaluated. Drop tests were performed by spotting 106, 105, 104, 103, and 102 cells ml−1 onto YEPD or YM agar plates supplemented with 1.5 M sorbitol, 100 μg·ml−1 SDS, 1 M or 1.5 M NaCl, 5 mM or 10 mM H2O2, 0.5 M or 1 M CaCl2, 100 μg·ml−1 calcofluor, 100 μg·ml−1 Congo red, 1 μg·ml−1 itraconazole, 10 μg·ml−1 amphotericin B, or 2 μg·ml−1 fluconazole. Duplicate plates were incubated for 24 h at 37°C and 42°C, respectively.
Chlamydospore production and hyphal formation.
C. albicans strains were cultured on cornmeal agar and broth (Difco) supplemented with 1% (vol/vol) Tween 80 and 20 μg·ml−1 methionine at 25°C for 5 to 7 days or on rice agar-Tween medium (RAT medium; Biomérieux) for chlamydospore formation. Hyphal formation was induced using 10% (vol/vol) calf serum (GIBCO BRL) or RPMI 1640 at 37°C for 2 to 4 h. Hyphal formation and chlamydospore formation were examined using a Nikon optical microscope, and images were captured at magnifications of both ×40 and ×100 by using a Nikon 4500 digital camera.
Imaging of cell fluorescence by fluorescence microscopy.
Cells were cultured under the conditions described above for microscopic visualization of yeasts, hyphae, and chlamydospores. Aliquots of culture were transferred to sterile Eppendorf tubes and centrifuged (10,000 × g) for 3 min. Supernatants were discarded and the pellets resuspended in sufficient 10% (wt/vol) ammonium hydroxide. Subsequently, 7.5 μl of each suspension was dispensed into wells of separate multispot microscope slides (Hendley, United Kingdom); a coverslip was affixed; and the suspension was immediately viewed. For fluorescence microscopy, cells were examined using a Nikon Eclipse E600 epifluorescence microscope. UV autofluorescence of yeast cells was examined using the 40× and 100× objective lenses with excitation emission filter set FS1 fitted. Digital images were acquired using a Nikon CoolPix digital camera.
RESULTS AND DISCUSSION
C. albicans CYP56 sequence.
The entire CYP56 gene sequence was found to comprise 1,434 bases, encoding a protein of 477 amino acids (55,140 Da) with 46% identity to the S. cerevisiae (P21595) homologue. In S. cerevisiae, the CYP56 ORF is adjacent to the DIT1 gene, encoding a second enzyme involved in dityrosine formation. Analysis of the C. albicans locus revealed that this synteny was conserved, strengthening our hypothesis that this was a functional homologue of S. cerevisiae CYP56. The amino acid identity of >40% validates the placement of these CYP56 genes in the same CYP family; however, it does not allow placement in the same subfamily. Further bioinformatic analysis has been undertaken, revealing CYP56 homologues adjacent to a DIT1 gene in other fungi besides Saccharomyces and Candida species, including aspergilli. C. albicans CYP56 shared 44%, 36%, 38%, 37%, and 38% identities with putative CYP56 proteins from Candida glabrata (Q6FUL1), Aspergillus clavatus (A1CD47), Aspergillus flavus (AFL2G_06061), Aspergillus oryzae (Q2U8G8), and Aspergillus nidulans (Q5B9S4), respectively. Following the completion of this work, the DIT2 gene (ORF19.544) was annotated in the most recent assembly (assembly 21) of the C. albicans SC5314 genome sequence (http://www.candidagenome.org).
Biochemical analysis of recombinant C. albicans CYP56.
Following heterologous expression in E. coli, CYP56 was purified as described previously (17) and exhibited a slight red-brown band during purification through the Ni2+-NTA agarose matrix. Analysis by SDS-polyacrylamide gel electrophoresis indicated that the purified CYP56 protein with a histidine tag had a molecular mass of 58 kDa. A characteristic Soret peak at 448 nm was obtained when the Ni2+-NTA agarose-purified protein was reduced in the presence of carbon monoxide (Fig. 1). Control cells containing the expression vector plasmid only did not exhibit the peak at 448 nm when the membrane fraction was reduced in the presence of carbon monoxide.
FIG. 1.
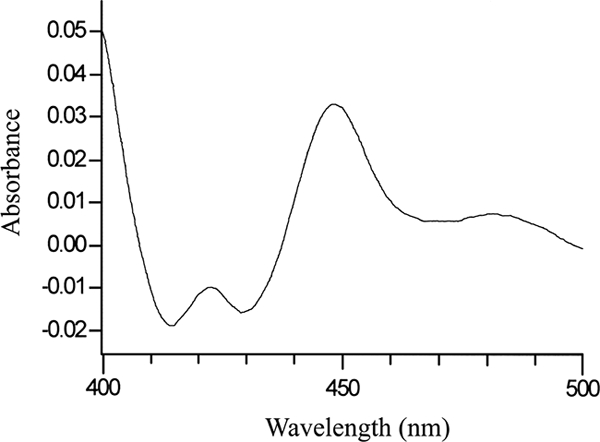
Carbon monoxide difference spectrum of CYP56. The carbon monoxide difference spectrum of Ni2+-NTA agarose-purified, reduced CYP56 (18.4 mg·ml−1) was determined using the method of Omura and Sato (22). A characteristic γ-Soret peak was observed at 448 nm when dithionite-reduced CYP56 bound carbon monoxide.
Spectral studies of CYP56 substrate binding.
The binding of N-formyl tyrosine to Ni2+-NTA agarose-purified CYP56 was monitored spectrophotometrically (320 to 470 nm). N-Formyl tyrosine was progressively titrated (26 to 253 μM) against a 2 μM solution of the CYP56 protein until saturation was observed (as indicated by no further increase in the absorbance difference between 390 and 412 nm). In the presence of N-formyl tyrosine, CYP56 produced a type I substrate binding spectrum (Fig. 2A) with a ks of 26 μM, indicating a high affinity for the N-formyl tyrosine substrate. Furthermore, the binding of N-formyl tyrosine to CYP56 appeared to be allosteric, since a Hill plot (Fig. 2B) of the data gave a straight line with an observed Hill number of 1.6, suggesting that two substrate molecules bound to the CYP56 enzyme. It was not possible to determine whether both enzyme-bound N-formyl tyrosine molecules were directly involved in the enzyme catalytic reaction or whether one molecule behaved as an allosteric effector regulating the binding of the second molecule to the CYP56 catalytic site.
FIG. 2.

N-Formyl tyrosine binding by C. albicans CYP56. (A) N-Formyl tyrosine was progressively titrated against Ni2+-NTA agarose-purified C. albicans CYP56 at 2 μM. A type I substrate binding spectrum for N-formyl tyrosine was observed when spectrophotometric determinations were made between 320 and 470 nm after each addition of N-formyl tyrosine. (B) Hill plot of the N-formyl tyrosine binding data. A plot of ΔA390-412 against the N-formyl tyrosine concentration (data not shown) was constructed to determine the observed ΔAmax value by using the Hill equation. The ΔAmax value was subsequently used to construct the Hill plot, from which the apparent Hill number and ks for N-formyl tyrosine were calculated.
Reconstitution of CYP56 activity.
In reconstituted enzymatic activity experiments, C. albicans CYP56 and S. cerevisiae Ncpr1p were incorporated into dilauroyl phosphatidylcholine micelles prior to the addition of N-formyl tyrosine and initiation of the reaction with NADPH. The production of N,N′-bisformyl dityrosine by CYP56 was demonstrated for the first time with purified CYP56. TLC and LC-MS-MS were employed for the identification of N,N′-bisformyl dityrosine bands produced in the reconstitution reaction. The elution of N,N′-bisformyl dityrosine (molecular mass, 416.4 Da) from the C18 HPLC column eluent was monitored by measuring the relative intensity of the 417 to 343 m/z SRM transition using MS-MS. The control horseradish peroxidase-synthesized N,N′-bisformyl dityrosine eluted with a retention time of 20.7 min (Fig. 3A). N,N′-Bisformyl dityrosine was detected in the CYP56 reconstitution assay product (Fig. 3B), eluting similarly at 21.4 min. The presence of dityrosine (molecular mass, 360.4 Da) in the eluent was also monitored by measuring the relative intensity of the 361 to 315 m/z SRM transition using MS-MS. No dityrosine was found in the CYP56 reconstitution assay product. Control experiments in which NADPH was omitted did not yield N,N′-bisformyl dityrosine. The reconstitution assay demonstrated that the CYP56 fraction converted N-formyl tyrosine to the oxidation product N,N′-bisformyl dityrosine (m/z 417) in the presence of Ncpr1p and NADPH. The retention time of this molecule under the HPLC conditions used was 21 min (Fig. 3). The turnover number observed for CYP56 N,N′-bisformyl dityrosine synthase activity was 21.6 min−1, which is similar to that of other CYPs engaged in biosynthetic functions.
FIG. 3.

Reconstitution of the N,N′-bisformyl dityrosine synthase catalytic activity of C. albicans CYP56. N,N′-Bisformyl dityrosine synthase activity assays were performed using 10 μM N-formyl tyrosine as the substrate. The assay product, N,N′-bisformyl dityrosine, was isolated by solvent extraction and TLC prior to analysis by LC-MS-MS along with a control horseradish peroxidase-synthesized sample of N,N′-bisformyl dityrosine. (A and B) N,N′-Bisformyl dityrosine was identified in the C18 column eluates of the control (A) and reconstitution assay (B) samples by MS-MS, with continuous monitoring of the relative abundance of the SRM transition from 417 to 343 m/z. (C) Full-scan MS of N,N′-bisformyl dityrosine confirmed the molecular mass ion to be 417 m/z. Other abundant mass ions present included a monohydroxylated N,N′-bisformyl dityrosine at 433.8 m/z and the molecular mass ion of formyl-tetratyrosine at 832.6 m/z.
Deletion of the C. albicans CYP56 gene.
Since C. albicans is diploid, the construction of a homozygous mutant defective in CYP56 required the deletion of two copies of the CYP56 gene (Fig. 4). A “knockout” mutant was constructed from strain SC5314 by using a cassette that contained the nourseothricin resistance marker and a Candida-adapted FLP recombinase-encoding gene that allowed the excision of the cassette following growth on maltose. Two rounds of integration and excision were necessary to create the homozygous mutant strain YNM11 (Fig. 4). A reconstituted strain was also constructed by integrating an intact copy of the CYP56 gene at the RP10 locus, yielding YNM12.
FIG. 4.

Disruption of the CYP56 gene. (A) Scheme of gene disruption of the SAT1 cassette contained in plasmid pSFS2. caFLP, C. albicans-adapted FLP gene; caSAT1, nourseothricin resistance marker; MAL, promoter; ACT1, transcription termination sequence; FRT, FLP recombination target. Correct integration was confirmed by Southern hybridization using probes from the DIT2KF and DIT2XR upstream region. (B) Southern hybridization analysis showed that all clones (lanes 4 to 13) had excised the SAT1 flipper by FLP recombination and had knocked out the second wild-type allele, inactivating the CYP56 gene. The upper band corresponds to the wild-type-copy of CYP56 and is absent in lanes 4 to 13. The lower band corresponds to the deleted copy of CYP56. Lanes: 1, SC5314; 2, heterozygous CYP56/cyp56Δ mutant; 3, positive-control DIT2 PCR product; 4 to 13, homozygous cyp56Δ/cyp56Δ mutants.
On standard YEPD agar, the homozygous cyp56Δ mutant strains exhibited a colony morphology similar to that of the wild-type parental strain. However, on minimal medium, the mutant exhibited a rough colony surface, in contrast to the smooth colonies exhibited by the wild-type strain (Fig. 5). Optical microscopy confirmed the presence of hyphae in the mutant strain and only yeast cells in the wild-type strain grown under these conditions (Fig. 5E and F). The homozygous cyp56Δ mutant strain also showed significantly less growth than the wild-type strain at 42°C in both YM and YEPD medium.
FIG. 5.

Visualization of the morphological features of strains grown on YM. (A and C) Smooth colonies shown by the wild-type (WT) strain SC5314; (B and D); rough colonies displayed by the homozygous cyp56Δ mutant YNM11. (E and F) Microscopic view of the yeast form in minimal medium broth. Optical microscopy confirmed the presence of yeast cells only in the WT strain (E) and the presence of hyphae in the mutant strain (F).
Antifungal binding and susceptibility testing.
Fluconazole, itraconazole, and voriconazole bound to CYP56, generating a type II binding spectrum with a minimum at 412 nm and a maximum at 430 nm due to coordination of the triazole N-3 with the heme as a sixth ligand. The binding spectra obtained with itraconazole are shown in Fig. 6, with the derived itraconazole saturation curve presented as an inset. The kd values for fluconazole, itraconazole, and voriconazole were determined from the azole saturation curves by nonlinear regression of the Hill equation (23). kd values of 0.23, 0.10, and 0.51 μM were obtained for fluconazole, itraconazole, and voriconazole, respectively.
FIG. 6.

Itraconazole binding by C. albicans CYP56. Itraconazole was progressively titrated against Ni2+-NTA agarose-purified C. albicans CYP56 at 2 μM. A characteristic type II azole-binding spectrum was observed when spectrophotometric determinations were made between 350 and 500 nm after each addition of itraconazole. (Inset) A plot of ΔA430-412 against the itraconazole concentration was constructed to determine the kd for itraconazole by using nonlinear regression (Levenberg-Marquardt algorithm) of the Hill equation.
The MICs of amphotericin B, fluconazole, and voriconazole for the wild-type strain were 1, 0.06, and 0.007 μg·ml−1, respectively. The MICs obtained for the YNM11 mutant were identical to those for the wild-type strain, indicating that the homozygous cyp56Δ mutant and its wild-type parental strain were equally susceptible to voriconazole and fluconazole.
The ability of CYP56 to bind azole antifungals suggested that it might contribute to the susceptibility of C. albicans to azole antifungal drugs. However, the MIC results indicate that the ability of CYP56 to bind azole drugs has no influence on the overall sensitivity of C. albicans to these agents. This is the first time such an investigation of the role of another CYP enzyme in the sensitivity of a fungus to CYP51 inhibitors has been carried out. In this case, the binding of azoles by CYP56 does not alter sensitivity, but such an effect cannot be excluded for other CYPs and in other fungi.
In contrast to its unchanged sensitivity to azole antifungals, the homozygous cyp56Δ mutant showed increased susceptibility to caspofungin, with a MIC of 0.015 μg·ml−1 compared to 0.03 μg·ml−1 for the wild-type parental strain and the reintegrant strain YNM12. This elevated sensitivity suggests an alteration in the cell wall, where glucan synthase is the target of echinocandin, and implies a role for dityrosine in the vegetative cell wall of C. albicans. A further implication is that inhibition of CYP56 activity could increase the efficacy of echinocandin in combination therapy.
Responses to osmotic and other stresses.
Since dityrosine is a component of the cell wall, assays to determine the effect of deleting the CYP56 gene on the integrity of the cell wall were performed. The homozygous deletion mutant was tested for susceptibility to a range of cell wall-perturbing agents and other agents whose effects have previously been associated with altered cell walls (8, 13, 25). The susceptibility of cells inoculated onto YEPD or YM agar plates supplemented with metabolic inhibitors was also investigated. Cells of the parental and mutant strains grown on YEPD agar plates did not reveal any difference in their susceptibilities to 1.5 M sorbitol, 100 μg·ml−1 SDS, 1 M or 1.5 M NaCl, 5 mM or 10 mM H2O2, 0.5 M or 1 M CaCl2, 100 μg·ml−1 calcofluor, or 100 μg·ml−1 Congo red when exposed at 37°C for 48 h.
Chlamydospore production and hyphal formation.
In cornmeal broth supplemented with 1% (vol/vol) Tween 80, wild-type C. albicans produced abundant pseudohyphae and chlamydospores after 72 h of static growth (without shaking) at room temperature (21). However, the homozygous cyp56Δ mutant produced abundant pseudohyphae but failed to produce chlamydospores under the same conditions. The mutant produced terminal buds on pseudohyphae but failed to produce mature, spherical, thick-walled spores (Fig. 7). Reintroduction of the CYP56 gene on plasmid CIp20 in the homozygous cyp56Δ mutant restored its ability to produce chlamydospores in cornmeal broth. Furthermore, chlamydospores of the wild-type strain fluoresced brightly when stimulated with UV light, in contrast to the cyp56Δ homozygous mutant, which did not produce chlamydospores (Fig. 7). This fluorescence was also restored in the complemented strain, suggesting that dityrosine production was restored. Chlamydospore formation is a developmental process that is clearly disrupted by the absence of CYP56, and this may be due to the alteration of cell wall composition caused by the reduced levels of dityrosine.
FIG. 7.

Imaging of cell fluorescence by fluorescence microscopy. (A and C) Light microscopic images (magnification, ×100); (B and D) fluorescence microscopic images. The wild-type (WT) strain, SC5314, produced chlamydospores on cornmeal broth (A), while the homozygous cyp56Δ mutant strain, YNM11, did not (C).
After the first report of the presence of dityrosine in C. albicans in 1995 (27), few studies have addressed the role of dityrosine in the biology of C. albicans. This is surprising, since components of the cell wall of C. albicans have proven to be excellent targets for antifungal drugs. The role of the novel CYP56 present in C. albicans was investigated here, and this enzyme was shown to be capable of producing N,N′-bisformyl dityrosine in vitro. CYP56 was also shown to bind two molecules of the N-formyl tyrosine substrate, raising the possibility that the C-C bond coupling of the two phenol rings occurs within the CYP56 catalytic site, followed by product release. This assertion will need to be verified by further experimentation, because at present we cannot distinguish between an allosteric regulatory role and a catalytic role for the second substrate molecule. The effects of deleting the CYP56 gene on C. albicans growth suggest that dityrosine plays an important role in the biology of this species. The precise nature of this role and the importance of dityrosine production by CYP56 in virulence are currently under investigation.
Acknowledgments
We are grateful to the CAPES—Brazilian ministry scholarship, the Irish Health Board, and the EU FP6 program EURESFUN for partial support. Work undertaken in the TCD Dental School Microbiology Unit was supported by Health Research Board grant RP/2005/2.
Footnotes
Published ahead of print on 28 July 2008.
REFERENCES
- 1.Amadò, R., R. Aeschbach, and H. Neukom. 1984. Dityrosine: in vitro production and characterization. Methods Enzymol. 107:377-388. [DOI] [PubMed] [Google Scholar]
- 2.Barnes, H. J., M. P. Arlotto, and M. R. Waterman. 1991. Expression and enzymatic activity of recombinant cytochrome P450 17α-hydroxylase in Escherichia coli. Proc. Natl. Acad. Sci. USA 88:5597-5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bogengruber, E., T. Eichberger, P. Briza, I. W. Dawes, M. Breitenbach, and R. Schricker. 1998. Sporulation-specific expression of the yeast DIT1/DIT2 promoter is controlled by a newly identified repressor element and the short form of Rim101p. Eur. J. Biochem. 258:430-436. [DOI] [PubMed] [Google Scholar]
- 4.Briza, P., M. Eckerstorfer, and M. Breitenbach. 1994. The sporulation-specific enzymes encoded by the DIT1 and DIT2 genes catalyze a two-step reaction leading to a soluble ll-dityrosine-containing precursor of the yeast spore wall. Proc. Natl. Acad. Sci. USA 91:4524-4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calderone, R. A., and P. C. Braun. 1991. Adherence and receptor relationships of Candida albicans. Microbiol. Rev. 55:1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giaever, G., A. M. Chu, L. Ni, C. Connelly, L. Riles, S. Veronneau, S. Dow, A. Lucau-Danila, K. Anderson, B. Andre, A. P. Arkin, A. Astromoff, M. El-Bakkoury, R. Bangham, R. Benito, S. Brachat, S. Campanaro, M. Curtiss, K. Davis, A. Deutschbauer, K. D. Entian, P. Flaherty, F. Foury, D. J. Garfinkel, M. Gerstein, D. Gotte, U. Guldener, J. H. Hegemann, S. Hempel, Z. Herman, D. F. Jaramillo, D. E. Kelly, S. L. Kelly, P. Kotter, D. LaBonte, D. C. Lamb, N. Lan, H. Liang, H. Liao, L. Liu, C. Luo, M. Lussier, R. Mao, P. Menard, S. L. Ooi, J. L. Revuelta, C. J. Roberts, M. Rose, P. Ross-Macdonald, B. Scherens, G. Schimmack, B. Shafer, D. D. Shoemaker, S. Sookhai-Mahadeo, R. K. Storms, J. N. Strathern, G. Valle, M. Voet, G. Volckaert, C. Y. Wang, T. R. Ward, J. Wilhelmy, E. A. Winzeler, Y. Yang, G. Yen, E. Youngman, K. Yu, H. Bussey, J. D. Boeke, M. Snyder, P. Philippsen, R. W. Davis, and M. Johnston. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387-391. [DOI] [PubMed] [Google Scholar]
- 7.Gillum, A. M., E. Y. Tsay, and D. R. Kirsch. 1984. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol. Gen. Genet. 198:179-182. [DOI] [PubMed] [Google Scholar]
- 8.Gow, N. A., S. Bates, A. J. Brown, E. T. Buurman, L. M. Thomson, and C. Westwater. 1999. Candida cell wall mannosylation: importance in host-fungus interaction and potential as a target for the development of antifungal drugs. Biochem. Soc Trans. 27:512-516. [DOI] [PubMed] [Google Scholar]
- 9.Hull, C. M., R. M. Raisner, and A. D. Johnson. 2000. Evidence for mating of the “asexual” yeast Candida albicans in a mammalian host. Science 289:307-310. [DOI] [PubMed] [Google Scholar]
- 10.Jefcoate, C. R. 1978. Measurement of substrate and inhibitor to microsomal cytochrome P-450 by optical-difference spectroscopy. Methods Enzymol. 52:258-279. [DOI] [PubMed] [Google Scholar]
- 11.Kelly, S. L., D. C. Lamb, A. J. Corran, B. C. Baldwin, L. W. Parks, and D. E. Kelly. 1995. Purification and reconstitution of activity of Saccharomyces cerevisiae P450 61, a sterol Δ22-desaturase. FEBS Lett. 377:217-220. [DOI] [PubMed] [Google Scholar]
- 12.Kelly, S. L., D. C. Lamb, C. J. Jackson, A. G. Warrilow, and D. E. Kelly. 2003. The biodiversity of microbial cytochromes P450. Adv. Microb. Physiol. 47:131-186. [DOI] [PubMed] [Google Scholar]
- 13.Konno, N., M. Ishii, A. Nagai, T. Watanabe, A. Ogasawara, T. Mikami, and T. Matsumoto. 2006. Mechanism of Candida albicans transformation in response to changes of pH. Biol. Pharm. Bull. 29:923-926. [DOI] [PubMed] [Google Scholar]
- 14.Lamb, D. C., M. Cannieux, A. G. Warrilow, S. Bak, R. A. Kahn, N. J. Manning, D. E. Kelly, and S. L. Kelly. 2001. Plant sterol 14α-demethylase affinity for azole fungicides. Biochem. Biophys. Res. Commun. 284:845-849. [DOI] [PubMed] [Google Scholar]
- 15.Lamb, D. C., D. E. Kelly, W. H. Schunck, A. Z. Shyadehi, M. Akhtar, D. J. Lowe, B. C. Baldwin, and S. L. Kelly. 1997. The mutation T315A in Candida albicans sterol 14α-demethylase causes reduced enzyme activity and fluconazole resistance through reduced affinity. J. Biol. Chem. 272:5682-5688. [DOI] [PubMed] [Google Scholar]
- 16.Lamb, D. C., D. E. Kelly, K. Venkateswarlu, N. J. Manning, H. F. Bligh, W. H. Schunck, and S. L. Kelly. 1999. Generation of a complete, soluble, and catalytically active sterol 14α-demethylase-reductase complex. Biochemistry 38:8733-8738. [DOI] [PubMed] [Google Scholar]
- 17.Lamb, D. C., S. Maspahy, D. E. Kelly, N. J. Manning, A. Geber, J. E. Bennett, and S. L. Kelly. 1999. Purification, reconstitution, and inhibition of cytochrome P-450 sterol Δ22-desaturase from the pathogenic fungus Candida glabrata. Antimicrob. Agents Chemother. 43:1725-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magee, B. B., and P. T. Magee. 2000. Induction of mating in Candida albicans by construction of MTLa and MTLα strains. Science 289:310-313. [DOI] [PubMed] [Google Scholar]
- 19.Magee, B. B., and P. T. Magee. 2005. Recent advances in the genomic analysis of Candida albicans. Rev. Iberoam. Micol. 22:187-193. [DOI] [PubMed] [Google Scholar]
- 20.National Committee for Clinical Laboratory Standards. 2002. Reference method for broth dilution antifungal susceptibility testing of yeasts. NCCLS document M27-2A. NCCLS, Wayne, PA.
- 21.Nobile, C. J., V. M. Bruno, M. L. Richard, D. A. Davis, and A. P. Mitchell. 2003. Genetic control of chlamydospore formation in Candida albicans. Microbiology 149:3629-3637. [DOI] [PubMed] [Google Scholar]
- 22.Omura, T., and R. Sato. 1964. The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J. Biol. Chem. 239:2379-2385. [PubMed] [Google Scholar]
- 23.Ouellet, H., L. M. Podust, and P. R. de Montellano. 2008. Mycobacterium tuberculosis CYP130: crystal structure, biophysical characterization, and interactions with antifungal azole drugs. J. Biol. Chem. 283:5069-5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reuss, O., A. Vik, R. Kolter, and J. Morschhauser. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119-127. [DOI] [PubMed] [Google Scholar]
- 25.Ruiz-Herrera, J., M. V. Elorza, E. Valentin, and R. Sentandreu. 2006. Molecular organization of the cell wall of Candida albicans and its relation to pathogenicity. FEMS Yeast Res. 6:14-29. [DOI] [PubMed] [Google Scholar]
- 26.Shepherd, M. G. 1987. Cell envelope of Candida albicans. Crit. Rev. Microbiol. 15:7-25. [DOI] [PubMed] [Google Scholar]
- 27.Smail, E. H., P. Briza, A. Panagos, and L. Berenfeld. 1995. Candida albicans cell walls contain the fluorescent cross-linking amino acid dityrosine. Infect. Immun. 63:4078-4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smail, E. H., and J. M. Jones. 1984. Demonstration and solubilization of antigens expressed primarily on the surfaces of Candida albicans germ tubes. Infect. Immun. 45:74-81. [DOI] [PMC free article] [PubMed] [Google Scholar]