

Abstract
Current clinical and microbiological information on acne fails to demonstrate a clear association between particular species, including Propionibacterium acnes, and disease, and the disease continues to be a considerable problem. To test if acne is associated with hitherto uncultured bacteria residing in diseased skin follicles, sequencing and phylogenetic analysis of approximately 5,700 amplified and cloned 16S rRNA genes were used to determine the microbial diversity in follicles from acne patients and healthy individuals and from the superficial skin of acne patients. Follicles from healthy skin were exclusively colonized by P. acnes, whereas the follicular microbiota of acne patients included, in addition, Staphylococcus epidermidis and minor proportions of other species. In comparison, samples from superficial skin showed a complex microbiota represented by 12 to 16 bacterial species. The findings of the study exclude the possibility that acne is associated with yet-uncultured bacteria and shows that healthy skin follicles constitute a remarkably exclusive habitat allowing colonization only by P. acnes.
Acne is a significant clinical problem with severe social, psychological, and emotional implications. The disease affects up to 80% of teenagers and frequently continues into adulthood. Clinically, acne is characterized by distended sebaceous follicles seen as open or closed comedones and inflamed lesions presenting as papules, pustules, and cysts (2, 19). The pathogenesis of acne appears to be multifactorial, although it is yet only partly understood. A mainly genetically determined host response pattern (3) combined with bacterial “triggering” is generally accepted as being important for the apparently unbalanced inflammatory activity.
Propionibacterium acnes, a regular inhabitant of human skin and sebaceous follicles, has been suspected to be an important triggering agent (4-6, 16). A significant increase in P. acnes colonization density is observed at puberty, concurrent with the onset of the disease (25). A preference for anaerobic conditions and the production of lipid-degrading lipases are believed to make P. acnes well suited to colonize the sebaceous follicles and to sustain this harsh environment (26). In addition, several studies have shown that P. acnes has potent immunostimulating properties, reflected in its ability to induce several proinflammatory cytokines and defensins through Toll-like receptors on keratinocytes, sebocytes, and monocytes (21, 29, 30, 37).
Although some indirect evidence supports the role of P. acnes as the causative agent of acne, other findings are in apparent conflict with this hypothesis. Some researchers have suggested that the involvement of P. acnes in the inflammation is relatively minor and that the predominance of P. acnes in the sebaceous ducts may be a side effect of the inflammation rather than the cause. P. acnes colonizes the skin and follicles of both acne-affected and healthy individuals, and no correlation has been observed between the absolute number of bacteria and the severity of disease (8, 9, 23, 25). It has even been reported that not all comedones or early inflamed lesions harbor microorganisms (23, 24). In addition, beta-lactam antibiotics are ineffective against acne, although P. acnes is fully sensitive to these drugs (36). Finally, prolonged periods of treatment with tetracyclines and macrolides are often necessary for the treatment of acne, in contrast to the shorter courses used for standard infections (9, 34). Studies that have attempted to explain these divisive findings have shown variations in host reactions to P. acnes colonization (37), differences among P. acnes strains (20, 27, 30, 31), and the ability of these bacteria to form local biofilms with reduced susceptibility to antimicrobials (7). In any case, the current treatment options are far from ideal. The use of broad-spectrum antibiotics has led to widespread resistance, and the alternative treatment, isotretinoin, is highly teratogenic and induces pronounced subjective side effects when it is used at therapeutic doses (11). A better understanding of the etiology of acne is essential to develop more efficient treatment options with fewer side effects.
Recent application of molecular techniques has demonstrated that the microbial diversity of most habitats is significantly underestimated by conventional culture-based methods. By amplification and sequencing of cloned libraries of the phylogenetically informative 16S rRNA gene, numerous hitherto unknown species were demonstrated in complex habitats such as the human oral cavity (1, 32) and intestines (12). Therefore, the possibility exists that previously uncultured bacteria resident in the sebaceous follicles play an etiologic role in the development of acne. This hypothesis was evaluated in the present study by mapping the microbial diversity of sebaceous follicles of acne-affected and healthy individuals. The study was based on sequencing of approximately 5,700 cloned PCR amplicons of the 16S rRNA gene generated from samples collected from diseased and healthy individuals.
MATERIALS AND METHODS
Patients.
Five patients and three healthy controls were recruited in Aarhus, Denmark, in 2006 and 2007 by advertising in local newspapers and by contact with local dermatologists. All patients were diagnosed and scored by the same specialist in dermatology (H.B.L.). The severity of disease was assessed by using the Leeds scoring system and grading of closed comedones, open comedones, papules, pustules, nodules and scar tissue into four grades: none (score 0), slight (score 1), moderate (score 2), or severe (score 3). Records of disease and medication were obtained, while the use of cosmetic products was not recorded. The clinical data are summarized in Table 1. None of the subjects included in the study were prescribed any form of acne therapy. All subjects were included on the basis of informed consent. The study protocol was approved by the Ethics Committee of the County of Aarhus, and the study was conducted according to the principles of the Declaration of Helsinki.
TABLE 1.
Patient characteristicsa
| Subject | Age (yr) | Gender | Leeds score | Grade
|
||||
|---|---|---|---|---|---|---|---|---|
| Closed comedones | Open comedones | Papules | Pustles | Scarring | ||||
| 1P | 15 | M | 3 | 2 | 1 | 2 | 2 | 0 |
| 2P | 33 | F | 3 | 3 | 1 | 3 | 1 | 3 |
| 3P | 23 | M | 5 | 1 | 1 | 2 | 3 | 2 |
| 4P | 36 | F | 5 | 1 | 1 | 3 | 1 | 1 |
| 5P | 17 | M | 2 | 1 | 2 | 2 | 2 | 0 |
| 1C | 25 | F | 0 | 0 | 0 | 0 | 0 | 0 |
| 2C | 30 | F | 0 | 0 | 0 | 0 | 0 | 0 |
| 3C | 24 | M | 0 | 0 | 0 | 0 | 0 | 0 |
Abbreviations: F, female; M, male; P, acne patient; C, healthy control.
Sample processing.
Follicular bacteria were sampled from acne-affected skin areas of the face by cyanoacrylate biopsy (17, 28). In control subjects, follicles on the nose were chosen as the target for two reasons. First, two of the patients had acne on the nose (the remaining were sampled from the skin of one cheek), and second, follicles on other areas of facial skin of healthy subjects were too few and small to generate samples with a sufficient number of bacteria. A thin layer of cyanoacrylate adhesive (Sikabond-585; Sika Danmark A/S Lynge, Denmark) was applied to a sterilized microscope glass slide and spread uniformly on an area of approximately 1.5 cm2 by pressing it against the skin for 90 s before removal. Seven to 15 follicular casts were dissected with a scalpel under a stereo microscope and pooled into a microcentrifuge tube containing lysis buffer (20 mM Tris-HCl, pH 8.0, 2 mM EDTA, 1.2% Triton X-100, 20 mg/ml lysozyme, 625 U/ml mutanolysin). The follicles were disrupted, and homogenates were prepared by the use of micropestles (Eppendorf AG, Hamburg, Germany). By using this sampling technique, we attempted to cover the full spectrum of acne follicles with regard to the age and the types of the lesions in the individual patient.
As a control for the ability of the method to map a complex microbiota, an acne-affected area of superficial skin on the cheeks of two patients was sampled by the detergent scrub technique of Williamson and Kligman (38) over an area of approximately 6 cm2. Two milliliters of wash fluid (0.075 M sodium phosphate [NaH2PO4·H2O], pH 7.9, 0.1% Triton X-100) was used for each sample. After the skin was scrubbed, the wash fluid was transferred to a microcentrifuge tube. The bacteria were harvested by centrifugation and were resuspended in lysis buffer. DNA from all samples was extracted with a DNeasy tissue kit (Qiagen, Hilden, Germany). The manufacturer's protocol for the isolation of genomic DNA from gram-positive bacteria was followed, with minor modifications. Incubation times were prolonged to 1.5 h for the lysis step and 1 h for the proteinase K degradation step. Samples were eluted in 50 μl of AE elution buffer.
PCR amplification of 16S rRNA genes.
An internal fragment corresponding to positions 28 to 1491 of the 16S rRNA genes (coordinates according to the Escherichia coli 16S rRNA gene) was amplified by universal bacterial primers 8F (forward primer; 5′-AGA GTT TGA TYM TGG CTC AG-3′) and 1492R (reverse primer; 5′-GGY TAC CTT GTT ACG ACT T-3′). These primers were used in a study of the microbiota of healthy skin reported by Gao et al. (13), with the exception that the reverse primer was modified to obtain an optimal coverage of bacteria according to Probe Match, the Ribosomal Database Project II (RDP II) database (http://rdp8.cme.msu.edu/docs/probe_match_doc.html). The PCR amplification reactions were carried out in a total volume of 25 μl containing 5 μl extracted template DNA, 1.2 μM of each primer, molecular-grade water (Eppendorf), and Hot Master mix, as recommended by the manufacturer (Eppendorf). The amplification comprised 5 min of incubation at 94°C, followed by 30 cycles of 1 min at 94°C, 1 min at 55°C, and 2 min at 72°C. A final extension was performed for 10 min at 72°C. Amplification products were visualized on a 1% agarose gel supplemented with ethidium bromide. To eliminate contaminating bacterial genomic DNA, the same batches of buffers and DNA extraction reagents were used for all samples and were tested for contamination by PCR amplification of a negative extraction control. For all PCRs, a negative control that included only the PCR reagents was examined in parallel by using procedures identical to those used for the follicle and skin samples. For none of these preparations was a visible band identified after electrophoresis and ethidium bromide staining.
16S rRNA gene clone libraries.
The PCR amplification products were ligated directly into the pCR4-TOPO cloning vector (Invitrogen) and cloned into One Shot TOP10 chemically competent E. coli cells (Invitrogen). Cloning and transformation reactions were performed as described by the supplier. Clones were checked for the presence of an insert of the correct size by vector-target PCR with universal primers M13F and M13R. A total of 576 clones from each library underwent sequence analysis with primers M13F and M13R, resulting in sequence reads of approximately 700 to 800 bp. Plasmid purification and sequencing were performed at the Beijing Genomics Institute (Beijing, China).
Sequence analysis.
Vector trimming and primer removal were performed manually. Alignment of the sequences amplified from each of the individuals was performed by use of the ClustalX program (35). The sequences were manually edited, and for identification purposes, the sequences encoding the first 500 bp of the 16S rRNA gene were obtained. All sequences were checked by use of the Chimera Check program of the RDP II database (http://rdp.cme.msu.edu/index.jsp). A total of 87 chimeric sequences were excluded from the phylogenetic analysis. Corrected and trimmed sequences were grouped into clusters of sequences at least 98% similar to members of their cluster, and representative sequences were determined with the FastgroupII program, accessible at http://biome.sdsu.edu/fastgroup/. The phylogenetic associations of representative sequences were determined with the help of the Sequence Match program, available at the RDP II and the GenBank (http://www.ncbi.nlm.nih.gov/BLAST/) databases, by using the standard nucleotide-nucleotide BLAST program to determine their closest relatives among designated type strains. The phylogenetic tree was constructed by the minimum evolution method, and evolutionary distances were calculated with the Kimura two-parameter algorithm integrated in the MEGA program (version 3.1) (22).
Estimation of microbial diversity.
The rarefaction curve and richness estimations were calculated with the EstimateS program (version 8.0), available at http://viceroy.eeb.uconn.edu/EstimateS. The sampled-based Mao Tau method was used to compute the rarefaction curve. The bias-corrected Chao1 estimator of species richness was calculated after 100 randomizations of sampling without replacement.
RESULTS
Clone analysis.
We analyzed a total of ca. 5,700 cloned bacterial 16S rRNA gene sequences from gene libraries generated from samples collected from healthy individuals and acne-affected patients. Approximately 4,600 of these clones were from samples of follicles from five acne patients with Leeds scores ranging from 2 to 5 and from three healthy subjects (Table 1). The remaining approximately 1,100 cloned sequences were from acne-affected areas of superficial skin from two acne patients. A sequence stretch corresponding to nucleotide positions 28 to 528 in the 16S rRNA gene of E. coli, which phylogenetically is the most informative part of the molecule, was analyzed and was used to determine identity or an approximate phylogenetic affiliation. The exclusion of truncated sequences and chimeras resulted in a total of 5,040 valid sequences, which were grouped into operational taxonomic units (OTUs). OTUs were defined as clusters of sequences that were at least 98% similar to members of their respective cluster. The taxonomic affiliation of OTUs was determined with the programs Sequence Match at the RDP II database and BLAST at NCBI, based on a comparison of the generated sequences to the 16S rRNA sequences of recognized species. Sequences of designated type strains were preferred as references, unless they were of insufficient quality.
Follicle samples.
After exclusion of unsuccessful and chimeric sequences, a total of 4,082 sequences generated from the follicles of acne patients and controls remained. Thus, a mean of 510 sequences (range, 475 to 537 sequences) from each of the eight subjects were analyzed. Compared to the bacterial community present on superficial skin, the bacterial community present in follicles was strikingly homogeneous. Thus, all sequences generated from follicles from the three healthy controls represented P. acnes. Although P. acnes was also dominant in the follicles from acne-affected patients, comprising 52.7 to 91.6% of all clones, additional species were detected. Staphylococcus epidermidis constituted 6.8 to 47.3% of the clones generated from the five patients. Minor proportions of additional species were detected in three of the patients (patients 2P, 3P, and 4P). Among these, Corynebacterium tuberculostearicum was detected in all three subjects and constituted 0.2 to 1.3% of the clones. In addition, patient 3P was colonized by a previously uncultured Actinobacterium sp. and by Staphylococcus hominis subsp. hominis. Patient 4P housed a yet uncultured Anaerococcus sp. and Propionibacterium granulosum.
Statistical analysis of the coverage showed that the number of species detected in the follicles covered the full spectrum of species present in the samples (data not shown).
Superficial skin samples.
To validate the nonculture technique, a total of 958 sequences obtained from acne-affected areas of the faces of two patients, patients 2P and 4P, were analyzed. The sequences clustered into 25 OTUs, 18 of which showed ≥98% similarity to the designated type strains of recognized species. The remaining OTUs showed ≥98% similarity to non-type strains of recognized species or to the PCR-generated 16S rRNA gene sequences of yet-uncultured bacteria. The taxonomic affiliations of the OTUs are listed in Table 2. The 25 OTUs identified belonged to four bacterial phyla, Actinobacteria, Firmicutes, Proteobacteria, and Bacteriodetes, and were distributed in 18 recognized genera and one yet-uncultured taxon belonging to the Betaproteobacteria. A total of 12 and 16 species were identified in samples from superficial acne-affected skin of the two subjects, respectively. As shown in Table 2, significant differences were observed between the two individuals. Only 3 of a total of 25 identified species were detected in both individuals. However, the three species carried by both of the acne patients, S. epidermidis, P. acnes, and Propionibacterium granulosum, accounted for 95% of all the sequences analyzed. In both subjects, the majority of sequences represented those of S. epidermidis, while P. acnes constituted the second most dominant species. Both species are known to be universal members of the bacterial community resident on human skin. Several of the species detected in minor proportions, e.g., Veillonella parvula, Streptococcus peroris, and Capnocytophaga gingivalis, are known to be regular residents of the oral cavity.
TABLE 2.
Bacterial diversity in follicles and superficial skin samples from acne-affected subjects and healthy controls
| Speciesa | No. of clones detected
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Healthy control follicles
|
Patient follicles
|
Patient scrub
|
Total | ||||||||
| 1C | 2C | 3C | 1P | 2P | 3P | 4P | 5P | 2P | 4P | ||
| Actinobacteria | |||||||||||
| Propionibacterium acnes (AB108480) | 485 | 517 | 522 | 283 | 362 | 471 | 402 | 454 | 192 | 95 | 3,783 |
| Propionibacterium granulosum | 3 | 4 | 31 | 38 | |||||||
| Corynebacterium tuberculostearicum | 6 | 1 | 6 | 14 | 27 | ||||||
| Actinobacterium sp. (AY770698) | 6 | 6 | |||||||||
| Micrococcus luteus | 2 | 2 | |||||||||
| Brevibacterium sanguinis | 1 | 1 | |||||||||
| Actinomyces sp. (AY349365) | 1 | 1 | |||||||||
| Nesterenkonia lacusekhoensis | 1 | 1 | |||||||||
| Corynebacterium rigelii | 1 | 1 | |||||||||
| Firmicutes | |||||||||||
| Staphylococcus epidermidis | 254 | 107 | 35 | 78 | 74 | 263 | 325 | 1136 | |||
| Uncultured Anaerococcus sp. (EF419345) | 15 | 2 | 17 | ||||||||
| Staphylococcus haemolyticus | 4 | 4 | |||||||||
| Staphylococcus hominis subsp. hominis | 1 | 1 | |||||||||
| Finegoldia magna (AB109770) | 1 | 1 | |||||||||
| Lactobacillus sp. (EU071482) | 2 | 2 | |||||||||
| Veillonella parvula | 1 | 1 | |||||||||
| Anaerococcus prevotii | 1 | 1 | |||||||||
| Streptococcus peroris | 1 | 1 | |||||||||
| Proteobacteria | |||||||||||
| Acinetobacter ursingii | 4 | 4 | |||||||||
| Acinetobacter lwoffii | 4 | 4 | |||||||||
| Acinetobacter johnsonii | 2 | 2 | |||||||||
| Enhydrobacter aerosaccus | 1 | 1 | |||||||||
| Neisseria bacilliformis | 1 | 1 | |||||||||
| Pseudoalteromonas nigrifaciens | 1 | 1 | |||||||||
| Uncultured Betaproteobacteria (AY225604) | 1 | 1 | |||||||||
| Kingella genomspecies P1 (DQ003616) | 1 | 1 | |||||||||
| Bacteriodetes | |||||||||||
| Capnocytophaga gingivalis | 1 | 1 | |||||||||
| Total | 485 | 517 | 522 | 537 | 475 | 514 | 504 | 528 | 490 | 468 | 5040 |
The GenBank accession number for the best matched sequence is cited in parentheses for non-type strains.
The total number of OTUs present on the superficial skin from the two acne-affected patients was estimated by the nonparametric estimator of total richness, Chao1 (Fig. 1). As expected, on the basis of the numerous species represented by a single clone (Table 2), the number of species present in the samples from superficial skin was not exhaustively revealed by the number of sequences analyzed. On the basis of these calculations, the combined bacterial communities in the samples from the two patients contained 43 OTUs (95% confidence interval ([CI]) range, 30 to 90). Thus, the 25 OTUs detected (95% CI range, 18 to 32) represent 58.1% (95% CI range, 35.5 to 60%) of the estimated number of species present in the superficial samples from acne-affected areas of the skin. Extrapolation of the Mao Tau graph in Fig. 1 indicates that sequencing of an additional ∼1,600 clones would be required to reveal the remaining OTUs, which were all present at proportions below 1%.
FIG. 1.
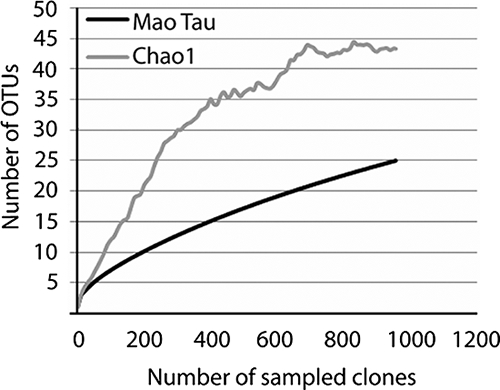
Rarefaction analysis of the combined bacterial 16S rRNA gene library of clones obtained from the superficial skin of two acne-affected individuals. The rarefaction curve, which plots the number of observed OTUs (Mao Tau) as a function of the number of clones, was computed by use of the EstimateS program, together with the corrected Chao1 estimator for species richness.
DISCUSSION
Our study evaluated the hypothesis that yet-uncultured bacteria play a role in the pathogenesis of acne. This was made possible by applying a PCR-based strategy with a sensitivity and a discriminatory power surpassing those of previously used culture-based methods. The 16S rRNA-based method was recently developed and has been applied in several studies of complex bacterial communities associated with humans, including the microbiota of healthy skin (1, 10, 12-14, 32) The 16S rRNA gene is present in all known bacteria and contains a combination of conserved regions suited as primer sites and variable regions ideal for species assignment. The sensitivity of the method depends on primer specificity and the number of sequences determined. The coverage of the primers used in our study was found to be significant, with the primers matching more than 55,000 bacteria representing 1,061 genera in the RDP II database. Similar primers were used in previous studies and revealed that the microbiota of the superficial skin of healthy individuals has considerable diversity (10, 13, 14).
As a validation of the method, the overall composition of the microbiota of superficial acne-affected skin that was demonstrated was very similar to that previously shown both for healthy human skin by a standard culture technique and for skin from the forearm and the forehead by the sequence-based technique used in this study (6, 10, 13). The results of these studies combined show that the skin microbiota of both healthy and acne-affected individuals contains a ubiquitous and dominant core of bacterial species, i.e., S. epidermidis and P. acnes. As an exception, Grice et al. (14) found Pseudomonas and Janthinobacterium species to be dominant in the more humid skin of the healthy human antecubital fossa, suggesting that significant regional differences may exist in the core bacterial species present on various skin surfaces. In addition, numerous species are present in minor proportions, and these proportions vary among individuals. The latter include bacteria that are recognized commensals of the oral cavity, which are presumably transiently present on the facial skin. In addition, our results and those reported by previous investigators (10, 13) clearly show that yet-uncultured phylotypes may be present but do not belong to the dominant core microbiota of the skin either in healthy individuals (10, 13) or in patients with acne (this study).
Sebaceous follicles, which are generally recognized to be the sites relevant to acne, must represent a uniquely harsh environment for bacteria, as evidenced by the surprising finding that only P. acnes colonized the follicles of healthy individuals. Our coverage analysis demonstrates that sequencing of more clones would not have revealed additional diversity. No other exposed microenvironment of the human body is known to selectively allow colonization by a single bacterial species. It is conceivable that the more heterogeneous microbiota found in previous culture-based studies of healthy follicles (15, 23, 33) was the result of differences in sampling methods. The cyanoacrylate method employed in the present study has the advantage, in addition to being noninvasive, that the upper part of the stratum corneum is captured in the applied cyanoacrylate, which minimizes the possibility of contamination with skin surface microorganisms when follicles are isolated by dissection. Microdissection of single follicles from biopsy punch specimens inevitably results in contamination with surface skin colonizers. Interestingly, Grice et al. (14) detected the same composition of skin microbiota on the antecubital fossa by use of swap, scrape, and punch biopsy techniques, all of which are likely to include surface bacteria.
The bacterial microbiota of follicles from acne-affected subjects showed more, although still very limited, diversity (Table 2). The microbiota was dominated by P. acnes and S. epidermidis, which were the only species consistently found. These results clearly exclude the possibility that yet-uncultured bacteria are associated with acne-affected skin follicles. Although the number of patients was limited, there was a complete absence of previously uncultured bacteria common to acne-affected follicles. It is unlikely that potential members of the microbiota present in numbers below the detection threshold of the method play a role in generating the intense inflammatory reaction typical of acne. We therefore conclude that yet-uncultured bacteria do not play an etiologic role in acne.
In contrast to previous culture-based studies (6, 15, 23), our molecular analysis revealed differences in the microbiota of follicles from healthy and acne-affected subjects. The main difference was the presence of S. epidermidis in acne-affected follicles (Table 2). There is evidence from histological studies with fluorescently labeled species-specific antibodies that the majority of staphylococci, when they are present, colonize the dilated openings of follicles, an area that is unlikely to be included by our method as a result of the previously mentioned effect of cyanoacrylate (18). Our detection of S. epidermidis and occasional other bacterial species exclusively in acne-affected follicles may be explained by the facilitated access of the bacteria to dilated, inflamed follicles and by the previous histological demonstration that staphylococci infiltrate tissues surrounding inflamed follicles (18). A possible alternative or additional explanation may be that the inflammatory reaction in the skin follicles of acne patients interferes with their apparently uniquely potent antimicrobial defense mechanisms.
The presence of S. epidermidis exclusively in acne-affected follicles raises the question of the potential role of this species. Previous studies excluded staphylococci as agents that play a role in the pathogenesis of acne on the basis of their rapid development of resistance to therapeutic antibiotics (15). It is possible that staphylococci are secondary colonizers, as inflamed and dilated follicles seem to be more conducive to colonization. However, on the basis of the findings of the present study, a reevaluation of the role of S. epidermidis may be advisable. It is widely accepted that microbes interact and that such interactions may influence their production of biologically reactive substances. Further studies are required to elucidate the potential symbiotic effects of the two species.
In conclusion, by using a genetically based strategy with a sensitivity and a discriminatory power surpassing those of culture-based methods, we demonstrated that the follicles of healthy skin constitute a uniquely selective habitat for P. acnes, which was the only species detected. A microbiota that was more heterogeneous but that had limited complexity was demonstrated in acne-affected follicles. The results exclude the possibility that yet-uncultured bacteria are associated with acne.
Acknowledgments
The study was supported by an unconditional grant from LEO Pharma, Ballerup, Denmark.
Footnotes
Published ahead of print on 20 August 2008.
REFERENCES
- 1.Aas, J. A., B. J. Paster, L. N. Stokes, I. Olsen, and F. E. Dewhirst. 2005. Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol. 435721-5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ayer, J., and N. Burrows. 2006. Acne: more than skin deep. Postgrad. Med. J. 82500-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bataille, V., H. Snieder, A. J. MacGregor, P. Sasieni, and T. D. Spector. 2002. The influence of genetics and environmental factors in the pathogenesis of acne: a twin study of acne in women. J. Investig. Dermatol. 1191317-1322. [DOI] [PubMed] [Google Scholar]
- 4.Brüggemann, H. 2005. Insights in the pathogenic potential of Propionibacterium acnes from its complete genome. Semin. Cutan. Med. Surg. 2467-72. [DOI] [PubMed] [Google Scholar]
- 5.Brüggemann, H., A. Henne, F. Hoster, H. Liesegang, A. Wiezer, A. Strittmatter, S. Hujer, P. Durre, and G. Gottschalk. 2004. The complete genome sequence of Propionibacterium acnes, a commensal of human skin. Science 305671-673. [DOI] [PubMed] [Google Scholar]
- 6.Chiller, K., B. A. Selkin, and G. J. Murakawa. 2001. Skin microflora and bacterial infections of the skin. J. Investig. Dermatol. Symp. Proc. 6170-174. [DOI] [PubMed] [Google Scholar]
- 7.Coenye, T., E. Peeters, and H. J. Nelis. 2007. Biofilm formation by Propionibacterium acnes is associated with increased resistance to antimicrobial agents and increased production of putative virulence factors. Res. Microbiol. 158386-392. [DOI] [PubMed] [Google Scholar]
- 8.Cogen, A. L., V. Nizet, and R. L. Gallo. 2008. Skin microbiota: a source of disease or defence? Br. J. Dermatol. 158442-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cove, J. H., W. J. Cunliffe, and K. T. Holland. 1980. Acne vulgaris: is the bacterial population size significant? Br. J. Dermatol. 102277-280. [DOI] [PubMed] [Google Scholar]
- 10.Dekio, I., H. Hayashi, M. Sakamoto, M. Kitahara, T. Nishikawa, M. Suematsu, and Y. Benno. 2005. Detection of potentially novel bacterial components of the human skin microbiota using culture-independent molecular profiling. J. Med. Microbiol. 541231-1238. [DOI] [PubMed] [Google Scholar]
- 11.Eady, E. A., M. Gloor, and J. J. Leyden. 2003. Propionibacterium acnes resistance: a worldwide problem. Dermatology 20654-56. [DOI] [PubMed] [Google Scholar]
- 12.Eckburg, P. B., E. M. Bik, C. N. Bernstein, E. Purdom, L. Dethlefsen, M. Sargent, S. R. Gill, K. E. Nelson, and D. A. Relman. 2005. Diversity of the human intestinal microbial flora. Science 3081635-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao, Z., C. H. Tseng, Z. Pei, and M. J. Blaser. 2007. Molecular analysis of human forearm superficial skin bacterial biota. Proc. Natl. Acad. Sci. USA 1042927-2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grice, E. A., H. H. Kong, G. Renaud, A. C. Young, G. G. Bouffard, R. W. Blakesley, T. G. Wolfsberg, M. L. Turner, and J. A. Segre. 2008. A diversity profile of the human skin microbiota. Genome Res. 181043-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holland, K. T. 1989. Microbiology of acne, p. 178-210. In W. J. Cunliffe (ed.), Acne. Martin Dunitz Ltd., London, United Kingdom.
- 16.Holland, K. T., W. J. Cunliffe, and C. D. Roberts. 1978. The role of bacteria in acne vulgaris: a new approach. Clin. Exp. Dermatol. 3253-257. [DOI] [PubMed] [Google Scholar]
- 17.Holland, K. T., and C. D. Roberts. 1974. A technique for sampling micro-organisms from the pilo-sebaceous ducts. J. Appl. Bacteriol. 37289-296. [DOI] [PubMed] [Google Scholar]
- 18.Imamura, S. 1975. The localization and distribution of gram-positive cocci in normal skin and in lesions of acne vulgaris. J. Investig. Dermatol. 65244-247. [DOI] [PubMed] [Google Scholar]
- 19.James, W. D. 2005. Clinical practice. Acne. N. Engl. J. Med. 3521463-1472. [DOI] [PubMed] [Google Scholar]
- 20.Jappe, U., R. Boit, M. D. Farrar, E. Ingham, J. Sandoe, and K. T. Holland. 2004. Evidence for diversity within Propionibacterium acnes: a comparison of the T-cell stimulatory activity of isolates from inflammatory acne, endocarditis and the laboratory. J. Eur. Acad. Dermatol. Venereol. 18450-454. [DOI] [PubMed] [Google Scholar]
- 21.Kim, J., M. T. Ochoa, S. R. Krutzik, O. Takeuchi, S. Uematsu, A. J. Legaspi, H. D. Brightbill, D. Holland, W. J. Cunliffe, S. Akira, P. A. Sieling, P. J. Godowski, and R. L. Modlin. 2002. Activation of Toll-like receptor 2 in acne triggers inflammatory cytokine responses. J. Immunol. 1691535-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar, S., K. Tamura, and M. Nei. 2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 5150-163. [DOI] [PubMed] [Google Scholar]
- 23.Leeming, J. P., K. T. Holland, and W. J. Cuncliffe. 1988. The microbial colonization of inflamed acne vulgaris lesions. Br. J. Dermatol. 118203-208. [DOI] [PubMed] [Google Scholar]
- 24.Leeming, J. P., K. T. Holland, and W. J. Cunliffe. 1984. The microbial ecology of pilosebaceous units isolated from human skin. J. Gen. Microbiol. 130803-807. [DOI] [PubMed] [Google Scholar]
- 25.Leyden, J. J., K. J. McGinley, O. H. Mills, and A. M. Kligman. 1975. Propionibacterium levels in patients with and without acne vulgaris. J. Investig. Dermatol. 65382-384. [DOI] [PubMed] [Google Scholar]
- 26.Marples, R. R. 1974. The microflora of the face and acne lesions. J. Investig. Dermatol. 62326-331. [DOI] [PubMed] [Google Scholar]
- 27.McDowell, A., S. Valanne, G. Ramage, M. M. Tunney, J. V. Glenn, G. C. McLorinan, A. Bhatia, J. F. Maisonneuve, M. Lodes, D. H. Persing, and S. Patrick. 2005. Propionibacterium acnes types I and II represent phylogenetically distinct groups. J. Clin. Microbiol. 43326-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mills, O. H., Jr., and A. M. Kligman. 1983. The follicular biopsy. Dermatologica 16757-63. [DOI] [PubMed] [Google Scholar]
- 29.Nagy, I., A. Pivarcsi, K. Kis, A. Koreck, L. Bodai, A. McDowell, H. Seltmann, S. Patrick, C. C. Zouboulis, and L. Kemeny. 2006. Propionibacterium acnes and lipopolysaccharide induce the expression of antimicrobial peptides and proinflammatory cytokines/chemokines in human sebocytes. Microbes Infect. 82195-2205. [DOI] [PubMed] [Google Scholar]
- 30.Nagy, I., A. Pivarcsi, A. Koreck, M. Szell, E. Urban, and L. Kemeny. 2005. Distinct strains of Propionibacterium acnes induce selective human beta-defensin-2 and interleukin-8 expression in human keratinocytes through Toll-like receptors. J. Investig. Dermatol. 124931-938. [DOI] [PubMed] [Google Scholar]
- 31.Oprica, C., L. Emtestam, J. Lapins, E. Borglund, F. Nyberg, K. Stenlund, L. Lundeberg, E. Sillerstrom, and C. E. Nord. 2004. Antibiotic-resistant Propionibacterium acnes on the skin of patients with moderate to severe acne in Stockholm. Anaerobe 10155-164. [DOI] [PubMed] [Google Scholar]
- 32.Paster, B. J., S. K. Boches, J. L. Galvin, R. E. Ericson, C. N. Lau, V. A. Levanos, A. Sahasrabudhe, and F. E. Dewhirst. 2001. Bacterial diversity in human subgingival plaque. J. Bacteriol. 1833770-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puhvel, S. M., R. M. Reisner, and D. A. Amirian. 1975. Quantification of bacteria in isolated pilosebaceous follicles in normal skin. J. Investig. Dermatol. 65525-531. [DOI] [PubMed] [Google Scholar]
- 34.Skidmore, R., R. Kovach, C. Walker, J. Thomas, M. Bradshaw, J. Leyden, C. Powala, and R. Ashley. 2003. Effects of subantimicrobial-dose doxycycline in the treatment of moderate acne. Arch. Dermatol. 139459-464. [DOI] [PubMed] [Google Scholar]
- 35.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 254876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tyrrell, K. L., D. M. Citron, Y. A. Warren, H. T. Fernandez, C. V. Merriam, and E. J. Goldstein. 2006. In vitro activities of daptomycin, vancomycin, and penicillin against Clostridium difficile, C. perfringens, Finegoldia magna, and Propionibacterium acnes. Antimicrob. Agents Chemother. 502728-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Webster, G. F. 1998. Inflammatory acne represents hypersensitivity to Propionibacterium acnes. Dermatology 19680-81. [DOI] [PubMed] [Google Scholar]
- 38.Williamson, P., and A. M. Kligman. 1965. A new method for the quantitative investigation of cutaneous bacteria. J. Investig. Dermatol. 45498-503. [DOI] [PubMed] [Google Scholar]