

Abstract
Extracellular Phr pentapeptides produced by gram-positive, spore-forming bacteria regulate processes during the transition from exponential- to stationary-phase growth. Phr pentapeptides are produced by cleavage of their precursor proteins. We determined the residues that direct this cleavage for the Bacillus subtilis Phr peptide, CSF, which is derived from the C terminus of PhrC. Strains expressing PhrC with substitutions in residues −1 to −5 relative to the cleavage site had a defect in CSF production. The mutant PhrC proteins retained a functional signal sequence for secretion, as assessed by secretion of PhrC-PhoA fusions. To determine whether the substitutions directly affected cleavage of PhrC to CSF, we tested cleavage of synthetic pro-CSF peptides that corresponded to the C terminus of PhrC and had an amino acid substitution at the −2, −3, or −4 position. The mutant pro-CSF peptides were cleaved less efficiently to CSF than the wild-type pro-CSF peptide whether they were incubated with whole cells, cell wall material, or the processing protease subtilisin or Vpr. To further define the range of amino acids that support CSF production, the amino acid at the −4 position of PhrC was replaced by the 19 canonical amino acids. Only four substitutions resulted in a >2-fold defect in CSF production, indicating that this position is relatively immune to mutational perturbations. These data revealed residues that direct cleavage of CSF and laid the groundwork for testing whether other Phr peptides are processed in a similar manner.
Gram-positive bacteria secrete small peptides into their environment that are used to self-monitor population density and/or the diffusivity of the environment, processes that are referred to as quorum sensing (3, 11, 19). The Phr peptides are pentamers that are secreted by gram-positive, endospore-forming bacteria to mediate quorum sensing or to control the timing of gene expression (26, 28). While much is known about the mechanisms involved in sensing and responding to the Phr peptides, there are still questions regarding the production of these peptides.
The Phr signaling peptides were first identified in Bacillus subtilis, where their functions include control of the development of genetic competence (the ability to take up exogenous DNA), sporulation (the formation of environmentally resistant spores), excision and transfer of a mobile DNA element, and production of extracellular degradative enzymes (2, 23, 26, 28). In Bacillus anthracis, Phr signaling peptides regulate sporulation, and in Bacillus cereus and Bacillus thuringiensis, a Phr-type signaling peptide regulates expression of virulence genes (5, 29). The functions of putative Phr signaling peptides encoded by the genomes of other bacteria have not been characterized (26, 28).
The B. subtilis Phr peptide, CSF, is a prototypical Phr peptide. This pentameric peptide (sequence, ERGMT) is derived from the C terminus of the PhrC precursor protein (31). PhrC has an N-terminal signal sequence for export through the Sec-dependent export pathway (28). When it is extracellular, PhrC is processed by one of three redundant proteases, subtilisin, Epr, or Vpr, to release CSF (15). At a critical extracellular concentration, CSF is transported into the cell by an oligopeptide permease (17). Once it is inside the cell, CSF interacts with cytoplasmic receptor proteins, RapC and RapB, to inhibit their activity (7, 25). RapC binds to and inhibits the DNA-binding activity of the ComA transcription factor (7), which regulates the expression of genes involved in extracellular and membrane functions, as well as genetic competence development (6, 24). By inhibiting RapC, CSF stimulates expression of ComA-controlled genes. ComA-controlled gene expression is similarly stimulated by several other Phr peptides, including PhrF, PhrG, PhrH, and PhrK (1, 4, 10, 30). However, CSF also inhibits ComA-controlled gene expression at higher concentrations by an incompletely understood mechanism (4, 16, 17). RapB, the other identified cytoplasmic receptor for CSF, dephosphorylates Spo0F, a response regulator protein required for sporulation (34). RapC, RapB, and the other identified cytoplasmic receptor proteins of Phr peptides are all members of the tetratricopeptide repeat domain family of proteins (7, 26).
Some Phr signaling peptides are derived from the C termini of their precursor proteins, whereas others are derived from internal portions (12, 31). The identity of the determinants of the cleavage site for release of the Phr pentapeptides is an important unanswered question. To address this question for B. subtilis Phr peptides, we previously aligned the B. subtilis Phr precursor proteins based on the known or predicted mature pentapeptide sequences (28). A loose consensus sequence was identified; this sequence was not a strict amino acid sequence but consisted of a string of amino acids with particular chemical characteristics. It was located at the five residues (residues −5 to −1) preceding the cleavage site; however, for 3 of the 13 Phr proteins insertion of a one-residue gap was necessary for alignment (Table 1). We hypothesized that these five residues could be important for directing the cleavage event. Consistent with this hypothesis, amino acid substitutions at the −1 and −3 positions relative to the cleavage site for PhrA, PhrE, and CSF decreased the expression of genes controlled by these Phr peptides (33). Here, we demonstrate that changes in any of the five residues preceding the cleavage site in PhrC reduced CSF production and directly affected cleavage of synthetic pro-CSF peptides.
TABLE 1.
Cleavage sites for B. subtilis Phr peptides
| Phr protein | Pro sequencea | Mature peptide | Reference(s) |
|---|---|---|---|
| PhrA | NTEGKTFHIA | ARNQT | 17, 25 |
| PhrC | NAEALDFHVT | ERGMT | 7, 31 |
| PhrE | SMYNGEMKEA | SRNVTb | 12 |
| PhrF | NAPTHQIEVA | QRGMI | 4 |
| PhrG | QTHSQEMKVA | EKMIG | 23 |
| PhrHc | LTNSGGFKEST | DRNTTb,d | 10 |
| PhrI | QSDHTEIKVAA | DRVGA | 2 |
| PhrK | TDSPSNIQVA | ERPVGb,d | 26 |
| Phr-pLS20 | ADSPGQFQVA | QKGMYd | 26, 28 |
| Phr-pTA1040 | IQQDSNVSVA | SRKATd | 28 |
| Phr-pPOD2000 | ATQDNSVSVA | SRNATd | 28 |
| Phr-pTA1060 | FVHHDEVSVAA | SRNAT | 14 |
| Phr-pPL10-1e | NHTETSMELA | IRFVTd | 26 |
| Consensus | ahcVA |
Sequences of the 10 or 11 amino acids preceding the N-terminal cleavage site for release of the mature Phr pentapeptides. The consensus sequence for the five residues preceding the cleavage site is (a/p)h(c/p)VA, where “a” is an acidic or polar residue, “h” is a hydrophobic residue, and “c” is a charged or polar residue. Bold type indicates residues that are the same as residues in the consensus sequence. Underlining indicates residues that are different than residues in the consensus sequence.
There are amino acids C terminal to the sequence of the mature Phr peptide in the precursor Phr protein.
The PhrH peptide fits the consensus least well, and the prediction for the mature pentapeptide sequence should be interpreted with caution.
Predicted sequence of a Phr peptide for which there is not direct experimental evidence. The predicted Phr peptide was chosen because its sequence was most similar to the sequence of a known Phr peptide of B. subtilis.
Phr-pPL10-1 is encoded on a Bacillus pumilus plasmid.
MATERIALS AND METHODS
Growth conditions.
B. subtilis cells were grown with shaking at 37°C in S7 minimal medium (35), except that the concentration of MOPS (morpholinepropanesulfonic acid) was 50 mM rather than 100 mM. This medium contained 1% glucose, 0.1% glutamate, and required amino acids (tryptophan, phenylalanine, and, when necessary, threonine) at a concentration of 50 μg/ml. When appropriate, 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added, and antibiotics were added at the following concentrations: ampicillin, 100 μg/ml; chloramphenicol, 5 μg/ml; erythromycin,0.5 μg/ml; neomycin, 5 μg/ml; spectinomycin, 100 μg/ml; and tetracycline, 12.5 μg/ml.
Strain and plasmid construction.
B. subtilis strains used in this study are listed in Table 2. The B. subtilis strains were constructed by transformation with chromosomal DNA or plasmids using standard protocols (9).
TABLE 2.
Strains used in this study
| Strain | Relevant genotypea | Reference or constructionb |
|---|---|---|
| BAL125 | amyE::(srfA-lacZΩ374 neo) ΔphrC::erm | 16 |
| BAL201 (AG1520) | ΔcomQ::spc | 20 |
| BAL218 (JH642) | trpC2 pheA1 | 27 |
| BAL376 (JRL358) | Δopp::erm (formerly Δspo0K::erm) | 18 |
| BAL397 | amyE::[Φ(rapA-lacZ)42 erm] (formerly gisA-lacZ) | 21 |
| BAL941 | ΔphrC::tet | 15 |
| BAL950 | Δopp::erm ΔcomQ::spc ΔphrC::tet | BAL201 + BAL376→BAL941 |
| BAL1147 | amyE::[Φ(rapA-lacZ)42 erm] ΔphrC::tet | BAL941→BAL397 |
| BAL1192 | ΔphrC::tet thrC::(Pspachyerm) | pBL112→BAL941 |
| BAL1191 | ΔphrC::tet thrC::(Pspachy-phrC erm) | pBL24→BAL941 |
| BAL1182 | Same as BAL1191 but phrCT35A | pBL129→BAL941 |
| BAL1183 | Same as BAL1191 but phrCT35K | pBL130→BAL941 |
| BAL1184 | Same as BAL1191 but phrCV34E | pBL291→BAL941 |
| BAL1185 | Same as BAL1191 but phrCH33A | pBL292→BAL941 |
| BAL1186 | Same as BAL1191 but phrCF32K | pBl293→BAL941 |
| BAL1187 | Same as BAL1191 but phrCD31A | pBL128→BAL941 |
| BAL2200 | ΔphrC::tet thrC::[Pspachy-Φ(phrC-′phoA) erm] | pBL483→BAL941 |
| BAL2199 | Same as BAL2200 but phrCRBS | pBL482→BAL941 |
| BAL2201 | Same as BAL2200 but phrCT35A | pBL484→BAL941 |
| BAL2202 | Same as BAL2200 but phrCT35K | pBL485→BAL941 |
| BAL2203 | Same as BAL2200 but phrCV34E | pBL486→BAL941 |
| BAL2204 | Same as BAL2200 but phrCH33A | pBL487→BAL941 |
| BAL2205 | Same as BAL2200 but phrCF32K | pBL488→BAL941 |
| BAL2206 | Same as BAL2200 but phrCD31A | pBL489→BAL941 |
| BAL2223 | amyE::[Φ(rapA-lacZ)42 ermC] ΔphrC::tet thrC::(Pspachy-phrC cat) | pBL495→BL1147 |
| BAL3012 | Same as BAL2223 but phrCF32P | pBL658→BL1147 |
| BAL3013 | Same as BAL2223 but phrCF32N | pBL660→BL1147 |
| BAL3014 | Same as BAL2223 but phrCF32E | pBL661→BL1147 |
| BAL3015 | Same as BAL2223 but phrCF32D | pBL663→BL1147 |
| BAL3016 | Same as BAL2223 but phrCF32I | pBL665→BL1147 |
| BAL3017 | Same as BAL2223 but phrCF32Q | pBL651→BL1147 |
| BAL3022 | Same as BAL2223 but phrCF32L | pBL666→BL1147 |
| BAL3023 | Same as BAL2223 but phrCF32Y | pBL667→BL1147 |
| BAL3024 | Same as BAL2223 but phrCF32C | pBL668→BL1147 |
| BAL3025 | Same as BAL2223 but phrCF32W | pBL669→BL1147 |
| BAL3026 | Same as BAL2223 but phrCF32H | pBL657→BL1147 |
| BAL3027 | Same as BAL2223 but phrCF32V | pBL662→BL1147 |
| BAL3028 | Same as BAL2223 but phrCF32M | pBL664→BL1147 |
| BAL3029 | Same as BAL2223 but phrCF32S | Backcrossed mutant from screen |
| BAL3030 | Same as BAL2223 but phrCF32G | Backcrossed mutant from screen |
| BAL3031 | Same as BAL2223 but phrCF32R | Backcrossed mutant from screen |
| BAL3032 | Same as BAL2223 but phrCF32A | pBL659→BL1147 |
| BAL3034 | Same as BAL2223 but phrCF32T | pBL670→BL1147 |
All strains are derivatives of BAL218 and contain trpC2 pheA1.
For strain construction an arrow indicates the direction of transfer of DNA into the recipient strain. The DNA is either genomic DNA from the indicated strain or the indicated plasmid.
Plasmid pBL495, containing the thrC::(Pspachy-phrC cat) allele, was constructed as follows. First, plasmid pBL354, containing the thrC::cat allele, was constructed by swapping the erm locus of plasmid pDG1664 (8) with the cat locus of pGEM-cat (36). All of pDG1664 except the erm locus was PCR amplified using Taq polymerase (Qiagen) and primers BL322 (5′-CAATTTCGTAATCGGAACGGTATCGG-3′) and BL323 (5′-CGTTACTAATCGCGAAGGGAATGTAG-3′), and the cat locus was amplified from pGEM-cat using Vent polymerase (New England Biolabs) and primers CM1 (5′-AAGCATGCGTTACCCTTATTATCAAGA-3′) and CM2 (5′-AAGCATGCGGAGCTGTAATATAAAAAC-3′). The ends of the pDG1664 PCR product were blunted using an end repair kit (Epicentre, Madison, WI), and then the two PCR products were ligated to generate plasmid pBL354, in which cat is transcribed in the same direction as thrC. pBL354 was then digested with EcoRI and BamHI and ligated to an EcoRI-BamHI fragment containing lacI and Pspachy-phrC from pBL24 (15) to create pBL495.
thrC::(Pspachy-phrC erm) alleles with mutations affecting the five residues preceding the cleavage site for CSF and thrC::(Pspachy-phrC cat) alleles with mutations affecting residue 32 (position −4) of phrC were constructed by site-directed mutagenesis of plasmids pBL24 (15) and pBL495, respectively, using a QuikChange site-directed mutagenesis kit (Stratagene). The phrC genes were then sequenced to confirm the presence of the desired mutations. The primers used for mutagenesis and the designations of the resulting plasmids are shown in Table S1 in the supplemental material. B. subtilis cells were transformed with these plasmids, and either erythromycin- or chloramphenicol-resistant transformants were selected as appropriate. Transformants were checked for replacement of the thrC locus by Pspachy-phrC based on the lack of spectinomycin resistance associated with the plasmids and auxotrophy for threonine.
thrC::[Pspachy-φ(phrC-′phoA) erm] alleles were constructed as follows. A portion of phoA encoding an alkaline phosphatase lacking the N-terminal signal sequence was PCR amplified from chromosomal DNA of Escherichia coli strain MC4100 with primers BL474 (5′-GAAACCCGGGTACCGTTACTGTTTACCC-3′) and BL466 (5′-GGTTAGATCTGCTAACAGCAAAAAAACCACCCG G-3′) containing SmaI and BglII sites (underlined), respectively. The amplified phoA gene was cloned into the corresponding sites of pBL112 (15). The various alleles of the phrC gene were released from pBL24 (15) and cloned into the HindIII and SmaI sites of the pBL112 plasmid containing phoA. Site-directed mutagenesis (Stratagene QuickChange mutagenesis kit) was then performed with primers BL580 (5′-GAGGAATGACGTTTACCCCTGTGACAAAAGCCCGGACACCAG-3′) and BL581 (5′-CTGGTGTCCGGGCTTTTGTCACAGGGGTAAACGTCATTCCTC-3′) to translationally fuse phrC and phoA. The constructed plasmids containing the phrC-′phoA fusions were designated pBL483 to pBL489.
A negative control strain was constructed, in which the phrC ribosome-binding site was fused to a truncated phoA gene, φ(phrCRBS-′phoA), which expressed a signal- sequence-less alkaline phosphatase. To obtain this construct, phoA was amplified using reverse primer BL466, which contained a BglII site, and forward primer BL475 (5′-GCGCAAGCTTAAAGGAGTGAAGGTTTGTATGTACTGTTTACCC-3′), which contained the 18 bp preceding phrC, including the ribosome-binding site (bold type), and a HindIII site (underlined). The PCR product was cloned into the corresponding sites of pBL112 (15) to generate plasmid pBL482.
phrC-′phoA fusion constructs of plasmids pBL482 to pBL489 were sequenced to confirm proper construction. B. subtilis cells were transformed with these plasmids, and erythromycin-resistant transformants were selected. Transformants were checked for replacement of the thrC locus with various Pspachy-φ(phrC-′phoA) erm alleles based on the lack of spectinomycin resistance associated with the plasmids and auxotrophy for threonine.
Isolation of PhrC-F32 substitution mutants.
To isolate phrC mutations that decreased production of CSF by altering the codon for residue 32 of PhrC (i.e., the −4 position relative to the cleavage site for CSF), we used site-directed mutagenesis to randomize this codon. Site-directed mutagenesis was performed on plasmid pBL495 using primers BL597 (5′-CTAATGCGGAAGCACTCGACNNNATGTGACAGAAAGAGGAATGACG-3′) and BL598 (5′-CGTCATTCCTCTTTCTGTCACATGNNNGTCGAGTGCTTCCGCATTAG-3′) (where N is any base). The resulting site-directed mutagenesis products were transformed into E. coli XL10-Gold Ultracompetent cells (Stratagene) with selection for ampicillin-resistant transformants. All transformant colonies were pooled in LB medium with ampicillin and grown at 37°C for 1 h, and then plasmid DNA was isolated. Isolated plasmid DNA was passaged though E. coli strain MC1061 (F′ lacIq lacZM15 Tn10) before it was transformed into B. subtilis strain BAL1147 (rapA-lacZ ΔphrC), and transformants were selected on agar plates containing Difco sporulation medium, chloramphenicol, IPTG, and 200 μg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside). After 18 h of incubation, white colonies were screened for further characterization. Under these growth conditions, a ΔphrC mutant strain was white, and a strain containing a wild-type copy of phrC was blue (data not shown). Putative phrC mutant strains were screened for a defect in endogenous CSF production, and the strains that showed a defect were backcrossed into BAL1147 by selecting for the chloramphenicol resistance associated with the thrC::Pspachy-phrC cat allele and rechecked for a defect in endogenous CSF production. The phrC gene of 25 mutant strains with a defect in CSF production was sequenced. BAL3029 with an F32S substitution (TTT-to-AGT codon change), BAL3030 with an F32G substitution (TTT-to-GGC codon change), and BAL3031 with an F32R substitution (TTT-to-CGA codon change) were obtained using this procedure. The remaining sequenced phrC mutants contained stop codons or nonoptimal usage codons, as determined in the study of Kanaya et al. (13).
We generated mutants in which residue 32 of PhrC was replaced by the remaining 15 amino acids that had not been tested yet at this position. Site-directed mutagenesis was performed with plasmid pBL495 using primers listed in Table S1 in the supplemental material, and the results were confirmed by sequencing. The plasmids generated using this mutagenesis procedure are listed in Table S1 in the supplemental material and were transformed into BAL1147.
Assay for endogenous CSF production by cells.
Cultures were grown to an optical density at 600 nm (OD600) of 0.2, and then 1 mM IPTG was added to induce phrC expression. At an OD600 of ∼0.7, a 7-ml sample was harvested. The cells were removed by centrifugation, and the culture supernatant was filtered (0.2 μm) and stored at −20°C. CSF in the supernatant was quantified using the biological assay described previously (15, 16).
In vitro pro-CSF cleavage assay.
The following peptides were synthesized by Bio-Synthesis Incorporated (Lewisville, TX): pro-CSF-WT (NAEALDFHVTERGMT), pro-CSF-F32K (NAEALDKHVTERGMT), pro-CSF-H33A (NAEALDFAVTERGMT), and pro-CSF-V34E (NAEALDFHETERGMT) (underlining indicates a substitution compared to the pro-CSF-WT peptide sequence). The identities and purities of the preparations were checked in-house by mass spectrometry. These synthetic peptides were tested for cleavage by either whole cells, a cell wall fraction, or subtilisin and Vpr as described previously (15). The levels of CSF produced after incubation of these peptides were assessed by using either a biological assay or a mass spectrometric assay, both of which have been described previously (15, 16).
Alkaline phosphatase assays.
Alkaline phosphatase (PhoA) activity assays were carried out essentially as described by Nicholson and Setlow (22). Strains were grown in minimal medium. IPTG (1 mM) was added when the OD600 of the cultures reached ∼0.2. Incubation was continued until the OD600 was ∼1. Culture supernatants were harvested by centrifugation and filtered (0.2 μm). One-milliliter aliquots were then each mixed with a 1-ml aliquot of freshly prepared substrate (1 g/liter p-nitrophenylphosphate in 1 M Tris [pH 8.1]). The reaction mixtures were incubated at room temperature until they were pale yellow. The reactions were then stopped by addition of 670 μl of 2 M NaOH, and the absorbance at 420 nm was recorded. Fresh minimal medium was used as a blank. Alkaline phosphatase specific activities were calculated as follows: A420/(incubation time in minutes × OD600 of culture at supernatant harvest time × volume of supernatant in milliliters). The level of alkaline phosphatase activity in each experiment was normalized to the level produced by the strain containing the wild-type PhrC-PhoA fusion.
RESULTS AND DISCUSSION
The five residues preceding the cleavage site for CSF are important for CSF production.
We sought to determine whether the conservation in Phr proteins of five residues preceding the cleavage site for release of the mature Phr pentapeptides was due to a requirement for these residues for directing the cleavage event. To this end, we constructed mutants with substitutions at positions −5 through −1 relative to the cleavage site in PhrC that releases CSF. Substitutions that moved a residue away from the identified consensus sequence were introduced (Fig. 1A). Site-directed mutagenesis of phrC was performed, and the mutant phrC genes were introduced into B. subtilis under the control of the IPTG-inducible Pspachy promoter in strains lacking endogenous phrC. The strains were grown to mid-exponential phase in minimal media, and then PhrC expression was induced. After this the cultures were grown for ∼2 cell doublings, and then culture supernatants were harvested. The levels of CSF that had accumulated in the culture supernatants were determined. Briefly, the CSF in the culture supernatants was partially purified using reverse-phase chromatography to separate CSF from other signaling peptides that affect ComA-controlled gene expression, and then it was quantified based on the ability of CSF to induce expression of a ComA-responsive reporter fusion, srfA-lacZ. Importantly, a strain lacking CSF had levels of activity either below or at the limit of detection (Fig. 1B) (15-17, 31, 32), indicating that any activity that is observed with other strains is CSF dependent.
FIG. 1.

Substitution of the five residues preceding the CSF cleavage site affects CSF production. (A) Amino acid sequence of the C-terminal 15 residues of PhrC. The five residues preceding the cleavage site are indicated by bold italics, and the mature signaling peptide (CSF) is underlined. The amino acid substitutions in PhrC are indicated below the PhrC sequence. The putative consensus sequence is indicated above the PhrC sequence; “a” indicates an acidic residue, “p” indicates a polar residue, “h” indicates a hydrophobic residue, and “c” indicates a charged residue. (B) Levels of CSF that accumulated in culture medium for strains BAL1191 (WT), BAL1192 (ΔphrC), BAL1187 (D31A), BAL1186 (F32K), BAL1185 (H33A), BAL1184 (V34E), BAL1183 (T35K), and BAL1182 (T35A). For each of three independent experiments, the CSF levels were normalized to the level produced by strain BAL1191. The bars indicate the means of the normalized values, and the error bars indicate the standard errors of the means.
The presence of charged or polar residues at the −5 and −3 positions was predicted based on the consensus sequence, and we substituted Ala for the Asp and His residues that occur at these positions in PhrC to remove the charge and polarity. These substitutions resulted in 40% (P = 0.006, n = 4) and 65% (P = 0.03, n = 3) decreases in CSF production for the Asp-to-Ala and His-to-Ala substitutions, respectively (Fig. 1B). These data support the notion that a charged or polar residue is important at the −5 and −3 positions relative to the cleavage site for CSF, although the −3 position appears to be more important. A Phe residue occurs at the −4 position of PhrC, and in the strain expressing PhrC with a Lys residue at this position there was a 95% (P = 0.0007, n = 3) reduction in CSF production (Fig. 1B). These data are consistent with the prediction based on the consensus sequence that a hydrophobic residue is required at this position. A Val residue is predicted to be important at the −2 position, and as we reasoned that other hydrophobic residues might be functional at this position, we changed the Val residue of PhrC to Glu. Interestingly, in the strain expressing the Glu variant there was only a 50% (P = 0.02, n = 3) reduction in CSF production (Fig. 1B). Thus, while Val appears to be more optimal for CSF production than Glu, CSF production was surprisingly tolerant of a change from Val to a charged residue. The findings for the −1 position of the consensus sequence indicate that an Ala residue is critical. Intriguingly, a Thr residue is at this position in PhrC. We replaced this Thr residue by Ala and found that the strains produced indistinguishable levels of CSF compared to the strains with wild-type PhrC (P = 0.8, n = 3) (Fig. 1B). The side chains of Ala and Thr are both small and at least partially hydrophobic. To test whether these side chains are important for CSF production, we replaced the Thr residue of PhrC with Lys, an amino acid with a large, charged side chain. CSF production was tolerant of such a radical change, and in strains having the Thr-to-Lys substitution there was only a 35% (P = 0.04, n = 3) decrease in CSF production (Fig. 1B). Although the effects of some of the amino acid substitutions were relatively small, together, the data obtained indicate that the five residues that precede the cleavage site in PhrC for CSF are required for normal CSF production.
The five residues preceding the CSF cleavage site are not required for a functional signal sequence.
A possible explanation for the decrease in CSF production caused by amino acid substitutions at positions −1 through −5 relative to the CSF cleavage site is that the substitutions affect an extended signal sequence necessary for secretion. To test this possibility, we created fusions of the mutant PhrC proteins to the E. coli alkaline phosphatase protein (PhoA), which lacked its own signal sequence. The PhrC-PhoA fusions were expressed in B. subtilis from the thrC locus under the control of the IPTG-inducible Pspachy promoter. PhrC-PhoA secretion was monitored by assaying the alkaline phosphatase activity in culture supernatants. As expected, a strain expressing PhoA lacking a signal sequence had no measurable alkaline phosphatase activity (Fig. 2). All of the strains expressing mutant PhrC-PhoA fusion proteins had alkaline phosphatase activities comparable to that of a strain expressing the wild-type PhrC-PhoA fusion protein (P > 0.25, n = 3) (Fig. 2). These data indicate that all of the mutant PhrC proteins contained a functional signal sequence and suggest that a secretion defect was not the cause of the reduced CSF production by strains expressing the mutant PhrC proteins.
FIG. 2.
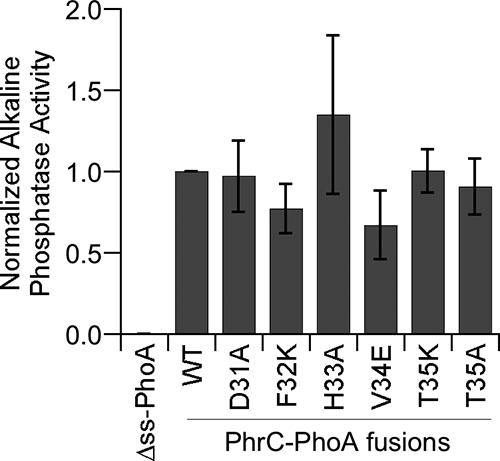
Mutant PhrC proteins are secreted at the same level as wild-type PhrC as measured by alkaline phosphatase activity for strains carrying PhrC-PhoA fusions. PhoA activity was measured using culture supernatants of strains BAL2199 (Δss-PhoA), BAL2200 (WT), BAL2201 (T35A), BAL2202 (T35K), BAL2203 (V34E), BAL2204 (H33A), BAL2205 (F32K), and BAL2206 (D31A). The normalized, mean levels of alkaline phosphatase activity from three independent experiments (indicated by bars) are plotted versus the strains assayed. The error bars indicate the standard errors of the means.
Synthetic pro-CSF peptides with amino acid substitutions are cleaved less efficiently to CSF.
The data described above indicated that strains expressing PhrC mutant proteins with amino acid substitutions at positions −5 through −1 relative to the CSF cleavage site produce less CSF and that the reduction in CSF production was not due to a reduction in secretion of PhrC. We reasoned that this reduction could have been due to reduced PhrC protein expression or reduced protease recognition and/or cleavage of PhrC to CSF. We measured the levels of phrC mRNA for the three mutant strains with the lowest levels of CSF production (i.e., the mutants with substitutions at the −2 to −4 positions) and observed no significant differences in the levels of phrC mRNA (see Fig. S1 in the supplemental material). There is no method to directly test the levels of PhrC inside cells at this time, and attempts to indirectly measure PhrC levels using a C-terminal epitope tag have been unsuccessful, possibly due to proteolytic removal of the tag extracellularly (data not shown). Given that the mutant PhrC-PhoA fusion proteins were expressed at levels similar to the levels of the wild-type fusion protein, it seemed unlikely that the defect in CSF production was due to a defect in PhrC expression.
To test directly the possibility that the mutant PhrC proteins were cleaved less efficiently to CSF, we synthesized peptides that corresponded to the portion of PhrC predicted to be C terminal to the signal sequence for secretion. The sequence of one peptide, pro-CSF-WT, was identical to the sequence of the C-terminal 15 residues of PhrC, and this peptide has been used previously in studies of CSF proteolytic processing (15). We also synthesized peptides that individually had the three amino acid substitutions that resulted in the greatest defects in CSF production, pro-CSF-F32K, pro-CSF-H33A, and pro-CSF-V34E. These peptides differed at the −2, −3, or −4 position relative to the CSF cleavage site (Fig. 3A).
FIG. 3.

Synthetic pro-CSF peptides with amino acid substitutions at the −4, −3, and −2 positions are cleaved less efficiently to mature CSF. (A) Sequence of synthetic pro-CSF peptides. (B) Synthetic pro-CSF peptides were incubated with cells of strain BAL950 (Δopp ΔcomQ ΔphrC), and the amount of CSF produced was determined using the biological assay. The normalized mean amounts of CSF produced in three independent experiments are indicated by bars, and the error bars indicate the standard errors of the means. (C) The pro-CSF peptides were incubated with cell wall material, and the ERGMT was quantified by LC-MS/MS-MRM. The intensity of the signal for the parent-to-fragment ion (m/z 386.5) transition is plotted versus the elution time for C18 columns. WT, pro-CSF-WT.
To test for cleavage of the synthetic pro-CSF peptides to CSF, the peptides were incubated with washed whole cells of B. subtilis strain BAL950, which lacks phrC and thus cannot produce any CSF. BAL950 also lacks the oligopeptide permease responsible for uptake of CSF from media and comQ, which encodes a protein needed to produce a signaling peptide with activity similar to that of CSF. All three of these mutations were included to increase the sensitivity of the assay for detection of CSF. Approximately 108 cells were incubated with pro-CSF for 40 min, a time sufficient for nearly complete cleavage of 100 pmol of pro-CSF-WT to CSF (15). After incubation, the cells were removed, and the culture medium was fractionated on a reverse-phase C18 column to separate pro-CSF from CSF. The amount of CSF that eluted from the column was then determined by determining the level of β-galactosidase specific activity after treatment of cells containing the ComA-controlled reporter fusion srfA-lacZ with dilutions of the eluates.
When the cells were incubated with pro-CSF-WT, CSF production was observed, but CSF production was not observed when the cells and pro-CSF-WT were incubated separately (Fig. 3B and data not shown). Compared to pro-CSF-WT, each of the mutant pro-CSF peptides produced less CSF when it was incubated with cells (Fig. 3B). Incubation with the pro-CSF-F32K peptide, with a substitution at the −4 position, yielded only 10% of the CSF produced with the pro-CSF-WT peptide (P = 0.01, n = 3), similar to the results obtained with the same amino acid substitution in the in vivo context (compare Fig. 3B and 1B). Incubation with the pro-CSF-V34E and pro-CSF-H33A peptides yielded 25% (P = 0.004, n = 3) and 60% (P = 0.02, n = 3) of the CSF produced with the pro-CSF-WT peptide, respectively. The magnitude of the defect caused by these substitutions was different than the magnitude observed when the same substitutions were encoded by phrC (compare Fig. 3B and 1B). This may have been because there was a slightly different profile of proteases able to cleave pro-CSF to CSF after cells were washed. Nevertheless, the defect in CSF production caused by the amino acid substitutions at positions −2 to −4 when they were part of an exogenously added peptide supports the hypothesis that these amino acid substitutions decreased the efficiency of cleavage of PhrC to CSF.
To confirm that the mutant peptides were cleaved less efficiently to the CSF peptide sequence ERGMT, we measured the amounts of CSF produced after incubation of the pro-CSF peptides using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay for CSF. This assay recorded the intensity of the transition of the doubly charged parent ion (m/z 297.2) to the singly charged fragment ion (m/z 386.5) under collisionally activated dissociation, multiple-reaction-monitoring (MRM) conditions, as previously described (15). Because whole bacterial cells would have interfered with the LC-MS/MS-MRM procedure, we used a cell wall-enriched fraction of B. subtilis cells, which we have previously shown to be the cellular fraction that contains the majority of pro-CSF processing activity (15). Under the prescribed chromatography conditions, the retention time of synthetic CSF was 14.5 min. Neither the pro-CSF peptides nor the cell wall fraction of B. subtilis separately resulted in a significant LC-MS/MS-MRM response for CSF (data not shown). In contrast, when the cell wall fraction was incubated with pro-CSF-WT, a significant MRM response was recorded at the appropriate retention time (Fig. 3C), indicating that pro-CSF-WT was cleaved into the CSF pentapeptide. When the cell wall fraction was incubated with the mutant pro-CSF peptides, the level of the MRM response was not more than 10% of the level observed with pro-CSF-WT (Fig. 3C). The greater defect in cleavage of pro-CSF to CSF for the mutant peptides determined by this assay than by the biological assays could have been due to the change in the profile or levels of pro-CSF processing proteases that occurred during preparation of the cell wall-enriched fraction of cells. The data obtained demonstrate that the amino acid substitutions at the −2, −3, and −4 positions significantly decreased proteolytic cleavage of ERGMT from a precursor peptide.
CSF processing proteases, subtilisin and Vpr, cleave mutant pro-CSF peptides less efficiently.
We previously showed that cells lacking the secreted serine proteases subtilisin, Vpr, and Epr had a defect in production of CSF and that purified subtilisin and Vpr were able to cleave synthetic pro-CSF to CSF (15). To further support the hypothesis that subtilisin and Vpr have a direct role in processing pro-CSF to CSF in vivo, we examined whether the amino acid substitutions in PhrC that decreased production of CSF in vivo similarly affected the cleavage of pro-CSF by subtilisin or Vpr. Purified subtilisin and Vpr were incubated separately with pro-CSF substrates having substitutions at positions −2 to −4 relative to the cleavage site, and the levels of CSF produced were determined using the biological assay for CSF (Fig. 4).
FIG. 4.

Cleavage of mutant pro-CSF substrates by subtilisin and Vpr. Purified subtilisin or Vpr was incubated with the pro-CSF substrates indicated. The level of CSF produced was normalized to the level of CSF produced after incubation with the wild-type pro-CSF substrate (WT). The bars indicate the averages of at least three independent experiments, and the error bars indicate the standard errors of the means. Under these conditions, incubation of subtilisin with pro-CSF and incubation of Vpr with pro-CSF resulted in statistically indistinguishable levels of CSF production.
The Phe-to-Lys substitution at the −4 position resulted in severely reduced cleavage of pro-CSF (98% reduction for subtilisin [P = <0.0001, n = 3] and 90% reduction for Vpr [P = 0.001, n = 3]). The His-to-Ala substitution at the −3 position had a more modest effect on cleavage (47% [P = 0.03, n = 3] and 79% [P = 0.003, n = 3] reductions in pro-CSF cleavage by subtilisin and Vpr, respectively). The Val-to-Glu substitution at the −3 position had the most disparate effects on pro-CSF cleavage (97% [P = <0.0001, n = 3] reduction in cleavage to CSF by subtilisin and 42% [P = 0.02, n = 4] reduction in cleavage by Vpr). Collectively, these data indicate that substitutions at positions −2 to −4 decreased the efficiency of cleavage of pro-CSF to CSF by subtilisin and Vpr, and they support the hypothesis that subtilisin and Vpr have a direct role in processing CSF in vivo.
Defining the amino acids that are tolerated at the −4 position of pro-CSF and allow cleavage.
To begin to determine the rules that govern what amino acid sequences can be recognized for cleavage that releases mature Phr pentapeptides, we changed the amino acid at the −4 position of pro-CSF to the other 19 canonical amino acids in order to determine which amino acids support cleavage that releases CSF. The −4 position was chosen for this analysis as substitutions at this position resulted in the greatest defects in CSF production. CSF production by strains individually expressing 1 of the 19 mutant PhrC-F32 proteins was assessed using the biological assay (Fig. 5).
FIG. 5.

Amino acid substitutions at the −4 position relative to the cleavage site of PhrC. The Phe at position 32 of PhrC was replaced by the other 19 canonical amino acids. The levels of endogenous CSF production by cells expressing the mutant PhrC proteins were determined and normalized to the level of CSF production by the strain expressing wild-type PhrC (amino acid F). The bars indicate the averages for at least three independent experiments, and the error bars indicate the standard errors of the means. The asterisks indicate mutant PhrC strains that were determined to be significantly different from the wild-type strain by the Student t test (P < 0.05).
The identified consensus sequence of the residues that precede the cleavage site for Phr peptides indicated that a hydrophobic residue is important at the −4 position. Consistent with this, the hydrophobic residues, such as Val, Ile, and Leu, supported levels of CSF production similar to the levels exhibited by the wild-type strain with a Phe residue at the −4 position. Furthermore, substitution of a hydrophobic Met residue at this position resulted in levels of CSF production that were 1.8-fold-higher than the levels observed with the Phe residue (P = 0.03, n = 4). The only exceptions to this were the hydrophobic Ala and Trp substitutions, which resulted in 20% (P = 0.03, n = 3) and 50% (P = 0.03, n = 3) decreases in CSF production, respectively. Trp is the largest amino acid, and the defect in CSF production may have been due to the large side chain sterically hindering cleavage. At the other extreme, Ala is the smallest hydrophobic amino acid, and the defect in CSF production may have been due to the small size of the side chain, which was not able to stabilize the interaction with pro-CSF processing proteases.
As further support for the hypothesis that a hydrophobic residue at the −4 position is important, some polar residues at this position decreased production of CSF. As noted above, a Lys substitution severely decreased CSF production; in addition, Arg, Cys, and Asp substitutions decreased CSF production 72% (P = 0.05, n = 3), 68% (P = 0.008, n = 3), and 44% (P = 0.03, n = 5), respectively. However, some polar amino acids could be tolerated at this position; a Glu, Asn, or Gln substitution did not result in a statistically significant difference in CSF production. The latter finding indicates that the prediction based on the consensus sequence that there is a hydrophobic residue at this position is not strictly correct. While there is a preference for hydrophobic residues at the −4 position, some polar residues can be tolerated.
Conclusions and implications.
In this study, we identified residues that are important for release of the CSF signaling peptide of B. subtilis from its precursor protein, PhrC. We previously identified a loose consensus sequence for five residues preceding the cleavage sites that produce the mature Phr pentapeptides of B. subtilis (28) (Table 1). Analysis of substitution of these five amino acid residues in PhrC, the precursor protein for the Phr peptide CSF, revealed the importance of these residues in directing cleavage of the precursor protein to release CSF. However, the amino acid substitution data also revealed that a relatively wide variety of sequences can be tolerated at these positions and that this toleration may be due to the fact that multiple proteases are able to cleave PhrC.
PhrC appears to tolerate a relatively wide variety of sequences in the residues preceding the cleavage site, resulting in CSF production that is relatively robust to mutational perturbation. For example, it was observed that only 4 of 19 amino acid substitutions at the −4 position resulted in a >2-fold defect in CSF production. This robustness to mutational perturbation appears to be due to the presence of multiple, redundant proteases that process CSF. If subtilisin were the only protease that processes PhrC to CSF, we would have observed that a Val-to-Glu substitution at the −2 position severely decreased CSF production. Instead, this substitution had a moderate effect on CSF production because Vpr is able to process PhrC with this substitution, when subtilisin cannot process it. It is interesting that a Glu residue occurs at the −2 position of PhrE and PhrH (with a one-residue gap allowed for the PhrH alignment, although it is difficult at this time to accurately predict the sequence of the mature PhrH peptide). Given the ability of Vpr to process a PhrC substrate with a Glu residue at the −2 position, we predict that Vpr and not subtilisin plays a significant role in processing of the PhrE and PhrH peptides. Further work could determine whether production of these Phr peptides exhibits robustness to mutational perturbation similar to that of CSF.
These studies contributed to our goal of identifying the proteases that process Phr peptides of B. subtilis and other bacteria. Even though CSF production was tolerant to many amino acid substitutions, identifying substitutions such as the Phe-to-Lys substitution at the −4 position relative to the cleavage site for CSF should allow us to test whether production of other Phr peptides is similarly disrupted by such a substitution. Of course, one method to test whether subtilisin, Vpr, or Epr has a role in cleaving other Phr peptides is to test these proteases in vitro to determine whether they have this processing activity, as we have done with PhrA (15). Showing that analogous amino acid substitutions that affect processing of CSF by subtilisin and Vpr in vitro also affect processing of a Phr peptide, such as PhrA, would provide in vivo support for the hypothesis that these Phr peptides are processed by the same proteases. One question that is important to answer before substitutional analyses of other Phr precursor proteins are performed is whether there is flexibility in the position of the residues that direct cleavage of Phr peptides. As shown in Table 1, in order to obtain maximal alignment of the Phr precursor proteins, it was necessary to introduce a one-residue gap between the cleavage site and the consensus sequence for a few of the Phr peptides. Future studies need to address how this additional amino acid affects the residues required for cleavage of the Phr precursor proteins. In the long term, these studies have laid the foundation for determining the mechanism of production of Phr peptides in B. subtilis and other bacteria.
Supplementary Material
Acknowledgments
This work was supported by NIH Public Health Service grant AI48616 to B.A.L. S.L. was supported in part by USPHS National Research Service award GM07104. The mass spectrometer used in this work was purchased in part with funds from the W. M. Keck Foundation.
Footnotes
Published ahead of print on 8 August 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Auchtung, J. M., C. A. Lee, and A. D. Grossman. 2006. Modulation of the ComA-dependent quorum response in Bacillus subtilis by multiple Rap proteins and Phr peptides. J. Bacteriol. 1885273-5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auchtung, J. M., C. A. Lee, R. E. Monson, A. P. Lehman, and A. D. Grossman. 2005. Regulation of a Bacillus subtilis mobile genetic element by intercellular signaling and the global DNA damage response. Proc. Natl. Acad. Sci. USA 10212554-12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bassler, B., P. Gibbons, and S. Roseman. 1989. Chemotaxis to chitin oligosaccharides by Vibrio furnissii, a chitinivorous marine bacterium. Biochem. Biophys. Res. Commun. 1611172-1176. [DOI] [PubMed] [Google Scholar]
- 4.Bongiorni, C., S. Ishikawa, S. Stephenson, N. Ogasawara, and M. Perego. 2005. Synergistic regulation of competence development in Bacillus subtilis by two Rap-Phr systems. J. Bacteriol. 1874353-4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bongiorni, C., R. Stoessel, D. Shoemaker, and M. Perego. 2006. Rap phosphatase of virulence plasmid pXO1 inhibits Bacillus anthracis sporulation. J. Bacteriol. 188487-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comella, N., and A. D. Grossman. 2005. Conservation of genes and processes controlled by the quorum response in bacteria: characterization of genes controlled by the quorum-sensing transcription factor ComA in Bacillus subtilis. Mol. Microbiol. 571159-1174. [DOI] [PubMed] [Google Scholar]
- 7.Core, L., and M. Perego. 2003. TPR-mediated interaction of RapC with ComA inhibits response regulator-DNA binding for competence development in Bacillus subtilis. Mol. Microbiol. 491509-1522. [DOI] [PubMed] [Google Scholar]
- 8.Guerout-Fleury, A. M., N. Frandsen, and P. Stragier. 1996. Plasmids for ectopic integration in Bacillus subtilis. Gene 18057-61. [DOI] [PubMed] [Google Scholar]
- 9.Harwood, C. R., and S. M. Cutting. 1990. Molecular biological methods for Bacillus. John Wiley & Sons Ltd, Chichester, England.
- 10.Hayashi, K., T. Kensuke, K. Kobayashi, N. Ogasawara, and M. Ogura. 2006. Bacillus subtilis RghR (YvaN) represses rapG and rapH, which encode inhibitors of expression of the srfA operon. Mol. Microbiol. 591714-1729. [DOI] [PubMed] [Google Scholar]
- 11.Hense, B. A., C. Kuttler, J. Muller, M. Rothballer, A. Hartmann, and J. U. Kreft. 2007. Does efficiency sensing unify diffusion and quorum sensing? Nat. Rev. Microbiol. 5230-239. [DOI] [PubMed] [Google Scholar]
- 12.Jiang, M., R. Grau, and M. Perego. 2000. Differential processing of propeptide inhibitors of Rap phosphatases in Bacillus subtilis. J. Bacteriol. 182303-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanaya, S., Y. Yamada, Y. Kudo, and T. Ikemura. 1999. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene 238143-155. [DOI] [PubMed] [Google Scholar]
- 14.Koetje, E. J., A. Hajdo-Milasinovic, R. Kiewiet, S. Bron, and H. Tjalsma. 2003. A plasmid-borne Rap-Phr system of Bacillus subtilis can mediate cell-density controlled production of extracellular proteases. Microbiology 14919-28. [DOI] [PubMed] [Google Scholar]
- 15.Lanigan-Gerdes, S., A. N. Dooley, K. F. Faull, and B. A. Lazazzera. 2007. Identification of subtilisin, Epr and Vpr as enzymes that produce CSF, an extracellular signalling peptide of Bacillus subtilis. Mol. Microbiol. 651321-1333. [DOI] [PubMed] [Google Scholar]
- 16.Lazazzera, B. A., I. G. Kurtser, R. S. McQuade, and A. D. Grossman. 1999. An autoregulatory circuit affecting peptide signaling in Bacillus subtilis. J. Bacteriol. 1815193-5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lazazzera, B. A., J. M. Solomon, and A. D. Grossman. 1997. An exported peptide functions intracellularly to contribute to cell density signaling in B. subtilis. Cell 89917-925. [DOI] [PubMed] [Google Scholar]
- 18.LeDeaux, J. R., and A. D. Grossman. 1995. Isolation and characterization of kinC, a gene that encodes a sensor kinase homologous to the sporulation sensor kinases KinA and KinB in Bacillus subtilis. J. Bacteriol. 177166-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyon, G. J., and R. P. Novick. 2004. Peptide signaling in Staphylococcus aureus and other Gram-positive bacteria. Peptides 251389-1403. [DOI] [PubMed] [Google Scholar]
- 20.Magnuson, R., J. Solomon, and A. D. Grossman. 1994. Biochemical and genetic characterization of a competence pheromone from B. subtilis. Cell 77207-216. [DOI] [PubMed] [Google Scholar]
- 21.Mueller, J. P., G. Bukusoglu, and A. L. Sonenshein. 1992. Transcriptional regulation of Bacillus subtilis glucose starvation-inducible genes: control of gsiA by the ComP-ComA signal transduction system. J. Bacteriol. 1744361-4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicholson, W. L., and P. Setlow. 1990. Sporulation, germination, and outgrowth, p. 391-429. In C. R. Harwood and S. M. Cutting (ed.), Molecular biological methods for Bacillus. John Wiley & Sons, Chichester, England.
- 23.Ogura, M., K. Shimane, K. Asai, N. Ogasawara, and T. Tanaka. 2003. Binding of response regulator DegU to the aprE promoter is inhibited by RapG, which is counteracted by extracellular PhrG in Bacillus subtilis. Mol. Microbiol. 491685-1697. [DOI] [PubMed] [Google Scholar]
- 24.Ogura, M., H. Yamaguchi, K. Yoshida, Y. Fujita, and T. Tanaka. 2001. DNA microarray analysis of Bacillus subtilis DegU, ComA and PhoP regulons: an approach to comprehensive analysis of B. subtilis two-component regulatory systems. Nucleic Acids Res. 293804-3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perego, M. 1997. A peptide export-import control circuit modulating bacterial development regulates protein phosphatases of the phosphorelay. Proc. Natl. Acad. Sci. USA 948612-8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perego, M., and J. A. Brannigan. 2001. Pentapeptide regulation of aspartyl-phosphate phosphatases. Peptides 221541-1547. [DOI] [PubMed] [Google Scholar]
- 27.Perego, M., and J. A. Hoch. 1988. Sequence analysis and regulation of the hpr locus, a regulatory gene for protease production and sporulation in Bacillus subtilis. J. Bacteriol. 1702560-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pottathil, M., and B. A. Lazazzera. 2003. The extracellular Phr peptide-Rap phosphatase signaling circuit of Bacillus subtilis. Front. Biosci. 832-45. [DOI] [PubMed] [Google Scholar]
- 29.Slamti, L., and D. Lereclus. 2002. A cell-cell signaling peptide activates the PlcR virulence regulon in bacteria of the Bacillus cereus group. EMBO J. 214550-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smits, W. K., C. Bongiorni, J. W. Veening, L. W. Hamoen, O. P. Kuipers, and M. Perego. 2007. Temporal separation of distinct differentiation pathways by a dual specificity Rap-Phr system in Bacillus subtilis. Mol. Microbiol. 65103-120. [DOI] [PubMed] [Google Scholar]
- 31.Solomon, J. M., B. A. Lazazzera, and A. D. Grossman. 1996. Purification and characterization of an extracellular peptide factor that affects two different developmental pathways in Bacillus subtilis. Genes Dev. 102014-2024. [DOI] [PubMed] [Google Scholar]
- 32.Solomon, J. M., R. Magnuson, A. Srivastava, and A. D. Grossman. 1995. Convergent sensing pathways mediate response to two extracellular competence factors in Bacillus subtilis. Genes Dev. 9547-558. [DOI] [PubMed] [Google Scholar]
- 33.Stephenson, S., C. Mueller, M. Jiang, and M. Perego. 2003. Molecular analysis of Phr peptide processing in Bacillus subtilis. J. Bacteriol. 1854861-4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tzeng, Y. L., V. A. Feher, J. Cavanagh, M. Perego, and J. A. Hoch. 1998. Characterization of interactions between a two-component response regulator, Spo0F, and its phosphatase, RapB. Biochemistry 3716538-16545. [DOI] [PubMed] [Google Scholar]
- 35.Vasantha, N., and E. Freese. 1980. Enzyme changes during Bacillus subtilis sporulation caused by deprivation of guanine nucleotides. J. Bacteriol. 1441119-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Youngman, P., H. Poth, B. Green, K. York, G. Olmedo, and K. Smith (ed.). 1989. Methods for genetic manipulation, cloning, and functional analysis of sporulation genes in Bacillus subtilis. American Society for Microbiology, Washington, DC.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.