

Abstract
A class of mutations that suppress the recombination defects of recB mutants in Salmonella enterica serovar Typhimurium strain LT2 activates the normally silent recET module of the Gifsy-1 prophage. Allele sbcE21 is a 794-bp deletion within the immunity region of the prophage. Concomitant with activating recET, sbcE21 stimulates Gifsy-1 excision, resulting in unstable suppression. Early studies found both recB suppression and its instability to depend on the presence of the related Gifsy-2 prophage elsewhere in the chromosome. In cells lacking Gifsy-2, the sbcE21 allele became stable but no longer corrected recB defects. Here, we show that a single Gifsy-2 gene is required for Gifsy-1 recET activation in the sbcE21 background. This gene encodes GtgR, the Gifsy-2 repressor. Significantly, the sbcE21 deletion has one end point within the corresponding gene in the Gifsy-1 genome, gogR, which in strain LT2 is a perfect duplicate of gtgR. The deletion truncates gogR and places the Gifsy-1 left operon, including the recET and xis genes, under the control of the gogR promoter. The ability of GtgR to trans-activate this promoter therefore implies that GtgR and GogR normally activate the transcription of their own genes. Consistent with the symmetry of the system, a similar deletion in Gifsy-2 results in a Gifsy-1-dependent sbc phenotype (sbcF24). Two additional Gifsy-1 deletions (sbcE23 and sbcE25) were characterized, as well. The latter causes all but the last codon of the gogR gene to fuse, in frame, to the second half of recE. The resulting hybrid protein appears to function as both a transcriptional regulator and a recombination enzyme.
Some classic contributions to the study of homologous recombination in Escherichia coli have come from the analysis of mutations that bypass the requirement for the RecBCD enzyme in the recombinational repair of damaged DNA. This approach led to the discovery of recombination proteins and was critical to the development of the concept that recombination can proceed through different pathways, depending on the nature of the DNA substrates (9, 29, 36). Mutations were selected as suppressors of the hypersensitivity of recB or recC mutants to mitomycin C and hence were designated sbc (for suppressor of recB and recC mutation) (2). The first class of alleles, sbcA, was mapped in the Rac prophage and a defective prophage found in the chromosomes of many E. coli strains (28). sbcA mutations cause the constitutive activation of the prophage's recombination module, which comprises the recE and recT genes. The RecE protein (deoxyexonuclease VIII) is a double-stranded-DNA-specific exonuclease, which degrades linear DNA in a 5′-to-3′ direction (23, 24). Thus, the enzyme leaves a protruding 3′ single-stranded tail that presumably acts in the strand invasion step of homologous recombination. The RecT protein binds single-stranded DNA and, acting in conjunction with RecE, can promote homologous pairing and strand exchange in vitro (21, 26). Therefore, the recET module is functionally equivalent to the α and β genes of the bacteriophage λ red operon (37). Physical characterization of DNA obtained from sbcA mutants showed them to result from a variety of changes, including deletions that place the recET module under the control of a distant promoter (25, 30), transposon insertions that provide a promoter (8, 10), and point mutations that remain uncharacterized (30).
During a study of conditional gyrase mutants of Salmonella enterica serovar Typhimurium several years ago, Gari and coworkers noticed that some of these strains derepressed the SOS regulon when grown at the semipermissive temperature of 37°C (20). Inactivation of the RecBCD enzyme completely abolished SOS induction and at the same time prevented growth at 37°C. Selection of mutants that recovered the ability to grow at 37°C yielded two classes of clones: one that no longer expressed SOS and another in which SOS induction was restored. All of the members of the first class carried changes linked to the initial gyr allele and were presumed to be true revertants. In contrast, the members of the second group carried unlinked mutations suppressing the recB defect. Thus, the presence of the latter class suggested that the gyr mutants depended on recombination to grow at a semipermissive temperature (20). Some of the suppressor mutations mapped to a novel locus, at 57 centisomes of the Salmonella chromosome, which was designated sbcE (20). Sequence analysis showed allele sbcE21 to be a deletion of 794 bp within the presumptive regulatory region of a prophage that was unknown at the time, Gifsy-1 (17). Significantly, one end point of the deletion was in the proximity of a recE gene ortholog, suggesting that sbcE21 somehow activated the expression of this gene. Thus, sbcE mutations appeared to be the Salmonella equivalent of sbcA mutations of E. coli. Still, strains harboring sbcE21 or other sbcE alleles displayed some peculiar properties. They were genetically unstable and segregated at high-frequency derivatives that had lost the suppressor phenotype. Two classes of segregants could be identified. The first class, by far the majority, comprised isolates that had lost the entire Gifsy-1 prophage by precise excision. These segregants could be easily accounted for by postulating that sbcE21 activated the Gifsy-1 excisionase (xis) gene concomitant with recE. Isolates from the second class still contained Gifsy-1 and the associated sbcE21; curiously, however, the mutation was no longer able to suppress the recB defect. When examined in detail, these segregants were found to result from the excision of a second unknown prophage, Gifsy-2, at 24 centisomes of the Salmonella chromosome (17). These findings suggested that the sbcE21-dependent induction of Gifsy-1 recE and xis activities required one or more factors encoded by the Gifsy-2 prophage. In the work described here, we verified this hypothesis and elucidated the mechanism underlying the peculiar Gifsy-1/Gifsy-2 complementation. The study offered the opportunity to begin characterizing Gifsy phage regulation, and the results obtained represent a first step toward the understanding of such regulation.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
All strains used in this study are derivatives of S. enterica serovar Typhimurium. Their genotypes are listed in Table 1. The bacteria were cultured in LB broth (4) solidified by the addition of 1.5% (wt/vol) Difco agar when needed. When appropriate, the LB medium was supplemented with 0.2% (wt/vol) arabinose. Antibiotics (Sigma) were included at the following final concentrations: chloramphenicol, 10 μg/ml; kanamycin monosulfate, 50 μg/ml; sodium ampicillin, 75 μg/ml; spectinomycin dihydrochloride, 80 μg/ml; and tetracycline hydrochloride, 25 μg/ml. LB plates containing 40 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) (Sigma) were used to monitor lacZ expression in bacterial colonies. Liquid cultures were grown in New Brunswick gyratory shakers, and growth was monitored by measuring the optical density at 600 nm with a Milton-Roy Spectronic 301 spectrophotometer.
TABLE 1.
Strains used in this work
| Straina | Genotypeb | Source or referencec |
|---|---|---|
| TT8793 | nadB227::MudA | J. R. Roth |
| JS130 | zjg-8103::pir | J. Slauch |
| MA2582 | din-1001::MudJ gyrA208 zej-754::Tn10 recB546::Tn10dCm sbcE23 | 20 |
| MA2684 | din-1001::MudJ Δ[argA-recBD]1742 gyrB652 zid-6782::Tn10dCm sbcE21 | 20 |
| MA2715 | din-1001::MudJ Δ[argA-recBD]1742 gyrA208 zej-6790::Tn10dCm sbcF24 | 20 |
| MA2716 | din-1001::MudJ Δ[argA-recBD]1742 gyrA208 zej-6790::Tn10dCm sbcE25 | 20 |
| MA3397 | Gifsy-1{gfoO8157::Tn10dTc (formerly zfh-8157::Tn10dTc) sbcE21} | 17 |
| MA3409 | Gifsy-1{−} | 17 |
| MA4398 | Gifsy-1{zfh-8163::MudF gfoO8157::Tn10dTc sbcE21} | 17 |
| MA4587 | Gifsy-1{−} Gifsy-2{−} | 17 |
| MA6057 | nadB227::MudA pncB150::Tn10 | |
| MA6215 | Gifsy-1{zfh-8179::MudJ} | |
| MA6735 | Gifsy-1{sbcE21 gfoO8157::Tn10dTc zfh-8165::MudF} zjg-8103::pir | |
| MA7153 | Gifsy-1{−}::pNFB9 | |
| MA7320 | Gifsy-2{−} | |
| MA7457 | Gifsy-2{ΔrecE59::lacZ aph} Gifsy-1{-} | |
| MA7489 | Gifsy-1{Δ[int-recE]97::lacZ aph} Gifsy-2{−} | |
| MA7507 | Gifsy-1{Δ[int-recE]97::lacZ aph gfoO8157::Tn10dTc sbcE21} Gifsy-2{−} | |
| MA7508 | Gifsy-1{Δ[int-recE]97::lacZ aph gfoO8157::Tn10dTc sbcE21} | |
| MA7525 | Gifsy-1{Δ[int-recE]97::lacZ aph gfoO8157::Tn10dTc} | |
| MA7526 | Gifsy-1{Δ[int-recE]97::lacZ aph gfoO8157::Tn10dTc} Gifsy-2{−} | |
| MA7612 | Gifsy-1{Δ[int-recE]97::lacZ-scarpNFB19gfoO8157::Tn10dTc sbcE21} Gifsy-2{Δ[int-STM1011]91::aph} | |
| MA7618 | Gifsy-1{Δ[int-recE]97::lacZ aph gfoO8157::Tn10dTc sbcE21} Gifsy-2{Δ[STM1013-sseI]89::scarpKD3} | |
| MA7653 | Gifsy-1{Δ[int-recE]97::lacZ aph gfoO8157::Tn10dTc sbcE21} Gifsy-2{Δ[int-STM1019]92::cat} | |
| MA7655 | Gifsy-1{Δ[int-recE]97::lacZ aph gfoO8157::Tn10dTc sbcE21} Gifsy-2{Δ[gftC-sseI]90::scarpKD3} | |
| MA7712 | Gifsy-1{zfh-8179::MudJ Δ21[STM2631-gogR]} | |
| MA7719 | Gifsy-2{Δ[int-STM1056]98::gtgR aph} | |
| MA7720 | Gifsy-1{Δ[int-recE]97::lacZ cat gfoO8157::Tn10dTc sbcE21} Gifsy-2{Δ[int-STM1056]98::gtgR aph} | |
| MA7782 | Gifsy-1{Δ[int-recE]97::lacZ cat gfoO8157::Tn10dTc sbcE21} Gifsy-2{-} ΔaraBAD99::gtgR aph | |
| MA7842 | Gifsy-1{-} Gifsy-2{Δ[STM1010-gtgR]21 Δ[STM1013-sseI]89::lacZY aph (pCE36) ΔaraBAD99::gtgR scarpKD13 | |
| MA9551 | Gifsy-1{zfh-8179::MudJ} Gifsy-2{−} | |
| MA9555 | Gifsy-1{zfh-8179::MudJ gfoO8157::Tn10dTc sbcE21} | |
| MA9556 | Gifsy-1{zfh-8179::MudJ gfoO8157::Tn10dTc sbcE21} Gifsy-2{−} | |
| MA9557 | Gifsy-1{zfh-8179::MudJ gfoO8157::Tn10dTc sbcE25} | |
| MA9558 | Gifsy-1{zfh-8179::MudJ gfoO8157::Tn10dTc sbcE25} Gifsy-2{−} |
All strains were derived from S. enterica serovar Typhimurium strain LT2, except for strains JS130, MA6735, and MA7153, which are derived from strain ATCC 14028.
Curly brackets following a prophage name define the genotype of that prophage. A minus sign signifies absence of the prophage. The term “scar” denotes the DNA sequence that remains after excision of the antibiotic resistance cassette. Superscript indicates the plasmid used as DNA templates in amplifying the cassette. MudA, MudF, and MudJ are transposition-defective derivatives of phage Mu (17, 22).
Where not specified, the source of the strain is this work.
Genetic techniques.
Generalized transduction was carried out using the high-frequency transducing mutant of phage P22, HT 105/1 int-201 (35). Typically, P22 lysates were used at a 1:50 dilution, mixed with aliquots from overnight cultures of recipient bacteria in a 1:2 ratio, and incubated for 30 min at 37°C prior to being plated on selective media. Transductant colonies were purified by two sequential passages on selective plates and verified to be phage free by streaking them on Evans Blue Uranine plates (5).
Chromosomal engineering was carried out by the λ red recombination method (12, 39) as implemented previously (12, 38). Donor DNA fragments were generated by PCR using plasmid or chromosomal DNA templates (Table 2). Amplified fragments were electroporated into strains harboring the conditionally replicating plasmid pKD46, which carries a λ red operon under the control of the PBAD promoter (12). Bacteria carrying pKD46 were grown at 30°C in the presence of ampicillin (100 μg/ml) and exposed to arabinose (10 mM) 3 hours prior to preparation of electrocompetent cells. Electroporation was carried out using a Bio-Rad MicroPulser under the conditions specified by the manufacturer. Recombinant colonies selected on LB plates containing the appropriate antibiotic were verified by PCR and, in most instances, by DNA sequencing. When needed, the antibiotic resistance cassette was excised by transforming strains with plasmid pCP20, which encodes the Flp recombinase (7). The template plasmids pNFB19 and pNFB22 (see below) were constructed in the course of this work. Briefly, a DNA fragment spanning the lacZ coding sequence of plasmid pJB20 (3) was amplified with primers pp266 (ACACACCCCGGGCTTTACACTTTATGCTTCCGGCTCGTATAATGTGTGGACACACAGGAAACAGCTATGAC) and pp267 (ACACAACAACCCGGGTTATTATTATTTTTGACACC), cleaved with XmaI restriction endonuclease, and ligated to XmaI-cut DNA from plasmids pSU312 and pSU313 (38), In the resulting recombinant plasmids, pNFB19 and pNFB22, respectively, the lacZ gene is oriented in the opposite direction to the aph gene (pNFB19) or in the same direction as the cat gene (pNFB22).
TABLE 2.
Oligonucleotides used as PCR primers for DNA deletion/replacement
| Primera | Sequence (5′-3′) | Templateb | Allele (simplified)c |
|---|---|---|---|
| pp198 (l) pp321 (r) | CATCAGGCCAGAAAGACGCTGTACTCTGGTGCAGTGATGCCGTCGTTTTACAACGTCGTG TTTGTGTCCACCAGTGCGTCCATATCCTATGATGGACACAATGTAGGCTGGAGCTGCTTC | pNFB19 pNFB22 | Gifsy-1{recE97::lacZ} |
| pp198 (l) pp199 (r) | CATCAGGCCAGAAAGACGCTGTACTCTGGTGCAGTGATGCCGTCGTTTTACAACGTCGTG TATCGTCCAGGGTGACGCGGTACTTATCGTGCAGTTCGTCGCTCTGCCAGTGTTACAACC | pUT lacZ | Gifsy-2{recE59::lacZ} |
| le13 (l) pp140 (r) | CCTCACGAGATTGACCCAGCAGCATACCCAAACCCAACCGTGTGTAGGCTGGAGCTGCTTC AAGAAAACCACCTCACCCTCATAACTCAGTAAGCGTCCCGCATATGAATATCCTCCTTAG | pKD3 | Gifsy-2{Δ89} |
| le18 (l) pp140 (r) | AAAGAGGTGCTACCTGTATGGATGAGTTCTTTGGTTCTATTGTGTAGGCTGGAGCTGCTTC AAGAAAACCACCTCACCCTCATAACTCAGTAAGCGTCCCGCATATGAATATCCTCCTTAG | pKD3 | Gifsy-2{Δ90} |
| le24 (l) le21 (r) | TCTGCGCTCTTCTCTGACATCCACTTTCCATACACCTTGTTGTAGGCTGGAGCTGCTTCG TGATCTAATTAACGACCTCAAAAAATAGAACCAAAGAACTATTCCGGGGATCCGTCGACC | pKD13 | Gifsy-2{Δ91} |
| le24 (l) pp342 (r) | TCTGCGCTCTTCTCTGACATCCACTTTCCATACACCTTGTTGTAGGCTGGAGCTGCTTCG TAGCCGCCACAAGGGACATGGCAAACCAAAGTTGCTTCATCATATGAATATCCTCCTTAG | pKD3 | Gifsy-2{Δ92} |
| le31 (l) le30 (r) | AGCGATGCGACTGCTAACCC AAAAATACTGTATATACAAACAGTTATTAGCGAAACACGTCGGTTGGGTTTGGGTATGCT | MA7612 | Gifsy-2{Δ98}::gtgR |
| le38 (l) le37 (r) | TGCTGCATGTCGGGCAACTGCGGCGCAAGCTGGCGGCAGACCACTTTCCATACACCTTGTT ACTGTTTCTCCATACCTGTTTTTCTGGATGGAGTAAGACGATGAACAAAAATCTTCATCCCAT | MA7720 | Gifsy-2{−} PBAD-gtgR |
| le33 (l) le32 (r) | TAACGCCTGAGACTTTCTTTTCGCAATTGAATAAAGCAATTGTGTAGGCTGGAGCTGCTTC GCGGTGACCGGAAGGCCTGAATATTGGTTCTTCATGGAGCCATATGAATATCCTCCTTAG | pKD3 | Gifsy-1{Δ21}::cat |
Primers are defined as “left” (l) or “right” (r) based on the orientation of the prophage map.
Template plasmids pKD3 and pKD13 are described in reference 12; plasmid pUT-lacZ is described in reference 13; plasmids pNFB19 and pNFB22 were constructed in the course of this work (see Materials and Methods). The portions of primers annealing to template plasmids are shown in italics.
Curly brackets following a prophage name define the genotype of that prophage. A minus sign indicates absence of the prophage
Construction of lacZ fusions.
Replacement constructs fusing the recE gene to the lacZ gene were obtained by λ red-mediated recombination. Donor fragments spanned the lacZ coding sequence and an adjacent antibiotic resistance marker for selection. Two sets of constructs were made, differing in the length of the phage material removed. Donor fragments obtained with primers pp198 and pp199 on plasmid pUT-lacZ (Table 2) produced an internal recE deletion in either Gifsy-1 or Gifsy-2 (ΔrecE59::lacZ), while fragments obtained with the pp198/pp321 pair on pNFB19 or pNFB22 deleted material up to the left end of the Gifsy-1 prophage (Δ[recE-int]97::lacZ) (Table 2 and Fig. 1).
FIG. 1.

Schematic representation of phage genome modifications. (A) Changes in the Gifsy-1 prophage. A lacZ-aph module amplified from plasmid pNFB19 DNA (primers pp198 and pp321) (Table 2) replaces the left portion of the prophage genome, generating an in-frame lacZ fusion to the 28th codon of recE. The sbcE21 deletion and the gftO::Tn10dTc insertion were previously described (17). (B) Changes in the Gifsy-2 prophage. Selected segments of the prophage genome were replaced with FRT-flanked antibiotic resistance cassettes (Table 2). In the construction of deletions Δ89 and Δ90, the cassette (cat) was removed by exposing cells to a Flp-expressing pCP20 plasmid (Materials and Methods). (C) Effects of Gifsy-2 deletions (panel B) on the expression of a Gifsy-1-borne recE-lacZ fusion in the presence of sbcE21 (panel A). The values of β-galactosidase activity (in Miller units) are the means of at least three independent measurements with duplicate cultures. The standard deviation was always below 15% and is omitted for clarity.
A lac operon fusion to the STM1013 locus of Gifsy-2 was derived from the Δ89[STM1013-sseI]::cat construct by Flp-mediated excision of the cat cassette and insertion of the pCE36 plasmid into the “scar” sequence, as described previously (14).
Isolation of zfh-8179::MudJ.
Prior to the development of the λ red method, lac fusions to Gifsy prophage genes were isolated using Mu-derived transposons. The strategy to identify prophage-linked inserts relied on the proximity of Gifsy-1 and Gifsy-2 prophages to genes involved in NAD metabolism (nadB and pncB, respectively) (17) and on the inability of nadB pncB double mutants to grow in media unsupplemented with a NAD precursor (33). Typically, phage P22 lysates prepared on pools of MudJ transposon mutants (22) were used to transduce strain MA6057 (nadB227::MudA [Apr] pncB150::Tn10 [Tcr]), selecting for growth on LB plates. Transductants were of two classes: one resulting from crossover events correcting the nadB allele (Aps) and the other resulting from correction of the pncB allele (recognizable by the Tcs phenotype). Replica plating on LB plates supplemented with kanamycin identified isolates carrying nadB- or pncB-linked MudJ insertions. Transductional tests on X-Gal indicator plates showed the lac genes in one of the nadB-linked inserts to be specifically activated in the presence of sbcE21. DNA sequence analysis showed this MudJ element, zfh-8179::MudJ, to be located immediately downstream from the recT ortholog in the distal portion of the Gifsy-1 left operon.
β-Galactosidase assays.
The activity of β-galactosidase was measured in toluene-permeabilized cells as described by Miller (32) and is expressed in Miller units. Bacteria (grown in LB medium) were centrifuged and resuspended in saline solution prior to the assay.
RESULTS
Allele sbcE21 activates the transcription of the Gifsy-1 prophage “left” operon only in the presence of the Gifsy-2 prophage.
The overall gene organization of the Gifsy-1 and Gifsy-2 prophages is typical of the lambdoid phage family. One can infer the existence of two main transcription units diverging from a site approximately 7 kb from the left end of the prophage map (6, 31). The recET module makes up the majority of the “left” operon. Allele sbcE21 is a 794-bp deletion 365 bp upstream from the beginning of the recE coding sequence of Gifsy-1 (Fig. 1). To study the effects of the mutation on recE transcription, sbcE21 was transferred by transduction into a recBC+ background. A mini-Tn10 insertion, zfh-8157::Tn10dTc (17), in the proximity of sbcE21 was used as a selectable marker. The insertion falls within a gene encoding a putative replication protein O ortholog and is referred to as gfoO::Tn10dTc throughout this paper. Unless otherwise specified, gfoO::Tn10dTc is present in all of the strains used for the study. A Gifsy-1 segment spanning most of the recE gene and extending to a position near the left end of the prophage was replaced by a lacZ-aph module (Fig. 1) (see Materials and Methods). The replacement generated an in-frame recE-lacZ fusion and concomitantly deleted the xis gene, thus stabilizing the fusion even in sbcE21 cells. β-Galactosidase assays showed that recE-lacZ is expressed at a very low level in the presence of the sbcE+ allele and is activated nearly 500-fold by sbcE21. Remarkably, this activation is observed on condition that the Gifsy-2 prophage is present in the genetic background. In cells cured for Gifsy-2, sbcE21 causes only a marginal increase of LacZ levels (Fig. 1). Thus, these results closely match the recB suppression patterns (17). They indicate that recE activation by sbcE21 is not merely due to a relief of repression, but results from an active mechanism dependent on one or more functions supplied in trans by the Gifsy-2 genome.
The above data are consistent with the idea that Gifsy-1 instability in cells harboring sbcE21 reflects the upregulation of the prophage's xis gene (19). The Gifsy-1 int gene, whose product is also required for prophage excision, is directed in the opposite orientation and is expected to be expressed independently (Fig. 1) (17). To further assess the presence of Gifsy-1 excisionase activity in sbcE21 cells, we made use of plasmid pNFB9, a small pir-dependent replicon that carries the Gifsy-1 int gene and attP site and integrates at the Gifsy-1 chromosomal attachment site. The plasmid also contains a β-lactamase gene and confers resistance to ampicillin (Apr). When a phage P22 lysate made on a strain carrying the pNFB9 insert (MA7153) was used to transduce an sbcE21 strain carrying the pir gene (MA6735), Apr colonies arose at 10- to 20-fold-higher frequency than when the recipient was either a pir mutant or pir+ sbcE+. Significantly, most of the clones obtained with the sbcE21 pir+ strain carried pNFB9 as an extrachromosomal element. In contrast, none of the transductants tested from the other two crosses carried the free plasmid. The findings suggest that excisionase activity in sbcE21 cells promotes pNFB9 excision from the transducing fragment. No excision can occur in sbcE+ cells. Once excised, the plasmid is lost unless the host strain expresses the pir gene.
A single Gifsy-2 locus is necessary and sufficient for trans-activation of the Gifsy-1 left operon in sbcE21 mutant cells.
The location of sbcE21, in a presumptive regulatory region, suggested that the Gifsy-2 factor(s) responsible for trans-activating Gifsy-1 transcription originated from the corresponding portion of the Gifsy-2 genome. The right end point of the sbcE21 deletion is within locus STM2628 (31), whose hypothetical product shows similarity to the DicA repressor of the E. coli K-12 cryptic Qin prophage (15). The corresponding locus in the Gifsy-2 genome, STM1012, is a perfect duplicate of STM2628 as part of an 11.5-kb segment of nearly complete sequence identity between the two prophages. Thus, our initial efforts to define the Gifsy-2 gene(s) required for recE-lacZ activation in sbcE21 mutants concentrated around the STM1012 region. Two large deletions removing nearly the entirety of the prophage segment to the right of STM1012 were constructed; one, Δ89, ended 324 bp upstream from the STM1012 coding sequence, and the other, Δ90, ended immediately upstream from the AUG and thus removed the STM1012 promoter (Fig. 1). Δ89 did not interfere with recE-lacZ activation to any significant extent. In contrast, Δ90 completely abolished sbcE21-dependent transcription (Fig. 1). Combined with the results from deletions on the left side of the prophage, these data delimited the region responsible for activation to a 747-bp fragment spanning STM1012 and an unannotated open reading frame (ORF), named gftC in this study, at the beginning of the prophage's early right operon. Next, the entire Gifsy-2 genome was replaced by a DNA fragment carrying solely the STM1012 gene with its presumptive promoter region and the kanamycin resistance aph gene. As shown in Fig. 1, this module activates sbcE21-dependent transcription as efficiently as the whole prophage, providing convincing evidence for the role of STM1012 as a transcriptional activator. Finally, the STM1012 coding sequence was inserted in place of the ara operon under the control of the PBAD promoter. This construction caused recE-lacZ activation to become arabinose dependent (Fig. 2A). Still, the recE-lacZ expression levels in the presence of arabinose reach only approximately half of those observed when STM1012 is expressed from its own promoter (compare Fig. 2A to the β-galactosidase data in Fig. 1). We interpret this difference to suggest that the STM1012 gene product is less efficient at activating transcription when overproduced. Such an interpretation stems from the finding that recE activation is sharply reduced in sbcE21 mutant cells that overexpress the STM1012 gene from a multicopy plasmid (data not shown). Concentration-dependent switching between activating and repressing modes is the hallmark of phage λ repressor regulatory activity at the repressor gene maintenance promoter PRM (34).
FIG. 2.

Transcriptional activation and repression by GtgR. Strains carry the gtgR gene under the control of the chromosomal PBAD promoter and the lacZ gene fused to either the Gifsy-1 recE gene in the presence of sbcE21 (A) or to a Gifsy-2 STM1013 locus (B). (Fig. 1A and 3 show schematic representations of the constructs). Overnight cultures were diluted 1:200 in regular LB medium or in LB medium supplemented with 0.2% arabinose and grown at 37°C with aeration to an optical density at 600 nm of anywhere between 0.4 and 0.7. The cells were permeabilized with toluene and assayed for β-galactosidase activity, which is expressed in Miller units. The strains used were MA7782 (A) and MA7842 (B). The vertical bars represent standard deviations.
STM1012, alias GtgR, is the Gifsy-2 phage repressor.
In light of its involvement in transcriptional control, STM1012 was renamed gtgR (for Gifsy-2 gene regulator). The analogies with phage λ strongly suggested that the gtgR gene encoded the Gifsy-2 repressor. To test this, we constructed a lac operon fusion to STM1013, the second ORF in the putative early right operon, which encodes a protein reminiscent of phage λ CII. The ability of GtgR to repress the STM1013-lac fusion was assessed using the construct carrying the gtgR gene under the control of the chromosomal PBAD promoter (Fig. 3). To make this analysis possible, the strain (MA7842) carried two additional modifications: it lacked the entire Gifsy-1 prophage, and it had the gtgR copy in the Gifsy-2 genome deleted (Fig. 3). The results in Fig. 2B show that the levels of expression of STM1013-lac are reduced by a factor of 100 when MA7482 is grown in the presence of arabinose. Thus, GtgR can repress transcription of the Gifsy-2 right operon. Finally, a strain cured for both Gifsy prophages and carrying the PBAD-gtgR fusion was susceptible to infection by the Gifsy-1 phage when grown in the absence of arabinose but became completely refractory to infection when grown in the presence of the sugar. Arabinose had no effect on infection of a strain lacking gtgR (data not shown). Taken together, these data demonstrate that GtgR is the lysogenic repressor of prophage Gifsy-2. Due to the complete identity of the immunity regions of Gifsy-1 and Gifsy-2 in strain LT2, GtgR and its replica, GogR (STM2028), are both expected to act on the operator of either prophage. The ability of GtgR to activate transcription at the gogR promoter, revealed by the study of sbcE21, suggests that the two proteins normally autogenously regulate their own expression. Three imperfect sequence repeats with dyad symmetry upstream of the gogR and gtgR genes might constitute the GogR/GtgR binding sites (Fig. 4). The three sites overlap with two putative divergent promoter elements in an overall arrangement strongly reminiscent of the phage λ tripartite operator region. Consistent with the λ analogy, the first ORF to the right of the presumptive PR promoter encodes a hypothetical 79-amino-acid (aa) protein with similarity to Cro (gfoC in Gifsy-1; gftC in Gifsy-2). Finally, a second triplet of putative operator sites is found to the left of gogR-gtgR in close proximity to a sequence motif that can be tentatively identified as the PL promoter (Fig. 4).
FIG. 3.

Diagram of gene constructs in strain MA7842. A DNA segment spanning the gtgR gene and the adjacent FRT-flanked aph cassette was PCR amplified from strain MA7612 (Δ91 in Fig. 1B) with primers le30 and le31 (Table 2). Recombination of the fragment into the chromosome placed the gtgR initiation codon at the position normally occupied by the araB AUG. The aph cassette was subsequently removed by exposing recombinant cells to Flp recombinase. In a separate line of work, a λ red-generated deletion covering all of the Gifsy-2 genome portion to the right of gftC (Δ89 in Fig. 1B) was converted to a lac operon fusion as described previously (14). The above-mentioned constructs were combined by transduction into a strain carrying a λ red-generated gtgR deletion and lacking the Gifsy-1 prophage. For the complete genotype of strain MA7842, see Table 1.
FIG. 4.

(A) DNA sequence of the immunity region of Gifsy-1 and Gifsy-2 prophages in strain LT2. Imperfect repeat sequences with dyad symmetry (green shadowing) were identified by the MEME program (1). Purple shadowing marks putative promoter elements. Translation initiation and termination codons are underlined in green and red, respectively. The unusual proximity of the presumptive PRM promoter to the initiation codon of gtgR (only 6 bp) is a feature also found at the λ cI promoter (34). Likewise, the sequence of the −10 region of the putative PR promoter is 100% identical to that of λ PR (34). (B) Nucleotide sequence alignment of putative operator sites and representation of conserved motifs in the WebLogo format (11).
Both the activator and repressor functions of GtgR are required for the sbcE21 phenotypes.
A deletion with the same end points as sbcE21 (Δ[STM2631-gogR]21::cat, hereafter referred to as Δ21::cat) was constructed by the λ red method. The in vitro-made deletion can be readily transferred by transduction from one strain to another using the chloramphenicol resistance (Cmr) marker for selection. Because of its location within identical regions of the two prophages, the construct can recombine with either Gifsy-1 or Gifsy-2 DNA. Much to our surprise, we discovered that Δ21::cat could not be introduced into strains lacking one or the other of the prophages. In contrast, Cmr transductants were recovered at normal frequencies when wild-type LT2 was used as the recipient. These findings were particularly puzzling, given that the changes associated with the loss of Gifsy-2 in sbcE21 cells constituted the starting point of this study. How could sbcE21 be perfectly viable in a Gifsy-2-cured background when Δ21::cat was not? Looking closely, it became apparent that the study of sbcE21 had been done entirely with reconstructed strains carrying, besides sbcE21, the linked gfoO::Tn10dTc insertion (see above). We therefore postulated that gfoO::Tn10dTc suppressed the lethality that results from the loss of Gifsy-2 in an sbcE21 background. To test this idea, we repeated the transduction experiment using a Gifsy-2-cured strain carrying the gfoO::Tn10dTc insertion as a recipient. This time, Cmr transductants were obtained. Significantly, although no tetracycline was present in the selection plates, all of the transductants were found to be Tcr, indicating that only the recombination events between Δ21[STM2631-gogR]::cat and gfoO::Tn10dTc gave rise to viable progeny. Thus, gfoO::Tn10dTc allows Gifsy-2-cured cells to tolerate the Δ21 deletion. In sbcE21 Gifsy-2-cured cells, the absence of GtgR is expected to cause the derepression of the Gifsy-1 right operon. This might result in the production of toxic phage proteins. The gfoO::Tn10dTc insertion might suppress the toxicity by preventing transcription from reaching the genes responsible for toxicity. The model predicted that it should be possible to introduce the Δ21::cat construct into a Gifsy-2-cured strain provided that the strain contained a functional gtgR gene. Results from a transductional cross with strain MA7719, which lacks the entire Gifsy-2 genome except for gtgR and its promoter/operator region (Table 1), verified this prediction (data not shown). In summary, this study shows that GtgR plays a double role in the biology of the sbcE21 mutant: it activates transcription of the recET operon and at the same time allows the mutant to be viable by repressing the transcription of detrimental phage genes.
Characterization of additional sbc alleles.
The original genetic selection that gave rise to the sbcE21 mutant also yielded a strain with an sbcB mutation (sbcB22) (20) and additional isolates that remained uncharacterized (unpublished data). Recent mapping experiments showed that two of these strains carried mutations transductionally linked to the gfoO::Tn10dTc insertion. These isolates were therefore tentatively identified as sbcE mutants (sbcE23 and sbcE25). A third strain contained a mutation (sbc-24) that appeared to be only weakly linked to gfoO::Tn10dTc. We considered that this allele might be located in Gifsy-2, with which gfoO::Tn10dTc recombines at a lower frequency. PCR amplification experiments with primers annealing to sequences in the immunity-recombination regions of both the Gifsy-1 and Gifsy-2 prophages revealed all three sbc alleles to be deletions. In the case of sbcE25, repeated attempts with increasingly widely spaced primers were necessary before an amplification product was obtained, indicative of the large size of the deleted segment. The PCR products were subjected to DNA sequence analysis. The results showed the sbcE23 deletion to closely resemble sbcE21 (Fig. 5). In the analysis of sbc-24, the sequence data included one of the rare positions that differentiate Gifsy-1 from Gifsy-2 in strain LT2. The presence of a T:A base pair in place of an A:T base pair 16 bp to the left of the deletion join point implied that the deletion is located in Gifsy-2. The allele was thus renamed sbcF24. The deletion spans the same region covered by the sbcE21-sbcE23 alleles in Gifsy-1. It activates the recET module of Gifsy-2 by generating a transcriptional fusion to the gtgR promoter. By symmetry with the sbcE system, we expected that recET activation in the sbcF24 mutant would require the presence of Gifsy-1. To test this, the gfoO::Tn10dTc insertion was transferred onto Gifsy-2 (gftO::Tn10dTc) and used as a selectable marker to combine sbcF24 with a Gifsy-2-borne recE::lacZ fusion in strains carrying or lacking Gifsy-1. The recombinants grew as dark-blue colonies in the Gifsy-1+ background, but they were white in the Gifsy-1-cured strain, indicating the requirement for a Gifsy-1-encoded function in the sbcF24-mediated activation of recE::lacZ. We presume, but did not verify, that this function is the GogR repressor.
FIG. 5.

Prophage deletions in sbcE-sbcF mutants. Deletion boundaries were determined by sequencing DNA fragments amplified from strains harboring the indicated alleles. sbcE21, sbcE23, and sbcE25 alleles are located in Gifsy-1; sbcF24 is located in Gifsy-2. The sequences of the two prophages are 99% identical in the depicted segment.
The sequence data showed sbcE25 to be a 2,486-bp deletion extending from a position only 4 bp upstream of the gogR translation stop codon to a site beyond the middle of the recE gene. The deletion generates an in-frame fusion, and it is predicted to yield a hybrid protein carrying the entire GogR sequence, except for the last amino acid, joined to the carboxyl-terminal half of RecE. These findings raised the question of whether the hybrid protein could have GogR activity. To address this point, we set out to measure the transcription of the Gifsy-1 left operon in the sbcE25 mutant in the presence or absence of Gifsy-2. As the recE-lacZ fusion was inappropriate for this study (since it is included in the deleted segment), a MudJ insertion further to the left in the Gifsy-1 genome was chosen as the transcriptional reporter. The insertion, zfh-8179::MudJ, identified in a search for Gifsy-1-linked lac operon fusions activated by sbcE21 (see Materials and Methods), is located immediately downstream of the recT gene. Although less efficiently expressed than the recE-lacZ construct, zfh-8179::MudJ is nonetheless suitable for comparative measurements. The results in Fig. 6 show that while lacZ transcription is completely dependent on Gifsy-2 in the sbcE21 mutant, it decreases by only a factor of 2 in the sbcE25 mutant upon Gifsy-2 removal. It thus appears that the GogR moiety of the GogR-RecE hybrid can still function as a transcriptional activator.
FIG. 6.
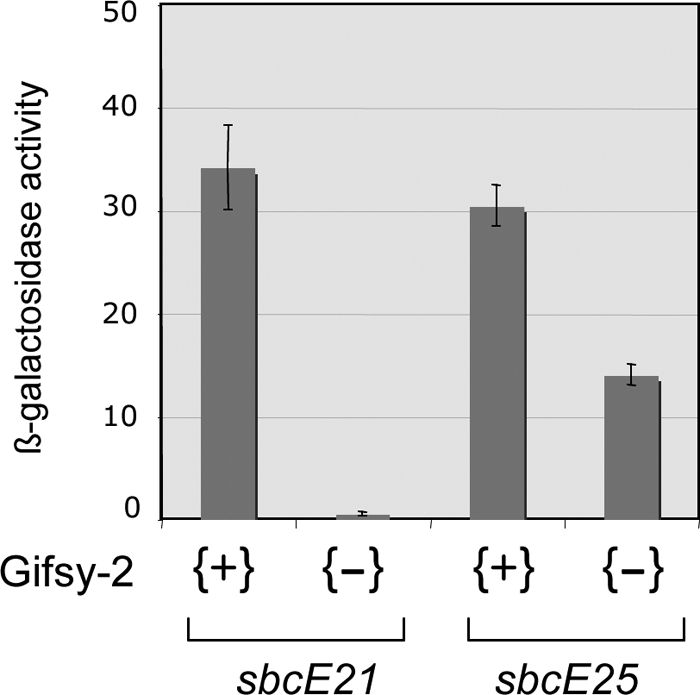
Role of Gifsy-2 in the activation of recE-lacZ expression by alleles sbcE21 and sbcE25. The strains used were MA9555, MA9556, MA9557, and MA9558. β-Galactosidase activity was determined in toluene-permeabilized cells as described previously (32).
DISCUSSION
In the present work, we characterized a class of spontaneous mutations that activate the recE pathway of homologous recombination in S. enterica serovar Typhimurium. Four independent alleles, isolated in strain LT2, result from deletions that derepress one or the other of the normally silent recET modules of the Gifsy-1 and Gifsy-2 prophages. Deletions sbcE21 and sbcE23 appear to act by the same mechanism, exemplified by the analysis of sbcE21. This allele truncates the Gifsy-1 repressor gene, gogR, generating a transcriptional fusion that places the recET module under the control of the gogR promoter. Expression of the gogR-recET fusion is made possible by the fact that Gifsy-2 encodes a perfect GogR homolog, GtgR, capable of activating the gogR promoter. Furthermore, in repressing Gifsy-1 rightward transcription, GtgR prevents the expression of deleterious phage genes. The roles are reversed in the recET activation by sbcF24. This deletion is located in Gifsy-2, and it derepresses transcription of the recET module of the latter by a mechanism dependent upon the presence of Gifsy-1. The Gifsy-1-encoded factor responsible for these effects was not directly identified here, but it is highly likely to be the GogR repressor/activator protein. Finally, sbcE25 is the sole allele that does not rely on complementation for recET activation. The sbcE25 deletion produces a GogR-RecE hybrid protein that is functional as a transcriptional activator. The fact that sbcE25 arose as a recB suppressor implies that the hybrid protein is also a functional recombination enzyme, despite lacking the N-terminal half of the RecE sequence. Similar findings were reported in E. coli, where up to 70% of the N-terminal portion of RecE was found to be dispensable for recombination activity (8). Curiously enough, alignment of the RecE protein sequences from E. coli and Salmonella shows the sequence conservation to be largely limited to the N-terminal domains, whereas the C-terminal domains diverge considerably. To date, the function of the RecE N-terminal domain remains unknown.
Concomitant with activating recET, the sbcEF changes derepress the xis genes, thus stimulating prophage excision. The latter phenotype proved very useful in the initial characterization of the two phages, as it allowed us to obtain cured strains that could be used as hosts for phage growth. Furthermore, the isolation of prophage-cured derivatives of virulent strains led to the discovery that Gifsy-2, and to a lesser extent Gifsy-1, contributes to S. enterica serovar Typhimurium virulence (16). Obtained prior to the completion of Salmonella genome sequencing, these findings hinted at the existence of virulence genes in the prophages' genomes. A number of such loci were subsequently identified and characterized (16, 18). Their overall contribution to Salmonella fitness in the host likely accounts for the wide distribution of Gifsy-1 and Gifsy-2 prophages among serovar Typhimurium strains (6). Sequence analysis of a growing number of isolates shows that the Gifsy-2 sequence is highly conserved; in contrast, Gifsy-1 structure appears highly polymorphic. In particular, the portion spanning the immunity module and the adjacent left-side region is hypervariable and in all cases differs completely from the corresponding region in the Gifsy-2 genome (27). Thus, the sequence identity between the regulatory regions of Gifsy-1 and Gifsy-2 in strain LT2 appears to be a peculiarity of the strain. Most likely, the duplication arose as a result of an intrachromosomal asymmetric recombination event that replaced a portion of the Gifsy-1 genome with the corresponding region from Gifsy-2. It is odd to consider that most of the alleles analyzed here, except for sbcE25, would not have existed if a strain other than LT2 had been used for selection. A further aspect where chance played a crucial role is in the segregation of Gifsy-2-cured cells by sbcE21 mutants. We know from the present study that these segregants should not be viable due to sbcE21 lethality in the absence of Gifsy-2-encoded GtgR. If they could be isolated in the original study, it is because the sbcE21 allele was being used in combination with the gfoO::Tn10dTc insertion that suppresses the lethality.
The segregation of Gifsy-2-cured cells in cultures of strains carrying sbcE21 occurs at a much lower rate than the segregation of Gifsy-1-cured cells, but it is nonetheless significant. While the latter can be readily accounted for by the sbcE21-induced derepression of the Gifsy-1 xis gene, the excision of Gifsy-2 remains unexplained. Possibly, the Gifsy-1 operator sites that become free following GogR inactivation compete with the Gifsy-2 sites for GtgR binding. This could relieve repression enough to result in the occasional production of Gifsy-2 excisionase. Alternatively, the GogR protein made in sbcE21 mutant cells could interfere with the functioning of GtgR. The sbcE21 deletion truncates the gogR gene halfway through the coding sequence, producing an in-frame fusion to the distal two-thirds of the STM2631 locus. The resulting hybrid protein might retain some ability to interact with GtgR monomers (GtgR exists as a dimer [our unpublished data]), effectively lowering the concentration of active repressors.
The DNA sequence upstream of the gogR-gtgR genes bears strong similarity to the region between the cI and cro genes in phage λ. The sequence includes three dyad symmetry repeats overlapping two putative promoter elements in opposite orientations. Interestingly, the −10 hexamer of the rightward promoter is identical to that of lambda PR. The analogy with lambda suggests that GogR/GtgR cooperative binding to the middle and right repeats inhibits the rightward promoter while activating gogR-gtgR transcription from the PRM promoter. Binding at the leftmost repeat, under conditions of excess GogR/GtgR accumulation, occludes the PRM promoter, thus helping to restore the optimal repressor concentration. In spite of the analogy in the switch design, the mechanisms activating the switch during prophage induction differ sharply between lambda and Gifsy-1 and -2. In λ, prophage induction results from the RecA-stimulated autocatalytic cleavage of the CI repressor. The repressors GogR/GtgR are much smaller than CI (136 aa versus 237 aa) and lack the typical cleavage motif. Indeed, our unpublished data show that GogR/GtgR are not cleaved during prophage induction; rather, they are inactivated by the binding of antirepressor proteins. The genes encoding these proteins are located outside the immunity regions and under the direct control of the LexA protein. Thus, Gifsy prophage regulation is an integral part of the SOS response. This is a general property of Gifsy prophages, as the heteroimmune Gifsy-1 variants found in different strains are regulated by a similar mechanism.
Acknowledgments
We are grateful to Nicholas Bergman for analyzing the DNA sequence of the Gifsy-1 immunity region with the MEME program and to David Friedman for encouragement and helpful discussions.
This work was supported by the French National Research Council (CNRS). S.L. was the recipient of fellowships from the French government (MENRT) and from the “Fondation pour la Recherche Medicale”.
Footnotes
Published ahead of print on 8 August 2008.
REFERENCES
- 1.Bailey, T. L., N. Williams, C. Misleh, and W. W. Li. 2006. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34W369-W373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barbour, S. D., H. Nagaishi, A. Templin, and A. J. Clark. 1970. Biochemical and genetic studies of recombination proficiency in Escherichia coli. II. Rec+ revertants caused by indirect suppression of rec− mutations. Proc. Natl. Acad. Sci. USA 67128-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beaber, J. W., B. Hochhut, and M. K. Waldor. 2002. Genomic and functional analyses of SXT, an integrating antibiotic resistance gene transfer element derived from Vibrio cholerae. J. Bacteriol. 1844259-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertani, G. 2004. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186595-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bochner, B. R. 1984. Curing bacterial cells of lysogenic viruses by using UCB indicator plates. BioTechniques 2234-240. [Google Scholar]
- 6.Bossi, L., and N. Figueroa-Bossi. 2005. Prophage arsenal of Salmonella enterica serovar Typhimurium., p. 165-186. In M. Waldor, D. Friedman, and S. Adhya (ed.), Phages: their role in bacterial pathognenesis and biotechnology. ASM Press, Washington, DC.
- 7.Cherepanov, P. P., and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1589-14. [DOI] [PubMed] [Google Scholar]
- 8.Chu, C. C., A. Templin, and A. J. Clark. 1989. Suppression of a frameshift mutation in the recE gene of Escherichia coli K-12 occurs by gene fusion. J. Bacteriol. 1712101-2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark, A. J., and K. B. Low. 1988. Pathways and systems of homologous recombination in Escherichia coli, p. 155-215. In K. B. Low (ed.), The recombination of genetic material. Academic Press, San Diego, CA.
- 10.Clark, A. J., L. Satin, and C. C. Chu. 1994. Transcription of the Escherichia coli recE gene from a promoter in Tn5 and IS50. J. Bacteriol. 1767024-7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crooks, G. E., G. Hon, J. M. Chandonia, and S. E. Brenner. 2004. WebLogo: a sequence logo generator. Genome Res. 141188-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 976640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Lorenzo, V., and K. N. Timmis. 1994. Analysis and construction of stable phenotypes in Gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol. 235386-405. [DOI] [PubMed] [Google Scholar]
- 14.Ellermeier, C. D., A. Janakiraman, and J. M. Slauch. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290153-161. [DOI] [PubMed] [Google Scholar]
- 15.Faubladier, M., and J. P. Bouché. 1994. Division inhibition gene dicF of Escherichia coli reveals a widespread group of prophage sequences in bacterial genomes. J. Bacteriol. 1761150-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Figueroa-Bossi, N., and L. Bossi. 1999. Inducible prophages contribute to Salmonella virulence in mice. Mol. Microbiol. 33167-176. [DOI] [PubMed] [Google Scholar]
- 17.Figueroa-Bossi, N., E. Coissac, P. Netter, and L. Bossi. 1997. Unsuspected prophage-like elements in Salmonella typhimurium. Mol. Microbiol. 25161-173. [DOI] [PubMed] [Google Scholar]
- 18.Figueroa-Bossi, N., S. Uzzau, D. Maloriol, and L. Bossi. 2001. Variable assortment of prophages provides a transferable repertoire of pathogenic determinants in Salmonella. Mol. Microbiol. 39260-271. [DOI] [PubMed] [Google Scholar]
- 19.Flanigan, A., and J. F. Gardner. 2007. Interaction of the Gifsy-1 Xis protein with the Gifsy-1 attP sequence. J. Bacteriol. 1896303-6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gari, E., N. Figueroa-Bossi, A. B. Blanc-Potard, F. Spirito, M. B. Schmid, and L. Bossi. 1996. A class of gyrase mutants of Salmonella typhimurium show quinolone-like lethality and require rec functions for viability. Mol. Microbiol. 21111-122. [DOI] [PubMed] [Google Scholar]
- 21.Hall, S. D., and R. D. Kolodner. 1994. Homologous pairing and strand exchange promoted by the Escherichia coli RecT protein. Proc. Natl. Acad. Sci. USA 913205-3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hughes, K. T., and J. R. Roth. 1988. Transitory cis complementation: a method for providing transposition functions to defective transposons. Genetics 1199-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joseph, J. W., and R. Kolodner. 1983. Exonuclease VIII of Escherichia coli. I. Purification and physical properties. J. Biol. Chem. 25810411-10417. [PubMed] [Google Scholar]
- 24.Joseph, J. W., and R. Kolodner. 1983. Exonuclease VIII of Escherichia coli. II. Mechanism of action. J. Biol. Chem. 25810418-10424. [PubMed] [Google Scholar]
- 25.Kaiser, K., and N. E. Murray. 1980. On the nature of sbcA mutations in E. coli K 12. Mol. Gen. Genet. 179555-563. [DOI] [PubMed] [Google Scholar]
- 26.Kolodner, R., S. D. Hall, and C. Luisi-DeLuca. 1994. Homologous pairing proteins encoded by the Escherichia coli recE and recT genes. Mol. Microbiol. 1123-30. [DOI] [PubMed] [Google Scholar]
- 27.Lemire, S., N. Figueroa-Bossi, and L. Bossi. 2008. Prophage contribution to Salmonella virulence and diversity, p. 159-192. In M. Hensel and H. Schmidt (ed.), Horizontal gene transfer in the evolution of bacterial pathogenesis.. Cambridge University, Cambridge, England.
- 28.Lloyd, R. G., and S. D. Barbour. 1974. The genetic location of the sbcA gene of Escherichia coli. Mol. Gen. Genet. 134157-171. [DOI] [PubMed] [Google Scholar]
- 29.Mahajan, S. K. 1988. Pathways of homologous recombination in Escherichia coli, p. 87-140. In R. Kucherlapati and G. R. Smith (ed.), Genetic recombination. American Society for Microbiology, Washington, DC.
- 30.Mahajan, S. K., C. C. Chu, D. K. Willis, A. Templin, and A. J. Clark. 1990. Physical analysis of spontaneous and mutagen-induced mutants of Escherichia coli K-12 expressing DNA exonuclease VIII activity. Genetics 125261-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McClelland, M., K. E. Sanderson, J. Spieth, S. W. Clifton, P. Latreille, L. Courtney, S. Porwollik, J. Ali, M. Dante, F. Du, S. Hou, D. Layman, S. Leonard, C. Nguyen, K. Scott, A. Holmes, N. Grewal, E. Mulvaney, E. Ryan, H. Sun, L. Florea, W. Miller, T. Stoneking, M. Nhan, R. Waterston, and R. K. Wilson. 2001. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413852-856. [DOI] [PubMed] [Google Scholar]
- 32.Miller, J. H. 1992. A short course in bacterial genetics. A laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 33.Penfound, T., and J. W. Foster. 1996. Biosynthesis and recycling of NAD, p. 721-730. In F. C. Neidhardt, J. L. Ingraham, K. B. Low, B. Magasanik, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella; cellular and molecular biology, 2nd ed. ASM Press, Washington, DC.
- 34.Ptashne, M. 1986. A genetic switch. Blackwell Scientific Publications, Cambridge, MA.
- 35.Schmieger, H. 1972. Phage P22-mutants with increased or decreased transduction abilities. Mol. Gen. Genet. 11975-88. [DOI] [PubMed] [Google Scholar]
- 36.Smith, G. R. 1989. Homologous recombination in E. coli: multiple pathways for multiple reasons. Cell 58807-809. [DOI] [PubMed] [Google Scholar]
- 37.Smith, G. R. 1989. Homologous recombination in prokaryotes: enzymes and controlling sites. Genome 31520-527. [DOI] [PubMed] [Google Scholar]
- 38.Uzzau, S., N. Figueroa-Bossi, S. Rubino, and L. Bossi. 2001. Epitope tagging of chromosomal genes in Salmonella. Proc. Natl. Acad. Sci. USA 9815264-15269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu, D., H. M. Ellis, E. C. Lee, N. A. Jenkins, N. G. Copeland, and D. L. Court. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 975978-5983. [DOI] [PMC free article] [PubMed] [Google Scholar]