

Abstract
A newly characterized archaeal rudivirus Stygiolobus rod-shaped virus (SRV), which infects a hyperthermophilic Stygiolobus species, was isolated from a hot spring in the Azores, Portugal. Its virions are rod-shaped, 702 (± 50) by 22 (± 3) nm in size, and nonenveloped and carry three tail fibers at each terminus. The linear double-stranded DNA genome contains 28,096 bp and an inverted terminal repeat of 1,030 bp. The SRV shows morphological and genomic similarities to the other characterized rudiviruses Sulfolobus rod-shaped virus 1 (SIRV1), SIRV2, and Acidianus rod-shaped virus 1, isolated from hot acidic springs of Iceland and Italy. The single major rudiviral structural protein is shown to generate long tubular structures in vitro of similar dimensions to those of the virion, and we estimate that the virion constitutes a single, superhelical, double-stranded DNA embedded into such a protein structure. Three additional minor conserved structural proteins are also identified. Ubiquitous rudiviral proteins with assigned functions include glycosyl transferases and a S-adenosylmethionine-dependent methyltransferase, as well as a Holliday junction resolvase, a transcriptionally coupled helicase and nuclease implicated in DNA replication. Analysis of matches between known crenarchaeal chromosomal CRISPR spacer sequences, implicated in a viral defense system, and rudiviral genomes revealed that about 10% of the 3,042 unique acidothermophile spacers yield significant matches to rudiviral genomes, with a bias to highly conserved protein genes, consistent with the widespread presence of rudiviruses in hot acidophilic environments. We propose that the 12-bp indels which are commonly found in conserved rudiviral protein genes may be generated as a reaction to the presence of the host CRISPR defense system.
Viruses of the hyperthermophilic crenarchaea are extremely diverse in their morphotypes and in the properties of their double-stranded DNA (dsDNA) genomes (reviewed in references 19 and 23). Moreover, some of the virion morphotypes are unique for dsDNA viruses from any domain of life. Many of these viruses have been classified into seven new families that include rod-shaped rudiviruses, filamentous lipothrixviruses, spindle-shaped fuselloviruses, and a bottle-shaped ampullavirus (reviewed in reference 24). The bicaudavirus Acidianus two-tailed virus (ATV) exhibits an exceptional two-tailed morphology and the unique viral property of developing long tail-like appendages independently of the host cell (11). Crenarchaeal viral research is still at an early stage of development, and insights into basic molecular processes, including infection, replication, packaging, and virus-host interactions, are limited. One of the main reasons for this lies in the high proportion of predicted genes with unknown functions (25).
At present, viruses of the family Rudiviridae are the most promising for detailed studies because they can be obtained in reasonable yields, and there are already some insights into their mechanisms of replication, transcriptional regulation, and host cell adaptation (4, 12, 13, 20, 21). To date, three rudiviruses have been characterized, all from the order Sulfolobales: the closely related Sulfolobus rod-shaped virus 1 (SIRV1), and SIRV2, isolated on Iceland, which infect strains of Sulfolobus islandicus (20, 22), and Acidianus rod-shaped virus 1 (ARV1), isolated at Pozzuoli, Italy, which propagates in Acidianus strains (34). Moreover, rudivirus-like morphotypes and partial rudiviral genome sequences have been detected in environmental samples collected from both acidic and neutrophilic hot aquatic sites (27, 29, 32).
All rudiviral genomes carry linear dsDNA genomes with long inverted terminal repeats (ITRs) ending in covalently closed hairpin structures with 5′-to-3′ linkages (4, 20). The terminal structure is important for replication, which presumably is initiated by site-specific single-strand nicking within the ITR, with the subsequent formation of head-to-head and tail-to-tail intermediates, and the conversion of genomic concatemers into monomers by a virus-encoded Holliday junction resolvase (20). This basic replication mechanism appears to be similar to that used by the eukaryal poxviruses, Chlorella virus and African swine fever virus, although there is no clear similarity between the sequences of the implicated archaeal and eukaryal proteins (20, 25).
The transcriptional patterns of rudiviruses SIRV1 and SIRV2 are relatively simple, with few temporal expression differences. An exception is the gene encoding the major structural protein that binds to DNA and, at an early stage of infection, is expressed as a polycistronic mRNA but appears as a single gene transcript close to the eclipse period (12). It has also been shown that rudiviral transcription can be activated by a Sulfolobus host-encoded protein, Sta1, that interacts specifically with TATA-like promoter motifs in the viral genome (13).
For SIRV1, a detailed study of the mechanism of adaptation to foreign hosts was conducted. Upon passage of the virus through closely related S. islandicus strains, complex changes were detected that were concentrated within six genomic regions (21, 22). These changes included insertions, deletions, gene duplications, inversions, and transpositions, as well as changes in gene sizes that often involved the insertion or deletion of what appeared to be “12-bp elements.” It was concluded that the virus generated a complex mixture of variants, one or more of which were preferentially propagated when the virus entered a new host (21).
Here we describe a novel rudivirus, Stygiolobus rod-shaped virus (SRV), isolated from the Azores, Portugal, a location geographically distant from the locations of the other characterized rudiviruses (20, 34). SRV shows sufficient differences from the other rudiviruses, both morphologically and genomically, to warrant its classification as a novel species. The structural and genomic properties of the rudiviruses are compared and contrasted, and new data on the conserved virion structural proteins are presented. Different rudiviruses were selected for these studies on the basis of the virion or protein yields that were obtained. Moreover, matches between the spacer regions of the crenarchaeal chromosomal CRISPR repeat clusters, which have been implicated in a viral defense system (18) involving processed RNA transcribed from one DNA strand (reviewed in references 16 and 17), and the rudiviral genomes are analyzed and their significance, and possible relationships to the 12-bp indels, are considered.
MATERIALS AND METHODS
Enrichment culture, isolation of viral hosts, and virus purification.
An environmental sample was taken from a hot acidic spring (93°C, pH 2) in the Furnas Basin on Saõ Miguel Island, the Azores, Portugal. The aerobic enrichment culture was established from the environmental sample and maintained at 80°C under conditions described previously for cultivation of members of the Sulfolobales (35). Single strains were isolated by plating on Gelrite (Kelco, San Diego, CA) containing colloidal sulfur (35) and grown in the medium of the enrichment culture. Cell-free supernatants of cultures were analyzed by transmission electron microscopy for the presence of virus particles.
SRV was isolated from the growth culture of its host strain Stygiolobus sp., which was colony purified as described above. After cells were grown to the late exponential phase and harvested by low-speed centrifugation (Sorvall GS3 rotor) (4,500 rpm), virions were precipitated from the supernatant by adding NaCl (1 M) and polyethylene glycol 6000 (10% [wt/vol]) and maintaining the mixture at 4°C overnight. They were purified further by CsCl gradient centrifugation (34).
Transmission electron microscopy.
Samples were deposited on carbon-coated copper grids, negatively stained with 2% uranyl acetate (pH 4.5), and examined in a CM12 transmission electron microscope (FEI, Eindhoven, The Netherlands) operated at 120 keV. The magnification was calibrated using catalase crystals negatively stained with uranyl acetate (28). Images were digitally recorded using a slow-scan charge-coupled-device camera connected to a PC running TVIPS software (TVIPS GmbH, Gauting, Germany). To some samples, 0.1% sodium dodecyl sulfate (SDS) was added, and those samples were maintained at 22°C for 30 min in order to study the stability of the virion particles. Electron tomography of intact, negatively stained virions was performed as described previously (10, 26). Visualization of the three-dimensional (3D) data was performed using Amira software (Visage Imaging, Fürth, Germany).
Protein analyses.
Proteins of SRV were separated in 13.5% SDS-polyacrylamide gels (14) and stained with Coomassie brilliant blue R-250 (Serva, Heidelberg, Germany). N-terminal protein sequences were determined by Edman degradation using a Procise 492 protein sequencer (Applied Biosystems, Foster City, CA).
SIRV2 proteins were separated in 4 to 12% SDS-polyacrylamide NuPAGE gradient gels by the use of MES (morpholineethanesulfonic acid) buffer (both from Invitrogen, Paisley, United Kingdom). The gels were stained with Sypro Ruby (Invitrogen). Protein bands were analyzed by peptide mass fingerprinting with matrix-assisted laser desorption ionization-time of flight mass spectrometry using a Voyager DE-STR biospectrometry workstation (Applied Biosystems, Framingham, MA) as described earlier (26). The analysis was performed in conjunction with the proteomic platform at the Pasteur Institute.
Cloning and heterologous expression of ARV1-ORF134b and purification of the recombinant protein and its self-assembly.
ARV1-ORF134b was amplified from purified viral DNA with primers ARV1ORF134F (GGAATTCCATATGATGGCGAAAGGACACACACC) and ARV1ORF134R (GGAATTCTCGAGACTTACGTATCCGTTAGGAC). The PCR product was purified (PCR purification kit; Roche, Mannheim, Germany) and cloned into pET30a expression vector (Novagen, Madison, WI) between restriction sites for EcoRI and XbaI. The protein was expressed overnight at 20°C in the Escherichia coli Rosetta(DE3)pLysS strain. Protein expression was controlled by SDS-polyacrylamide gel electrophoresis analysis and by performing a Western blot analysis using anti-His-tag-specific antibodies (Novagen). The native protein was purified on a Ni2+-nitrilotriacetic acid (Ni2+-NTA)-agarose column (Novagen) with elution buffers containing 50 to 500 mM imidazole. The accuracy of its sequence was confirmed. Self-assembly of the recombinant protein into filamentous structures was performed at 75°C and pH 3.5 and observed by electron microscopy.
Preparation of cellular and viral DNA and DNA sequencing.
DNA was extracted from Stygiolobus azoricus cells as described previously (2), and the 16S rRNA gene was amplified by PCR using primers 8aF and 1512 uR (6) and sequenced.
Viral DNA was obtained by disrupting SRV particles with 1% SDS for 1 h at room temperature and extraction with phenol-chloroform (9). A shotgun library was prepared by sonicating viral DNA to generate fragments of 2 to 4 kb and cloning these into the SmaI site of the pUC18 vector. DNA was purified from single colonies by the use of a Biorobot 8000 workstation (Qiagen, Westburg, Germany) and sequenced in MegaBACE 1000 sequenators (Amersham Biotech, Amersham, United Kingdom). The viral sequence was assembled using Sequencher 4.2 software (Gene Code, Ann Arbor, MI). PCR primers for gap closing and resolving sequence ambiguities were designed using Primers for Mac, version 1.0. Sequence alignments were obtained using MUSCLE software (7). Open reading frames (ORFs) were defined with the help of ARTEMIS software (30) and investigated in searches using the EMBL and GenBank (1), 3D-Jury (8), and SMART (15) databases. Genome maps were generated and compared using Mutagen software, version 4.0 (5).
Bioinformatical matching of crenarchaeal CRISPR spacers to rudiviral genomes.
CRISPRs were predicted for each of the 14 publicly available crenarchaeal genomes in GenBank (NC_000854 [Aeropyrum pernix K1], NC_002754 [Sulfolobus solfataricus P2], NC_003106 [Sulfolobus tokodaii strain 7], NC_003364 [Pyrobaculum aerophilum strain IM2], NC_007181 [Sulfolobus acidocaldarius DSM 639], NC_008698 [Thermofilum pendens Hrk5], NC_008701 [Pyrobaculum islandicum DSM 4184], NC_008818 [Hyperthermus butylicus DSM 5456], NC_009033 [Staphylothermus marinus F1], NC_009073 [Pyrobaculum calidifontis JCM 11548], NC_009376 [Pyrobaculum arsenaticum DSM 13514], NC_009440 [Metallosphaera sedula DSM 5348], NC_009676 [Cenarchaeum symbiosum], and NC_009776 [Ignicoccus hospitalis KIN4/I]). In addition, the six sequenced repeat clusters from Sulfolobus solfataricus P1 (16) were added to the data set as well as CRISPRs from five incomplete Sulfolobus islandicus genomes publicly available through the Joint Genome Institute (http://genome.jgi.doe.gov/mic_asmb.html) and unpublished genome sequences of Sulfolobus islandicus HVE10/4 and Acidianus brierleyi from the Copenhagen laboratory. The repeat cluster sequences were found using publicly available software (3, 7).
All predictions were curated manually. The orientation of each repeat cluster was inferred from the repeat sequence and by locating the low-complexity flanking sequence that generally resides immediately upstream from the cluster and contains the transcriptional leader (16). All unique spacer sequences of the repeat clusters, corresponding to the processed spacer transcript sequence (16), were aligned to the complete nucleotide sequences on each strand of all four rudiviral genomes (SRV [accession no. FM164764], SIRV1 [AJ414696], SIRV2 [AJ344259], and ARV1 [AJ875026]) by use of Paralign, an MMX-optimized implementation of the Smith-Watermann algorithm (31). Moreover, assuming that the spacer DNA can be incorporated into the oriented CRISPRs in either direction, we also translated the two strands of the spacer DNA into all the reading frames, yielding six amino acid sequences per spacer. Reading frames containing stop codons (ca. 50%) were omitted to make the subsequent search more specific. Each translation was aligned against the amino acid sequences of all the annotated ORFs in each of the four rudiviral genomes. Significant e-value cutoffs were determined for both the nucleotide and amino acid sequence searches using the genome sequence of Saccharomyces cerevisiae as a negative control (data not shown).
RESULTS
SRV isolation and structure.
The virus-producing strain was colony purified from an enrichment culture established from a sample collected from an acidic hot spring in the Azores (see Materials and Methods). Its 16S rRNA sequence represented the genus Stygiolobus of the Sulfolobales crenarchaeal order and was closely related to that of Stygiolobus azoricus. However, it differs from S. azoricus, the type species of the genus, in its capacity to grow aerobically, and a description of the new species is in preparation. The virus particles produced constituted flexible rods 702 (± 50) by 22 (± 3) nm in size, with three short fibers at each terminus (Fig. 1A to C; Table 1). A Fourier analysis of the virion (not shown) revealed the presence of regular features with a periodicity of (4.2 nm)−1, which probably reflect a helical subunit arrangement. This feature is also seen in the tomographic data set (Fig. 1D to H), which revealed more structural details. The helical arrangement in the virion core occurs in two different configurations. In the central region, a zigzag structure with dark contrast, probably arising from uranyl acetate staining, is surrounded by a protein shell (Fig. 1D and E). In contrast, in the terminal plug, which is about 50 nm in length, a helically arranged protein mass, with no obvious uranyl acetate inclusions, is seen (Fig. 1D to F). The three terminal fibers, anchored in the plug-like structure, appear to be built up of multiple subunits ordered in a linear array (Fig. 1D). The side view of the reconstructed virion particle (Fig. 1E), as well as cross-sections of the negatively stained virions obtained from the tomograms (Fig. 1G and H), shows that the virion particles are embedded in negative stain (Fig. 1G and H) and partially collapsed due to staining and air drying; the height of the particles was about half of the apparent diameter. Nevertheless, the accumulated central stain is clearly visible in the cross-section (Fig. 1G) of the central part of the virion (Fig. 1D), while this feature is absent from the plug (Fig. 1G and H). The rod-shaped morphology of SRV, with a regular helical core and tail fibers, is characteristic of rudiviruses.
FIG. 1.

Electron micrographs of SRV virions negatively stained with 3% uranyl acetate. (A) A full virion particle, with a discontinuous central line along the virion. (B) Six virions attached to liposome-like structures. (C) Enlargement of a portion of panel B displaying the terminal fibers. (D to H) Electron tomography images of an SRV virion. (D) Horizontal x-y slice (0.7 nm) showing the accumulated stain in the central part of the virion (white arrow). (E) Vertical y-z slice (0.7 nm) through the 3D data set of the reconstructed part of an SRV particle. (F) Visualization of the 3D data set using Amira software. (G and H) Vertical x-z slice (0.7 nm) through the tomogram showing that the virion particles are embedded in negative stain and that accumulated stain visible in panel D is absent from the plug (black arrows). Bars, 200 nm (A and B); 50 nm (C); 20 nm (D, E, G, and H).
TABLE 1.
Properties of the rudiviruses
To investigate further the fine structure of the virion, virion particles were incubated in buffer containing 0.1% SDS for 30 min at 22°C. Most of the virion remained undisturbed, with the particles showing the same diameter as native virions and the densely stained, helical core. However, in local regions the protein shell had dissociated (Fig. 2) and a fine fiber with a diameter of 3 to 4 nm that constituted either naked DNA or a DNA-protein complex was visible.
FIG. 2.
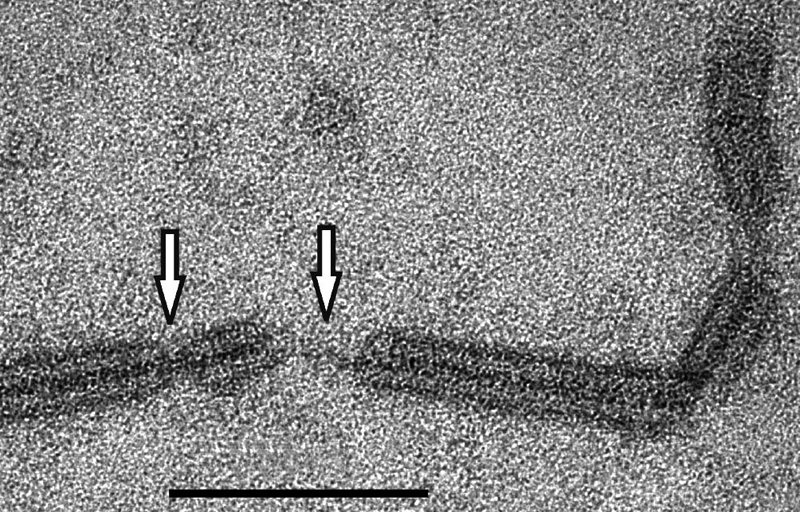
Electron micrograph of a portion of an SRV virion after treatment with 0.1% SDS for 30 min (see Materials and Methods). White arrows indicate DNA or DNA-protein fibers lacking the protein core. Bar, 100 nm.
Self-assembly of the major coat protein.
The major rudiviral structural protein is highly conserved in sequence and is glycosylated (20, 22, 32a, 34). In order to study its possible self-assembly properties, the ARV1 protein (ORF134b [34]) was expressed heterologously in E. coli (see Materials and Methods) and a His-tagged protein was purified to homogeneity on an Ni2+-NTA-agarose column. The protein was shown by transmission electron microscopy to self-assemble to produce filamentous structures of uniform widths and different lengths (Fig. 3). The optimal conditions for the assembly, 75°C and pH 3, were close to those of the natural environment, and no additional energy source was required for this process. The transmission electron microscopy analysis revealed that the filaments had structural parameters similar to those of the native virions, with a diameter of 21 (± 3) nm and a periodicity of (4.2 nm)−1. Thus, the data suggest that the single major coat protein alone can generate the body of the virion.
FIG. 3.

Electron micrograph images of the self-assembled major coat protein of ORF134 from ARV1 after negative staining with 3% uranyl acetate. Bar, 100 nm.
Minor rudiviral virion proteins.
To date, the major coat protein is the only rudiviral structural protein to have been characterized. Given the closely similar structures of the different rudiviruses, we attempted to identify minor structural proteins for the SIRV2 virus, which can be produced in high yields. Protein components of SIRV2 virions, separated on a polyacrylamide gel, yielded six distinct major bands (Fig. 4), and all except D2, which is the strongest band and corresponds to ORF134 (gp26), were analyzed by mass spectrometry. Their identities were as follows: band A contained ORF1070 (gp38), band B contained ORF488 (gp33), and band C contained ORF564 (gp39), while bands D1 and D3 both contained ORF134 (gp26), probably in a glycosylated or, in the case of D3, a proteolytically degraded form. Thus, three additional SIRV2 structural proteins were identified, each highly conserved in sequence in all rudiviruses (Table 2).
FIG. 4.
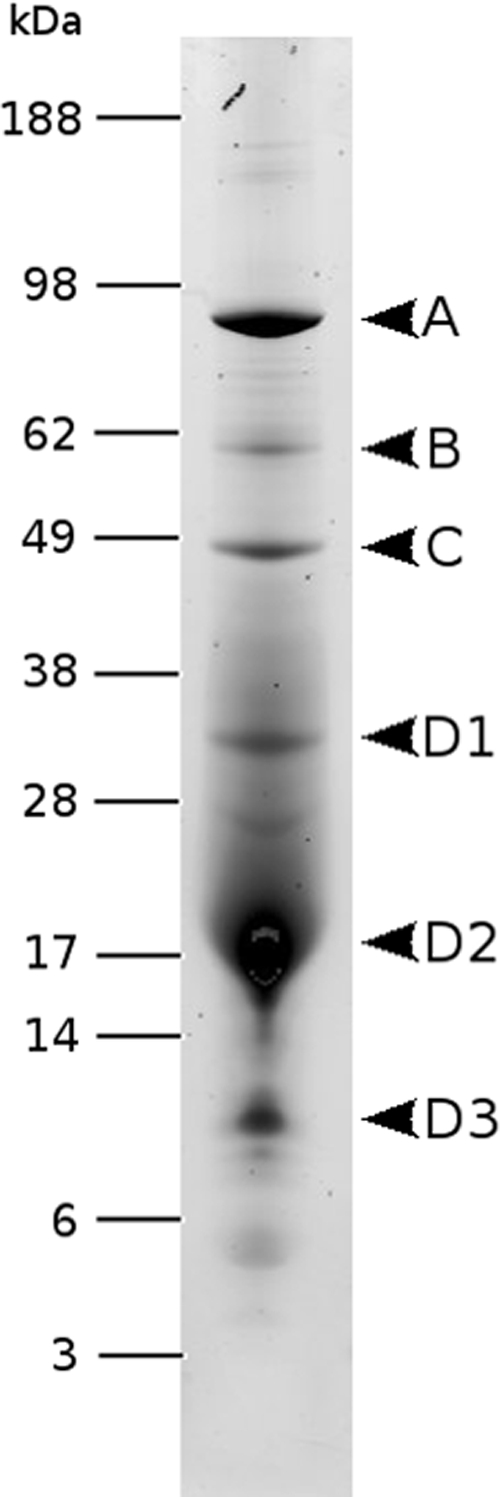
SIRV2 virion proteins separated by SDS-polyacrylamide gel electrophoresis and stained with Sypro Ruby. Molecular masses of protein standards are indicated in kilodaltons on the left.
TABLE 2.
Rudiviral proteins with predicted functions
| SRV ORF category | Rudiviral homolog(s) | Predicted function or description | Analysis tool | E-value or score | Other crenarchaeal virus(es) |
|---|---|---|---|---|---|
| Structural proteins | |||||
| ORF134 | All | Structural protein | |||
| ORF464 | All | Structural protein | |||
| ORF581 | All | Structural protein | |||
| ORF1059 | All | Structural protein | |||
| Transcriptional regulators | |||||
| ORF58 | All | RHH-1 | SMART | 2.0e-08 | Many |
| ORF95 | None | “Winged helix” repressor DNA binding domain | 3D-Jury | 64.00 | None |
| Translational regulator | |||||
| ORF294 | SIRV1 and -2 | tRNA-guanine transglycosylase | 3D-Jury | 167.57 | STSV1 |
| DNA replication | |||||
| ORF440 | All | RuvB Holliday junction helicase (Lon ATPase) | 3D-Jury | 53.71 | AFV1, AFV2 |
| ORF116c | All | Holliday junction resolvase (archaeal) | SMART | 2.4e-45 | |
| ORF199 | All | Nuclease | 3D-Jury | 63.86 | AFV1, SIFV |
| DNA metabolism | |||||
| ORF168 | SIRV1 and -2 | dUTPase | SMART | 1.5e-12 | STSV1 |
| ORF257 | ARV1 | Thymidylate synthase (Thy1) | SMART | 7.9e-46 | STSV1 |
| ORF159 | All | S-adenosylmethionine-dependent methyltransferase | 3D-Jury | 73.67 | SIFV |
| Glycosylation | |||||
| ORF335 | All | Glycosyl transferase group 1 | SMART | 6.7e-09 | |
| ORF355 | All | Glycosyl transferase | SMART | 5.1e-04 | |
| Other | |||||
| ORF419 | SIRV1 and -2 | 11 transmembrane regions | TMHMM |
SRV genome content.
A shotgun library of the viral genome was prepared, sequenced, and assembled (see Materials and Methods) to yield an approximately 10-fold coverage of a 26-kb contig. Since 1 to 2 kb of terminal sequence is always absent from shotgun libraries of linear viral genomes, these additional sequences were generated by primer walking using viral DNA, or using PCR products obtained therefrom, until subsequent rounds of walking yielded no further sequence. The total sequence obtained was 28,096 bp, with a G+C content of 29% and an ITR of about 1,030 bp (Table 1). An EcoRI restriction digest yielded fragments consistent with the genome size (data not shown).
Thirty-seven ORFs were predicted for which start codons were assigned on the basis of the upstream locations of TATA-like and transcription factor B-responsive element (BRE) promoter motifs and/or Shine-Dalgarno motifs. Details of the putative genes and operon structures are presented in Table S1 in the supplemental material, and a comparative genome map of SRV and rudiviruses SIRV1 and ARV1 is presented in Fig. 5; the genome map of SIRV2, which is closely similar to that of SIRV1, is not included (12, 20). SRV differs from the other rudiviruses in that fewer ORFs are organized in operons, and it has a lower level of gene order conservation (Fig. 5). Moreover, whereas for the other rudiviruses TATA-like motifs are often directly preceded by a conserved GTC triplet (12, 20, 34), in SRV the ensuing triplet sequence was GTA for 10 of the 30 putative TATA-like motifs (see Table S1 in the supplemental material).
FIG. 5.

Genome maps of SRV, SIRV1, and ARV1 showing the predicted ORFs and the ITRs (bold lines). SRV ORFs are identified by their amino acid lengths. Homologous genes shared between the rudiviruses are color-coded. Genes above the horizontal line are transcribed from left to right, and those below the line are transcribed in the opposite direction. Predicted functions or structural characteristics of the gene products are indicated as follows: sp, structural protein; rhh, ribbon-helix-helix protein; wh, winged helix protein; tm, transmembrane; tgt, tRNA guanine transglycosylase; hjh; Holliday junction helicase; hjr, Holliday junction resolvase; n, nuclease; du, dUTPase; ts, thymidylate synthase; sm, S-adenosylmethionine-dependent methyltransferase; gt, glycosyl transferase.
Homologs of 17 SRV ORFs are present in all rudiviruses, and a further 10 SRV ORFs are conserved in some rudiviruses. Each virus type carries a few genes which are unique, and these are generally clustered near the ends of the linear genomes and yield no matches to genes in public sequence databases. In SRV, these are ORF145, -116a, -109, -59, -108, -97b, and -92 (left to right in Fig. 5). Although for SIRV1 and SIRV2 some of these nonconserved ORFs have been shown to be transcribed (12), further work is necessary to establish whether they are all protein-coding genes. Some of the proteins carry predicted structural motifs, and putative functions could be assigned to some of the conserved ORFs on the basis of public database searches; most of these are encoded in other crenarchaeal genomes (Table 2).
The host-encoded transcriptional regulator Sta1, a winged helix-turn-helix protein, was shown to bind to some SIRV1 promoters, including those of ORF134 and ORF399, and to enhance their transcription (13). A similar regulation may occur also for SRV, since the promoter regions of the homologs of ORF134 and ORF399 contain putative Sta1 binding sites. In contrast, in ARV1 only the ORF134 homolog is present in an operon for which the first ORF is a putative transcriptional regulator, and its promoter does not carry Sta1 binding motifs.
Genomic features.
Sequence heterogeneities and other exceptional properties were detected in the SRV genome and in other rudiviral genomes that are described below.
(i) ITRs.
For SRV, the 1,030-bp ITR is perfect, except for a 36-bp insert at positions 799 to 834 at the left end and inverted tetramer sequences (AAAA [positions 425 to 428] and TTTT [positions 27672 to 27669]). It shows little sequence similarity to ITRs of the other rudiviruses, except for the 21-bp sequence (AATTTAGGAATTTAGGAATTT) located at the terminus that is predicted to be a Holliday junction resolvase binding site occurring in all sequenced rudiviruses (34). The ITRs of SRV and SIRV1 and -2 carry four to five degenerate copies of this direct sequence repeat, while that of ARV1 carries multiple degenerate copies of other diverse repeats of similar sizes.
(ii) Genome heterogeneity in SRV and 12-bp indels.
Sequence heterogeneities were detected in the SRV genome, within the 10-fold sequence coverage, and mutations were localized to groups of subpopulations, including one 180-bp deletion between positions 11896 and 12077 in two out of six clones. Moreover, a 48-bp insertion was observed in one variant (out of 18 clones) precisely at the C terminus of ORF533 (position 20285) that generated a third copy of a 16-amino-acid direct repeat. Some changes corresponding to 12-bp indels were also apparent in overlapping clones, and they are indicated in Table 3 together with those observed earlier for SIRV1 (4, 20, 21). Moreover, sequence comparison of highly conserved ORFs present in the four rudiviral genomes revealed several additional 12-bp indels. The locations of all the identified indels which occur in conserved rudiviral genes or sites corresponding to SRV ORF75, -104, -138, -163, -168, -197, -199, -286, -294, -419, -440, -464, -533, and -1059 (Fig. 5) are indicated in the SIRV1 genome map in Fig. 6.
TABLE 3.
Occurrence of the 12-bp indels in overlapping rudiviral clone libraries
FIG. 6.

CRISPR spacer sequence matches for SIRV1 are superimposed on the SIRV1 genome map. Protein-coding regions translated from left to right are shown above the line, and those translated from right to left are shown below the line. Highly conserved coding genes are presented in dark blue, while less-conserved or nonconserved genes are in light blue. The inverted terminal repeat is shaded in violet. Matches to spacers are shown as vertical lines and are color-coded as indicated. Matches to the upper DNA strand are placed above the genome, and those to the lower strand are located below the genome. The red vertical lines correspond to the nucleotide sequence matches, and the green vertical lines correspond to matching amino acid sequences, after translation of the spacer sequences from both DNA strands. In total, there were 106 matches to SIRV1 at the nucleotide level, some of them occurring more than once, and an additional 127 matches to SIRV1 ORFs at the amino acid level. The black arrowheads indicate the positions of the 12-bp indels that occur in one or more conserved rudiviral genes.
Rudiviral matches to CRISPRs.
The availability of four separate rudiviral genome sequences provided a basis for analyzing the frequency and distribution of the matches of CRISPR spacer sequences to the viral genomes. Therefore, we analyzed the repeat clusters of each of the available crenarchaeal genomes in the public EMBL/GenBank and JGI sequence databases and in our own unpublished genomes (see Materials and Methods). Fourteen complete genomes and 8 partial genomes were analyzed. In total, 82 repeat clusters from complete genomes and 44 clusters, some incomplete, from partial genomes yielded 4,283 spacer sequences. Subsequently, 278 sequences that are shared between S. solfataricus strains P1 and P2 (16) were omitted from the data set, yielding a total of 4,005 spacer sequences.
In the first analysis, each of the 4,005 spacer sequences was compared to the four rudiviral genomes at the nucleotide level. In total, 158 spacers yielded 268 rudiviral matches. The latter number exceeds the former because (i) some spacers match to more than one locus within repeat sequences of a given virus and (ii) some spacers match to more than one virus. Second, the analysis was performed at the protein level (see Materials and Methods). This analysis revealed 148 additional matching spacers and a further 427 rudiviral genome matches exclusively at the protein level. (An additional 105 matching spacer sequences from the latter analysis that overlapped, partially or completely, with 158 of those detected within rudiviral ORFs at the nucleotide level were not counted.) Only 6 of the 14 completed crenarchaeal genomes carried spacers yielding matches to rudiviral genomes, and they are listed, together with the results for the partial genomes, in Table 4. These results reinforced the choice of criteria employed for determining the significance of sequence matches (see Materials and Methods).
TABLE 4.
Number of CRISPR spacer sequences from complete and partial crenarchaeal genomes which match rudiviral genomesa
| Strain | No. of matching sequences at the indicated levelb
|
Accession no., reference, or source | |
|---|---|---|---|
| Nucleotide | Amino acid | ||
| Complete genomes | |||
| S. solfataricus P2 | 22 (14) | 31 (18) | NC_002754 |
| S. tokodaii 7 | 9 | 14 | NC_003106 |
| M. sedula | 5 | 15 | NC_009440 |
| S. acidocaldarius | 5 | 9 | NC_007181 |
| S. marinus F1 | 2 | 1 | NC_009033 |
| H. butylicus | 0 | 1 | NC_008818 |
| Incomplete genomes | |||
| S. solfataricus P1 | 20 (14) | 30 (18) | 16 |
| S. islandicus (5 strains) | 39/12/4/2/1 | 26/7/2/2/0 | See text |
| S. islandicus HVE10/4 | 36 | 11 | Unpublished |
| A. brierleyi | 15 | 14 | Unpublished |
All the acidothermophilic organisms from the family Sulfolobaceae have spacers matching those of the rudiviral genomes. However, the neutrophilic hyperthermophiles S. marinus and H. butylicus produced very few matches. Matches at the amino acid sequence level that overlapped with those at the nucleotide sequence level were excluded from the data.
Numbers in parentheses in columns 2 and 3 indicate the number of matches that arose from spacers shared by S. solfataricus strains P1 and P2 (16).
The locations of the spacer sequence matches are superimposed on the genome map of SIRV1 in Fig. 6. The matches are not evenly distributed along the genome; some genes have no matches, while others carry up to 18. Although there is no strict correlation between the level of gene sequence conservation and the number of matching spacers, the five most conserved genes, ORF440, ORF1059, ORF134, ORF355, and ORF581, exhibit the highest number of matches (18, 15, 14, 14, and 13, respectively) (Fig. 3 and 6).
DISCUSSION
The morphological and genomic data for SRV and the other characterized rudiviruses are summarized in Table 1. The conservation of their morphologies and genomic properties contrasts with that of other crenarchaeal viruses and, in particular, with that of the filamentous lipothrixviruses, which exhibit a variety of surface, envelope, and tail structures and much more heterogeneous genomes (24).
The virion length of SRV, 702 (± 50) nm, shows the same direct proportionality to genome size (28 kb) as those for the other rudivirus virions, which range in length from 610 (± 50) nm (ARV1) to 900 (± 50) nm (SIRV2) (Table 1). A superhelical core, with a pitch of 4.3 nm and a width of 20 nm, terminates in 45-nm-long nonhelical “plugs,” and it correlates with the internal structure observed earlier in electron micrographs of SIRV1 (22). In order to determine whether a single superhelical DNA can span the SRV virion length, we applied the following formulae to estimate the sizes and length of the superhelical DNA:
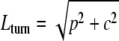 |
where Lturn represents the arc length of a turn, p represents the pitch, and c represents the cylinder circumference, and
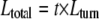 |
where t represents the number of turns and Ltotal represents the arc length of entire helix.
Calculations using structural parameters for B-form DNA yielded a genome size of 26 kbp without, and 30 kbp with, terminal “plugs”. The estimated width (20 nm) is an upper-limit estimate. A reciprocal calculation, with a 28-kbp genome, yields a diameter of 21.2 nm without, and 18.5 nm with, the “plugs.” Given that the major rudiviral coat protein is capable of self-assembly into filamentous structures similar in width to the native virion (Fig. 4), it is likely that the rod-shaped body consists of a single superhelical DNA embedded within this filamentous protein structure. Thus, the three newly identified minor structural proteins probably contribute to conserved terminal features of the virion; consistent with this, the largest structural protein (corresponding to SRV ORF1059) was localized within the virion tail fibers of SIRV2 by studying functional groups by the use of bioconjugation (Steinmetz et al., submitted).
We still have limited insight into functional roles of rudivirus-encoded proteins (Table 2). The glycosyl transferases have been implicated in the glycosylation of the structural proteins (34). Moreover, a few proteins have been linked to viral replication. Two of these, ORF440 and ORF199, lie within an operon and are conserved in phylogenetically diverse lipothrixviruses (33). The former yielded significant matches to RuvB, the helicase facilitating branch migration during Holliday junction resolution, while ORF199 yielded the best matches to nucleases, including Holliday junction resolvases (Table 2). Thus, they are likely to facilitate rudiviral replication, which, in SIRV1, involves site-specific nicking within the ITR, formation of head-to-head and tail-to-tail intermediates, and conversion of genomic concatemers to monomers by a Holliday junction resolvase (ORF116c) (20). In addition, SRV encodes a dUTPase and a thymidylate synthase, both of which are involved in thymidylate synthesis, whereas the other rudiviruses encode only one of these enzymes, both of which are considered helpful in maintaining of a low dUTP/dTTP ratio and thus in minimizing detrimental effects of misincorporating uracil into DNA. Two putative transcriptional regulators have been identified, together with the putative tRNA transglycosylase encoded by SRV, which has homologs in SIRV1 and -2 and in other crenarchaeal viruses (Table 2) and is distantly related to a tRNA-guanine transglycosylase implicated in archeosine formation.
The two approaches employed to analyze CRISPR spacers matching the four rudiviral genomes demonstrated that about 10% of the 3,042 unique acidothermophile spacers yielded positive matches. Employing alignments at the amino acid level considerably increased the number of positive matches detected, because nucleotide sequences diverge more rapidly. Thus, the genomes of SRV and SIRV1 share almost no (∼4%) similarity at the DNA level, whereas most homologous proteins show, on average, 47% sequence identity or similarity. When studying the distribution of the spacer matches in the rudiviral genomes, some trends are evident. First, there is no significant bias with regard to the DNA strand carrying the matching sequence. In SIRV1, for example, 122 matches occur on one strand and 111 on the other (Fig. 6). This is consistent with our assumption that the incorporation of viral or plasmid DNA into the orientated CRISPRs is nondirectional. Second, in accordance with earlier analyses (16), for matches to coding regions, there is no significant bias to matches occurring in a sense or antisense direction. Thus, for SIRV1, 39% of the matches are in the sense direction whereas 54% are antisense—the remaining 7% constitute nucleotide matches to non-protein-coding regions (Fig. 6). Third, when the latter nucleotide sequence-based matches are considered, the proportion of matches which occur in intergenic regions, as opposed to those occurring in protein-coding regions, is not significantly different from the overall coding percentage of the virus. For SIRV1 19% of the nucleotide matches fall within intergenic regions, whereas 20% of the genome is non-protein-coding. Finally, some genes have many matches whereas others have none at all. Five genes have 13 or more matches in SIRV1; these genes correspond to SRV ORF440, ORF1059, ORF134, ORF355, and ORF581. Apart from being conserved in each rudivirus, their gene products have important structural or functional roles (Table 2).
The results pose an important question as to how the host distinguishes between more important and less important genes when adding the spacers to its CRISPRs. Possibly, although the de novo addition of spacers may well be an unbiased process with respect to both viral genome position and direction, the selective advantage provided by some spacers would result in a population being enriched in hosts with CRISPRs carrying spacers targeting crucial viral genes.
The 12-bp viral indels were originally shown to occur commonly in SIRV1 variants that arose as a result of passage of an SIRV1 isolate through different closely related S. islandicus strains from Iceland, and it was inferred that this unusual activity reflected adaptation of the rudivirus to the different hosts (21). The positions of the 12-bp indels that have been identified in conserved rudiviral protein genes are shown together with the CRISPR spacer matches on the SIRV1 genome map in Fig. 6. Many of the sites are very close or overlap. This raises the possibility that lengthening or shortening of conserved protein genes by 12 bp could be a mechanism to overcome the host CRISPR defense system.
We conclude that the rudiviruses are excellent models for studying details of viral life cycles and virus-host interactions in crenarchaea. These viruses appear to be much more conserved in their morphologies and genomes than, for example, the equally ubiquitous lipothrixviruses. Moreover, they are relatively stably maintained in their hosts and can be isolated in reasonable yields for experimental studies.
Supplementary Material
Acknowledgments
We are grateful to Georg Fuchs for providing the environmental sample from Saõ Miguel Island, the Azores.
The research in Copenhagen was supported by grants from the Danish Natural Science Research Council, the Danish National Research Foundation, and Copenhagen University. The research in Paris was partly supported by grant NT05-2_41674 from Agence Nationale de Recherche (Programme Blanc).
Footnotes
Published ahead of print on 22 August 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 253389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bettstetter, M., X. Peng, R. A. Garrett, and D. Prangishvili. 2003. AFV1, a novel virus infecting hyperthermophilic archaea of the genus Acidianus. Virology 31568-79. [DOI] [PubMed] [Google Scholar]
- 3.Bland, C., T. L. Ramsey, F. Sabree, M. Lowe, K. Brown, N. C. Kyrpides, and P. Hugenholtz. 2007. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blum, H., W. Zillig, S. Mallok, H. Domdey, and D. Prangishvili. 2001. The genome of the archaeal virus SIRV1 has features in common with genomes of eukaryal viruses. Virology 2816-9. [DOI] [PubMed] [Google Scholar]
- 5.Brügger, K., P. Redder, and M. Skovgaard. 2003. MUTAGEN: multi-user tool for annotating genomes. Bioinformatics 192480-2481. [DOI] [PubMed] [Google Scholar]
- 6.Eder, W., W. Ludwig, and R. Huber. 1999. Novel 16S rRNA gene sequences retrieved from highly saline brine sediments of Kebrit Deep, Red Sea. Arch. Microbiol. 172213-218. [DOI] [PubMed] [Google Scholar]
- 7.Edgar, R. C. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ginalski, K., A. Elofsson, D. Fischer, and L. Rychlewski. 2003. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics 191015-1018. [DOI] [PubMed] [Google Scholar]
- 9.Häring, M., X. Peng, K. Brügger, R. Rachel, K. O. Stetter, R. A. Garrett, and D. Prangishvili. 2004. Morphology and genome organization of the virus PSV of the hyperthermophilic archaeal genera Pyrobaculum and Thermoproteus: a novel virus family, the Globuloviridae. Virology 323233-242. [DOI] [PubMed] [Google Scholar]
- 10.Häring, M., G. Vestergaard, K. Brügger, R. Rachel, R. A. Garrett, and D. Prangishvili. 2005. Structure and genome organization of AFV2, a novel archaeal lipothrixvirus with unusual terminal and core structures. J. Bacteriol. 1873855-3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Häring, M., G. Vestergaard, R. Rachel, L. Chen, R. A. Garrett, and D. Prangishvili. 2005. Virology: independent virus development outside a host. Nature 4361101-1102. [DOI] [PubMed] [Google Scholar]
- 12.Kessler, A., A. B. Brinkman, J. van der Oost, and D. Prangishvili. 2004. Transcription of the rod-shaped viruses SIRV1 and SIRV2 of the hyperthermophilic archaeon Sulfolobus. J. Bacteriol. 1867745-7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kessler, A., G. Sezonov, J. I. Guijarro, N. Desnoues, T. Rose, M. Delepierre, S. D. Bell, and D. Prangishvili. 2006. A novel archaeal regulatory protein, Sta1, activates transcription from viral promoters. Nucleic Acids Res. 344837-4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227680-685. [DOI] [PubMed] [Google Scholar]
- 15.Letunic, I., R. R. Copley, B. Pils, S. Pinkert, J. Schultz, and P. Börk. 2006. SMART 5: domains in the context of genomes and networks. Nucleic Acids Res. 34D257-D260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lillestøl, R. K., P. Redder, R. A. Garrett, and K. Brügger. 2006. A putative viral defence mechanism in archaeal cells. Archaea 259-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makarova, K. S., N. V. Grishin, S. A. Shabalina, Y. I. Wolf, and E. V. Koonin. 2006. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mojica, F. J., C. Diez-Villasenor, J. Garcia-Martinez, and E. Soria. 2005. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 60174-182. [DOI] [PubMed] [Google Scholar]
- 19.Ortmann, A. C., B. Wiedenheft, T. Douglas, and M. Young. 2006. Hot crenarchaeal viruses reveal deep evolutionary connections. Nat. Rev. Microbiol. 4520-528. [DOI] [PubMed] [Google Scholar]
- 20.Peng, X., H. Blum, Q. She, S. Mallok, K. Brügger, R. A. Garrett, W. Zillig, and D. Prangishvili. 2001. Sequences and replication of genomes of the archaeal rudiviruses SIRV1 and SIRV2: relationships to the archaeal lipothrixvirus SIFV and some eukaryal viruses. Virology 291226-234. [DOI] [PubMed] [Google Scholar]
- 21.Peng, X., A. Kessler, H. Phan, R. A. Garrett, and D. Prangishvili. 2004. Multiple variants of the archaeal DNA rudivirus SIRV1 in a single host and a novel mechanism of genomic variation. Mol. Microbiol. 54366-375. [DOI] [PubMed] [Google Scholar]
- 22.Prangishvili, D., H. P. Arnold, D. Gotz, U. Ziese, I. Holz, J. K. Kristjansson, and W. Zillig. 1999. A novel virus family, the Rudiviridae: structure, virus-host interactions and genome variability of the Sulfolobus viruses SIRV1 and SIRV2. Genetics 1521387-1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prangishvili, D., P. Forterre, and R. A. Garrett. 2006. Viruses of the Archaea: a unifying view. Nat. Rev. Microbiol. 4837-848. [DOI] [PubMed] [Google Scholar]
- 24.Prangishvili, D., and R. A. Garrett. 2005. Viruses of hyperthermophilic Crenarchaea. Trends Microbiol. 13535-542. [DOI] [PubMed] [Google Scholar]
- 25.Prangishvili, D., R. A. Garrett, and E. V. Koonin. 2006. Evolutionary genomics of archaeal viruses: unique viral genomes in the third domain of life. Virus Res. 11752-67. [DOI] [PubMed] [Google Scholar]
- 26.Prangishvili, D., G. Vestergaard, M. Häring, R. Aramayo, T. Basta, R. Rachel, and R. A. Garrett. 2006. Structural and genomic properties of the hyperthermophilic archaeal virus ATV with an extracellular stage of the reproductive cycle. J. Mol. Biol. 3591203-1216. [DOI] [PubMed] [Google Scholar]
- 27.Rachel, R., M. Bettstetter, B. P. Hedlund, M. Häring, A. Kessler, K. O. Stetter, and D. Prangishvili. 2002. Remarkable morphological diversity of viruses and virus-like particles in hot terrestrial environments. Arch. Virol. 1472419-2429. [DOI] [PubMed] [Google Scholar]
- 28.Reilin, A. 1998. Preparation of catalase crystals. University of Illinois at Urbana-Champaign, Urbana, IL. http://www.itg.uiuc.edu/publications/techreports/98-009.
- 29.Rice, G., K. Stedman, J. Snyder, B. Wiedenheft, D. Willits, S. Brumfield, T. McDermott, and M. J. Young. 2001. Viruses from extreme thermal environments. Proc. Natl. Acad. Sci. USA 9813341-13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rutherford, K., J. Parkhill, J. Crook, T. Horsnell, P. Rice, M. A. Rajandream, and B. Barrell. 2000. ARTEMIS: sequence visualization and annotation. Bioinformatics 16944-945. [DOI] [PubMed] [Google Scholar]
- 31.Sæbø, P. E., S. M. Andersen, J. Myrseth, J. K. Lærdahl, and T. Rognes. 2005. PARALIGN: rapid and sensitive sequence similarity searches powered by parallel computing technology. Nucleic Acids Res. 33W535-W539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Snyder, J. C., B. Wiedenheft, M. Lavin, F. F. Roberto, J. Spuhler, A. C. Ortmann, T. Douglas, and M. Young. 2007. Virus movement maintains local virus population diversity. Proc. Natl. Acad. Sci. USA 10419102-19107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32a.Steinmetz, N. F., A Bize, K. C. Findlay, G. P. Lomonossoff, M. Manchester, D. J. Evans, and D. Prangishvili. Site-specific and spatially controlled addressability of a new viral nanobuilding block: Sulfolobus islandicus rod-shaped virus 2. Adv. Funct. Mat., in press.
- 33.Vestergaard, G., R. Aramayo, T. Basta, M. Häring, X. Peng, K. Brügger, L. Chen, R. Rachel, N. Boisset, R. A. Garrett, and D. Prangishvili. 2008. Structure of the Acidianus filamentous virus 3 and comparative genomics of related archaeal lipothrixviruses. J. Virol. 82371-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vestergaard, G., M. Häring, X. Peng, R. Rachel, R. A. Garrett, and D. Prangishvili. 2005. A novel rudivirus, ARV1, of the hyperthermophilic archaeal genus Acidianus. Virology 33683-92. [DOI] [PubMed] [Google Scholar]
- 35.Zillig, W., A. Kletzin, C. Schleper, I. Holz, D. Janekovic, H. Hain, M. Lanzendörfer, and J. K. Kristjansson. 1994. Screening for Sulfolobales, their plasmids and their viruses in Icelandic solfataras. System. Appl. Microbiol. 16609-628. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.