

Abstract
Despite its high coding capacity, murine CMV (mCMV) does not encode functional enzymes for nucleotide biosynthesis. It thus depends on cellular enzymes, such as ribonucleotide reductase (RNR) and thymidylate synthase (TS), to be supplied with deoxynucleoside triphosphates (dNTPs) for its DNA replication. Viral transactivation of these cellular genes in quiescent cells of host tissues is therefore a parameter of viral fitness relevant to pathogenicity. Previous work has shown that the IE1, but not the IE3, protein of mCMV transactivates RNR and TS gene promoters and has revealed an in vivo attenuation of the mutant virus mCMV-ΔIE1. It was attractive to propose the hypothesis that lack of transactivation by IE1 and a resulting deficiency in the supply of dNTPs are the reasons for growth attenuation. Here, we have tested this hypothesis with the mutant virus mCMV-IE1-Y165C expressing an IE1 protein that selectively fails to transactivate RNR and TS in quiescent cells upon transfection while maintaining the capacity to disperse repressive nuclear domains (ND10). Our results confirm in vivo attenuation of mCMV-ΔIE1, as indicated by a longer doubling time in host organs, whereas mCMV-IE1-Y165C replicated like mCMV-WT and the revertant virus mCMV-IE1-C165Y. Notably, the mutant virus transactivated RNR and TS upon infection of quiescent cells, thus indicating that IE1 is not the only viral transactivator involved. We conclude that transactivation of cellular genes of dNTP biosynthesis is ensured by redundancy and that attenuation of mCMV-ΔIE1 results from the loss of other critical functions of IE1, with its function in the dispersal of ND10 being a promising candidate.
Major immediate-early (MIE) genes of cytomegaloviruses (CMVs) are governed by strong cis-acting transcriptional enhancers and code for proteins that perform major regulatory functions in promoting the viral transcriptional program in acute infection, as well as in the initiation of virus reactivation from latency. Several recent articles have reviewed what is known today of CMV enhancers and their cognate MIE genes and proteins (4, 12, 51-53, 71, 80, 81, 85). IE1 proteins of both human CMV (hCMV) and murine CMV (mCMV), in addition to their key regulatory roles in viral-gene expression, play major roles in the antiviral immune response (5, 9, 11, 26, 37, 43, 60, 65, 67, 82; reviewed in references 34 and 64). As shown by Sylwester and colleagues (82), MIE genes of hCMV contribute to HLA polymorphic immunity to a degree above the prediction made by their genomic sequence coverage, thus indicating a nonstochastic mechanism favoring a high quantitative representation of MIE-specific T cells in memory T-cell pools. As a possible explanation for this phenomenon, work by Holtappels and colleagues (35), confirmed by later work of Karrer et al. (38), showed an expansion of IE1 epitope-specific effector memory CD8 T cells during viral latency, suggesting frequent restimulation and “memory inflation” due to episodes of latency-associated MIE gene expression (29, 45, 77). Recent work has provided evidence for mCMV MIE gene expression constituting an immunological checkpoint where IE1 epitope-specific CD8 T cells can sense and terminate viral reactivation at an early stage, long before the assembly of infectious virions (76; for reviews, see references 69 and 79).
In the specific case of the mCMV MIE locus, the MIE enhancer region (19), which actually might represent a tandem of two bona fide enhancers (15), is flanked on the left and right by the transcription units ie1 to ie3 (m123-M122) and ie2 (m128), respectively, so that these MIE genes are arranged head to head on opposite strands of the viral genomic DNA and are thus transcribed in opposite directions (40, 41, 54, 56; reviewed in reference 69). Such a gene arrangement is reminiscent of bidirectional gene pair organization (77), a gene architecture frequently used in mammalian genomes for genes involved in important regulatory functions, with DNA repair being an example (1, 87). Interestingly, the mouse thymidylate synthase (TS) gene, which is critically involved in deoxynucleoside triphosphate (dNTP) biosynthesis (see below), is controlled by a bidirectional promoter (49). Why mCMV has adopted this host genome-related gene organization is a matter of speculation, but it is intriguing that it is used right at the MIE locus, a locus of such outstanding regulatory relevance for the viral life cycle. Notably, in hCMV, another crucial locus shows bidirectional gene pair organization, namely, oriLyt (90). In mCMV, the leftward and rightward genes are expressed stochastically, that is, either the leftward gene is on and the rightward gene is off or vice versa (77). As pointed out by Stinski and Isomura (80), similarly to the MIE locus of mCMV, the MIE region of hCMV can also be viewed as a bidirectional architecture, with the significant, though poorly understood, difference that expression of the rightward gene, UL127, is repressed by the unique region that functions as an insulator between UL127 and the hCMV enhancer. Notably, a gene transcribed rightward relative to the MIE enhancer exists in the English strain of rat CMV (88).
MIE transcription of mCMV gives rise to three spliced mRNAs: the leftward transcript IE1 (coding exons 2, 3, and 4) specifies the 89-kDa IE1 phosphoprotein and its 76-kDa posttranslational-modification product (42); the leftward transcript IE3 (coding exons 2, 3, and 5) encodes the 88- to 90-kDa IE3 protein (54), which is homologous to the hCMV IE2 protein; and finally, the rightward transcript IE2 (coding exon 3) specifies the 43-kDa IE2 protein (56). Whereas IE3 has been identified as the essential transactivator of early (E)-phase genes (2, 54), IE2 is a nonessential, promiscuous transactivator of unknown significance (13, 56). The situation is more complicated with IE1, as this protein is nonessential for virus replication in various cell types in cell culture but is attenuated for growth in vivo (25). As summarized in recent review articles (12, 51, 52, 85), mCMV IE1 performs an accessory function in the transactivation of E-phase gene promoters and appears to play a key role in breaking epigenetic host cell defense by disrupting repressive nuclear domains (ND10), a function that involves the interaction of IE1 with constituents of ND10, including Daxx, PML, and histone deacetylase 2 (HDAC-2). Specifically, inhibition of HDAC-2 by mCMV IE1 can contribute to gene desilencing by favoring a hypoacetylated open viral chromatin structure.
A particularly intriguing function attributed to mCMV IE1 is the transactivation of cellular genes involved in dNTP biosynthesis, such as TS (28) and ribonucleotide reductase (RNR) (48) genes. As mCMV, as far as we know, does not encode functional enzymes for nucleotide metabolism (47), transactivation of these cellular genes may be crucial for efficient viral growth in the contact-inhibited resting cells that for the most part constitute host tissues. In fact, as shown by Gribaudo et al. and Lembo et al., mCMV replication and viral-DNA synthesis in quiescent fibroblasts can be inhibited by ZD1694 and by hydroxyurea, inhibitors of TS and RNR, respectively (28, 48), indicating that provision of dNTPs is indeed required. Thus, transactivation of dNTP biosynthesis is likely to be an important pathogenicity factor, and loss of this function in an IE1 deletion mutant may well explain in vivo growth attenuation contrasting with unaltered replication in dividing cells in cell culture. Here, we have identified a point mutation in the IE1 protein that selectively abrogates its function in the transactivation of TS and RNR while maintaining its function in the dispersal of ND10. This allows for the first time an experimental distinction between these two functions of IE1 in studies of viral replicative fitness in quiescent cells in cell culture, as well as in host tissues.
(This work was part of the M.Sc. thesis of Vanessa Wilhelmi, in the degree program “Biomedicine” of the Johannes Gutenberg-University, Mainz, Germany.)
MATERIALS AND METHODS
Construction of plasmids.
Recombinant plasmids were constructed according to established procedures, and enzyme reactions were performed as recommended by the manufacturers.
(i) Shuttle plasmid for mutagenesis.
Shuttle plasmid pST76KIE1Y165C, encompassing the IE1 protein-encoding sequence, is equivalent to pST76KIE1Leu* (76), except for a spontaneous point mutation, TAT → TGT, at position 181055 (63) in exon 4 of the ie1 gene, which results in the amino acid Cys in place of Tyr at amino acid position 165 of the IE1 protein. The revertant shuttle plasmid pST76KIE1C165Y was constructed as follows. A 1,471-bp ApaLI fragment carrying a point mutation of nucleotide A→T at the wobble position of the 176-Leu codon CTA (76) was generated by mutagenesis PCR with pST76KIE1Y165C as a template DNA and with primers rIE1-revert and Nona-Leu-f*, as well as Nona-Leu-r* and fIE1-revert (for primer sequences, see reference 76). The fusion reaction was performed with primers rIE1-revert and fIE1-revert. pST76KIE1Y165C was digested with ApaLI, and the 1,471-bp ApaLI fragment was replaced by the PCR-generated 1,471-bp ApaLI fragment. The fidelity of PCR-based cloning steps and cloning crossings was verified by sequencing.
(ii) Plasmids for dual-luciferase reporter (DLR) gene assays.
The recombinant plasmid pIE1-Y165C (pMut) was constructed as follows. Plasmid pST76KIE1Y165C was first cleaved with XbaI and Eco47III and then digested with BsmI to eliminate vector sequences. The resulting 1,809-bp fragment, encoding the mutated IE1 protein Y165C, was cloned into the XbaI- and Eco47III-digested plasmid pWT (identical to pIE100/1[54]). This plasmid contains a genomic fragment of mCMV that encodes the authentic IE1 protein. The recombinant plasmid pIE1-C165Y (pRev) was constructed in an analogous manner, except that pST76KIE1C165Y and pMut provided the insert and the backbone sequence, respectively. Plasmids pTLG (3) and pGL3R2 1.5 (14) contain the mouse TS and the mouse RNR 2 (R2) promoter, respectively, each linked to an intronless luciferase reporter gene. Plasmid pRL-TK (GenBank accession no. AF025846; Promega catalog no. E2241), encoding Renilla luciferase, was used to standardize for transfection efficacy.
Generation of recombinant viruses. (i) BAC mutagenesis.
Mutagenesis of the full-length mCMV genome, cloned as bacterial artificial chromosome (BAC) plasmid pSM3fr (89), was performed in Escherichia coli strain DH10B (Invitrogen) by using a two-step replacement method (7, 55, 58) with modifications described previously by Wagner et al. (89) and Borst et al. (8). The shuttle plasmid pST76KIE1Y165C was used to generate the BAC plasmid C3XIE1Y165C, which contains the mutated codon TGT corresponding to the amino acid point mutation Y165C in the IE1 protein sequence. To restore Tyr in position 165 of IE1, E. coli DH10B carrying the BAC plasmid C3XIE1Y165C was used for recombination with the shuttle plasmid pST76KIE1C165Y, encompassing the Tyr codon TAT.
(ii) Integrity and sequence analysis of recombinant mCMV BAC plasmids.
The physical integrity of the BAC plasmids was routinely verified by restriction enzyme fragment analysis as described in detail previously (76). The point mutations in the recombinant mCMV BAC plasmids C3XIE1Y165C and C3XIE1C165Y were verified by sequencing. Purified BAC DNA served as a template and was sequenced in both directions with primers IE1-ex4F1-IR and IE1-ex4R1-IR (76).
(iii) Viruses.
The reconstitution of viruses by transfection of BAC plasmid DNA, as well as the routine elimination of BAC vector sequences, was performed as described previously (24, 25, 76, 89). Only PCR-verified BAC vector-free virus clones were used to prepare high-titer stocks of the sucrose gradient-purified viruses (46, 62) mCMV-IE1-Y165C (7.1 × 108 PFU/ml) and mCMV-IE1-C165Y (5 × 107 PFU/ml). Other viruses used in this study were BAC-derived mCMV-WT.BAC MW97.01 (89) and the ie1 gene deletion mutant mCMV-Δie1 (25), referred to here as mCMV-ΔIE1. Inactivated virions of mCMV-WT.BAC were generated by exposure to high-intensity radiation in the UVC range (low-heat UV sterilizer UVC-CRL 400; Umwelt und Technik GmbH, Horb, Germany) for 5 min. Irradiation was performed just prior to use, and the inactivated virion preparations were kept permanently on ice. Inactivation was verified by the absence of intranuclear IE1 protein expression in otherwise highly permissive mouse embryo fibroblasts (MEF) inoculated with a dose that was equivalent to a multiplicity of infection (MOI) of 4 (see below).
Cell culture conditions, growth arrest, and infection of cells.
NIH 3T3 murine fibroblasts were grown as monolayers in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% newborn calf serum (PAA Laboratories, Cölbe, Germany). BALB/c MEF were propagated similarly in minimal essential medium (MEM) (Invitrogen) supplemented with 10% fetal calf serum (FCS) (PAA Laboratories). Using the method described in detail by Gribaudo et al. and Lembo et al. (28, 48), growth arrest of NIH 3T3 cells and of MEF was achieved by three wash steps with low-serum starvation medium (0.5% newborn calf serum and 0.5% FCS, respectively), followed by incubation in this starvation medium for exactly 48 h. Successful growth arrest was confirmed by immunofluorescence analysis specific for Ki-67 antigen (see below). Second-passage MEF (regularly cultured or growth arrested) were usually infected under conditions of centrifugal enhancement of infectivity with 0.2 PFU of purified virions per cell, which corresponds to an MOI of 4 (62). To optimize presentation of the IE1 peptide for the enzyme-linked immunospot (ELISPOT) assay (see below), viral-gene expression was arrested in the IE phase and MIE gene expression was enhanced by infection in the presence of cycloheximide (50 μg/ml) for 3 h, which was then replaced by actinomycin D (5 μg/ml) for 2 h (62). For multistep viral growth curves, quiescent MEF in six-well culture dishes were centrifugally infected with 0.02 PFU per cell, which corresponds to an MOI of 0.4. Virus titers in the cell culture supernatants were determined by a virus plaque assay in MEF indicator cultures under conditions of centrifugal enhancement of infectivity (62).
Assay for antigenic peptide presentation.
The ELISPOT assay was used to detect the presentation of endogenously processed antigenic IE1 peptide 168-YPHFMPTNL-176 in infected cells on the basis of gamma interferon secretion by sensitized cytolytic T lymphocytes (CTL) of an IE1 epitope-specific cell line (IE1-CTLL) characterized previously (5, 60). The peptide-presenting stimulator cells were BALB/c (H-2d) MEF infected with the respective recombinant viruses under conditions of selective and enhanced expression of IE-phase proteins (see above). The ELISPOT assay was performed essentially as described previously (references 37 and 60 and references therein), with 105 stimulator cells per assay culture and with 100 IE1-CTL effector cells seeded in triplicate.
Immunofluorescence analysis of cell quiescence.
MEF (see Fig. 5A) or NIH 3T3 cells (not shown) from confluent monolayers were trypsinized and seeded onto acetone-cleaned glass coverslips in 24-well plates at a density of ∼4 × 104 cells per coverslip in MEM-10% FCS. After an initial 24-h period of incubation, allowing adherence and growth, the cells were growth arrested for 48 h as described above. In parallel, cells intended to be used as positive controls were refed with MEM-10% FCS. Thereafter, the cells were washed twice with phosphate-buffered saline (PBS) and fixed in chilled (−20°C) 70% methanol for 90 min at 4°C. The fixed cells were stored in PBS at 4°C and were incubated prior to use with 50 μl of blocking buffer (PBS supplemented with 0.3% Triton X-100 and 15% FCS) for 60 min at room temperature. The coverslips were then incubated overnight in a humid chamber with 50 μl of blocking buffer containing the primary antibody, namely, rabbit anti-Ki-67 monoclonal antibody (MAb) (1:200; catalog no. KI681C01; Innovative Diagnostik-Systeme, Hamburg, Germany). After being washed with PBS, the coverslips were incubated for 1 h with the secondary antibody Alexa Fluor 488 (green fluorescence)-conjugated goat anti-rabbit immunoglobulin G (heavy plus light chains; catalog no. A11008; Invitrogen), as well as with Texas red-conjugated phalloidin (catalog no. T7471; Invitrogen), both diluted in blocking buffer (1:200). The incubations for staining and all subsequent steps were performed in the dark. The coverslips were washed five times in PBS before they were mounted. Fluorescence images were acquired using a Zeiss LSM-510 laser scanning microscope and Zeiss software.
FIG. 5.
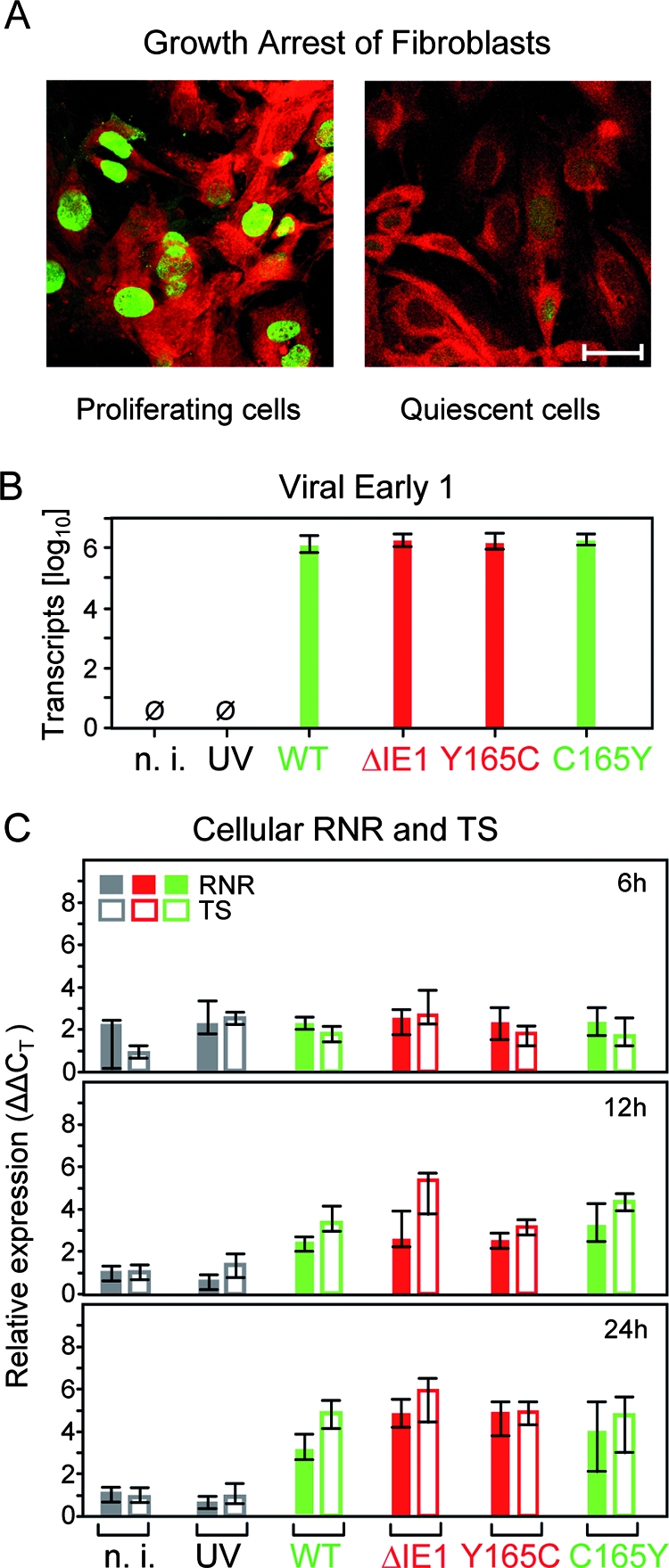
Transactivation of cellular genes of dNTP synthesis during infection of quiescent fibroblasts in cell culture. (A) Verification of growth arrest. MEF were cultured for 48 h either in regular cell culture medium (left) or in low-serum starvation medium (right), followed by immunofluorescence staining of the proliferation-associated nuclear antigen Ki-67 (Alexa Fluor 488 label; green fluorescence) and of F-actin (Texas red-conjugated phalloidin; red fluorescence). Shown are CLSM images of representative areas. The bar marker represents 20 μm. (B) Control for equivalence of infection. Growth-arrested MEF (see above) were infected for 6 h at an MOI of 4 with the viruses indicated, and E1 transcripts representing the E phase of viral-gene expression were determined by real-time RT-PCR. The data refer to 50 ng of total RNA, corresponding to ∼5,000 cells. n. i., uninfected MEF; UV, MEF exposed to UV light-inactivated mCMV-WT.BACUV at a dose corresponding to an MOI of 4; WT, MEF infected with mCMV-WT.BAC. The bars represent the median values of triplicate cultures. The error bars indicate the range. Green and red labeling of the bars highlights the absence and presence of mutations, respectively. (C) Relative quantitation of RNR and TS transcripts by real-time RT-PCR in MEF infected for 6 h, 12 h, and 24 h with the viruses indicated. Shown are expression levels (ΔΔCT values; see Materials and Methods) relative to GAPDH transcripts. The bars (filled bars, RNR; open bars, TS) represent the median values of triplicate cultures; the error bars indicate the range. Green and red labeling of the bars highlights the absence and presence of mutations, respectively.
Transient transfections and reporter gene assays.
Transient transfections and reporter gene assays in resting cells were performed as described in detail previously (76). In essence, the DLR assay (catalog no. E1910; Promega) was employed to standardize the transfection efficacy. NIH 3T3 cells were cotransfected with the Renilla luciferase-encoding standardization plasmid pRL-TK, a firefly luciferase-encoding reporter plasmid (pTLG or pGL3R2), and a transactivator donor plasmid encoding authentic (pWT or pRev) or mutated (pMut) IE1 protein. The vector plasmid pUC19 served as a negative control. Transfection was performed with PolyFect transfection reagent (catalog no. 301107; Qiagen, Hilden, Germany). The transactivating effect of the authentic and mutated IE1 proteins on the R2 (48) and TS (28) gene promoters was tested in growth-arrested NIH 3T3 cells. Luciferase activity was measured by the DLR assay using 20-μl samples of cleared cell lysate. Luminescence, expressed as relative luminescence units (RLU), was measured with a single-sample luminometer (Lumat LB 9507; Berthold, Bad Wildungen, Germany).
Detection of nuclear domains (ND10).
Detection and quantitation of promyelocytic leukemia (PML) bodies (ND10) by confocal laser scanning microscope (CLSM) analysis was performed essentially as described in detail previously (76). In brief, MEF grown on glass coverslips in 24-well plates at a density of ∼8 × 104 cells per coverslip were centrifugally infected with mCMV at an MOI of 4. After 4 h of incubation, the infected MEF were washed, fixed, and incubated in blocking buffer for 30 min at room temperature. For the double-immunofluorescence studies, each coverslip was incubated overnight in a humidity chamber with blocking buffer containing the primary antibodies, namely, affinity-purified rabbit anti-PML polyclonal antibody H-238 (1:200; catalog no. sc-5621; Santa Cruz Biotechnology, Inc.) combined with either mouse anti-IE1 MAb (Croma 101; 1:200) or with mouse anti-E1 MAb (Croma 103; 1:100). After being washed, the coverslips were incubated with the appropriate secondary antibody diluted in blocking buffer. The secondary antibodies used were an Alexa Fluor 488-conjugated goat anti-mouse immunoglobulin G (H+L) antibody (catalog no. A11001; Invitrogen) for staining of IE1 or E1 and an Alexa Fluor 546-conjugated goat anti-rabbit antibody (catalog no. A11010; Invitrogen) for staining of PML bodies. The coverslips were washed and stored at 4°C in the dark until the measurements were performed. Throughout, immunofluorescence analyses were followed by blue fluorescence staining of the cell nuclei for 5 min at room temperature with 4′-6-diamidino-2-phenylindole (Hoechst 33342 dissolved in PBS; Invitrogen). Images were acquired by using a Zeiss LSM-510 laser scanning microscope and Zeiss software. For the counting of intranuclear PML bodies and exclusion of signals from extranuclear PML protein, three-dimensional scanning of nuclei was performed with a 100-fold magnification lens and a Z-step size of 0.3 μm.
Western blot analysis of viral-protein expression.
The expression of authentic and mutated IE1 proteins, as well as of the E-phase protein m164/gp36.5 (Torsten Däubner, unpublished data), in infected MEF was analyzed by Western blotting as described in detail previously (36). In essence, lysates of infected MEF were prepared, and 30-μg aliquots of total protein were subjected to separation by 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by Western blotting onto polyvinylidene difluoride membranes and staining of the proteins of interest with the respective antibodies. Specifically, MAb Croma 101 and polyclonal affinity-purified rabbit antibodies directed against a C-terminal peptide (33) were used for the detection of the proteins IE1 (pp89/76) and m164 (gp36.5), respectively. Antibody binding was visualized by enhanced chemiluminescence using the ECLplus Western blotting detection system (catalog no. RPN2132; Amersham Biosciences, Little Chalfont, United Kingdom) and Lumi-Film (catalog no. 11666657001; Roche Applied Sciences, Mannheim, Germany). Comparable protein loading of the polyvinylidene difluoride membranes was verified by staining them with Coomassie blue G-250 (catalog no. 9598.1; Roth, Karlsruhe, Germany).
Experimental mCMV infection of mice.
BALB/cJ (H-2d haplotype) mice were immunodepleted and infected essentially as described in detail previously (62). In brief, hematoablative conditioning of 8- to 9-week-old female mice was achieved by total-body gamma irradiation with a single dose of 6.5 Gy. Intraplantar infection at the left hind footpad was performed ∼2 h later with 105 PFU of the viruses under test. The mice were bred and housed under specific-pathogen-free conditions in the Central Laboratory Animal Facility of the Johannes Gutenberg-University. Animal experiments were approved according to German federal law under permission number 177-07-04/051-62.
Preparation of hepatocyte and NPLC suspensions.
Hepatocytes and nonparenchymal liver cells (NPLCs) were isolated from mouse livers by a modified two-step perfusion method as described previously (73, 74). A 22G permanent venous catheter was inserted in the portal vein. Thereafter, the posterior vena cava was opened to allow an outflow of perfusion buffers. The liver was perfused first with 100 ml of calcium-free buffer solution (142 mM NaCl, 6.7 mM KCl, 10 mM HEPES, pH 7.4), followed by a second perfusion step with 50 ml collagenase buffer (66.7 mM NaCl, 6.7 mM KCl, 4.76 mM CaCl2·2H2O, 100 mM HEPES, 0.05% collagenase A [Roche], pH 7.6). The flow rates for both buffers were adjusted to 20 ml/min. After perfusion, the livers were excised, mechanically dissected in washing buffer (142 mM NaCl, 6.7 mM KCl, 1.2 mM CaCl2·2H2O, 10 mM HEPES), filtered over a 100-μm cell strainer, and centrifuged gently for 5 min at only 40 × g (with the centrifuge brake turned off) to pelletize the hepatocytes. The supernatant was used for preparation of NPLCs. The hepatocytes and NPLCs were resuspended in washing buffer and centrifuged for 5 min at 40 × g (with the brake off) and for 10 min at 400 × g (with the brake on), respectively. Each sample was resuspended in a total of 10 ml PBS for cell counting. For isolation of total RNA from hepatocytes and NPLCs, the cells were pelletized by centrifugation for 5 min at 400 × g and resuspended in an appropriate amount of buffer RLT (Qiagen).
IE1- and MCP-specific IHC analysis of liver tissue infection.
Samples of livers were embedded in paraffin and processed for the preparation of 2-μm tissue sections and subsequent detection of viral proteins in infected tissue cells according to standard histological procedures (62). Immunohistochemical (IHC) detection of intranuclear IE1 protein pp76/89 by black staining using the peroxidase-diaminobenzidine-nickel method was performed as described in detail previously (30, 62). For the detection of major capsid protein (MCP) (M86), heat-induced epitope retrieval was achieved by incubation in 10 mM trisodium-citrate-dihydrate buffer (pH 6) for 5 min in a microwave oven at 95°C. Specific labeling of MCP was performed by using a modified alkaline phosphatase anti-alkaline phosphatase method with custom-made (Peptide Specialty Laboratories GmbH, Heidelberg, Germany) polyclonal rabbit antibodies directed against both N-terminally His- and Arg-tagged synthetic MCP peptides His-329-ADRIQQSSQKDLPESQFLDQRL-351 and Arg-1171-YPNNPRGRSSSMLGVDPYDEDA-1193, respectively, predicted by antigenicity algorithms. The peptides were coupled to N-terminal cysteine-to-maleimide-activated keyhole limpet hemocyanin (Pierce Biotechnology, Rockford, IL), and a mixture of the resulting conjugates was used to immunize rabbits. Antipeptide antibodies were purified according to the method of Harlow and Lane (31) by protein A affinity chromatography, followed by affinity chromatography on columns with the immunizing peptides coupled to Sulfo-link coupling gel (Pierce Biotechnology). The liver tissue sections were overlaid with goat serum (diluted 1:10 in Tris-buffered saline [TBS]) for 20 min at ∼22°C to block nonspecific antibody-binding sites. Specific labeling was achieved by incubating the samples for 18 h at ∼22°C with the MCP-specific antibodies (see above) diluted 1:2,000 in TBS. Sections were then incubated for another 20 min at ∼22°C with the “bridging antibody” alkaline phosphatase-conjugated mouse anti-rabbit antibody (catalog no. A2306; Sigma) diluted 1:25 in TBS. Finally, alkaline phosphatase rabbit anti-alkaline phosphatase soluble complex (catalog no. A9811; Sigma) was added in a 1:10 dilution in TBS, followed by a further incubation for 30 min at ∼22°C prior to red staining by using the DakoCytomation Fuchsin Substrate-Chromogen kit (catalog no. K0624; Dako, Hamburg, Germany). Light-blue counterstaining was achieved with hematoxylin added for just a few seconds.
Quantification of viral genomes in infected cells and host tissues. (i) Isolation of DNA from infected MEF.
DNA was extracted with the DNeasy Blood and Tissue kit (catalog no. 69506; Qiagen) as described in the manufacturer's spin column protocol (62a) from step 1c onward. In brief, the cells were trypsinized, collected with a cell scraper, sedimented by centrifugation, and frozen at −70°C. For DNA extraction, the samples were lysed by proteinase K digestion, followed by DNA binding to the DNeasy Mini spin column, washing steps, and elution with 200 μl of elution buffer, yielding 1 to 2 μg of DNA per 1 × 106 cells.
(ii) Isolation of DNA from infected host tissues.
Lungs (∼190 to 210 mg in total) and spleen (∼100 to 160 mg in total) were homogenized in an appropriate amount of buffer ATL (catalog no. 19076; Qiagen) (in 2.34 ml and 1.45 ml, respectively) by using TissueRuptor (catalog no. 9001272; Qiagen) for at least 30 s at full speed. DNA was extracted from 200-μl aliquots of lysates with the DNeasy Blood and Tissue kit (Qiagen).
(iii) M55/gB-specific real-time PCR.
Viral genomes were quantified by real-time PCR specific for the viral gene M55/gB normalized to 106 cells on the basis of real-time PCR specific for the diploid cellular gene pthrp essentially as described previously (72, 76, 78), using the QuantiTect Sybr green PCR kit (catalog no. 204243; Qiagen). In brief, 100-ng aliquots of the DNA served as template DNA to amplify a 135-bp fragment of M55/gB with primers LCgB-forw and LCgB-rev, as well as a 142-bp fragment of the cellular pthrp gene with primers LCPTHrP-forw and LCPTHrP-rev. Standard curves for quantification were established by using graded numbers of linearized plasmid pDrive_gB_PTHrP_Tdy as a template (78).
Isolation of RNA from infected cells and host tissues. (i) Isolation of total RNA from quiescent MEF.
RNA was isolated by using the RNeasy Micro kit (catalog no. 74004; Qiagen) as described in the manufacturer's animal cell protocol (62b) from step 2 onward. MEF were lysed directly in the cell culture vessel with buffer RLT and collected with a cell scraper. The lysate was homogenized by using Qiashredder spin columns (catalog no. 79656; Qiagen). RNA was bound to the RNeasy MinElute spin column, followed by digestion of contaminating DNA with DNase using an RNase-Free DNase set (catalog. no. 79254; Qiagen). After the reaction was stopped and after the washing steps, the bound RNA was eluted with 14 μl of elution buffer, yielding 1 to 2 μg of total RNA per 1 × 105 cells. Samples were stored at −70°C.
(ii) Isolation of total RNA from liver tissue.
Livers were shock frozen in liquid nitrogen and were then transferred without delay into a Falcon tube (50 ml) containing 15 ml of lysis buffer RLT (catalog no. 79216; Qiagen) per whole liver (∼1 g). The tissue was homogenized with the TissueRuptor (Qiagen) for 30 s at full speed. Total RNA was isolated from the lysates using the RNeasy Mini kit (catalog no. 74106; Qiagen) as described in the manufacturer's animal tissue protocol (62c). For this, only 600 μl of the whole liver lysate (i.e., of ∼15 ml) was used. In brief, RNA was bound to the RNeasy spin column, followed by digestion of contaminating DNA with DNase using an RNase-Free DNase set (Qiagen). After the reaction was stopped and after the washing steps, bound RNA was eluted in two steps with 30 μl elution buffer each, yielding 30 to 60 μg of total RNA in 60 μl. The collected samples were stored at −70°C.
Quantification of transcripts.
Real-time reverse transcription (RT)-PCR specific for the spliced transcript of the viral E-phase gene e1 (M112-M113), resulting in a 200-bp amplificate, was performed as described previously by using E1 in vitro transcripts in the standard titration for an absolute quantification (76). Relative quantification of cellular transcripts from genes encoding RNR and TS was performed by real-time RT-PCRs with the appropriate primers and probes (see Table 2). The transcript specifying GAPDH (glyceraldehyde-3-phosphate dehydrogenase) served as the reference transcript.
TABLE 2.
Primers and probes used in RT-PCRs specific for transcripts of cellular genes involved in dNTP synthesis
| Primer or probe | Description
|
|
|---|---|---|
| RNR gene | TS gene | |
| Primer | ||
| Forward | 5′ 3900 to 3917 3′; exon 7 (accession no. X15666); TCAAGAAACGGGGGCTGA | 5′ 582 to 600 3′; exon 1 (accession no. J02617); CAAGAAGGAGGACCGCACG |
| Reverse | 5′ 4847 to 4870 3′; exon 8 (accession no. X15666); CTTGTGTACCAGGTGCTTGAACAT | 5′ 63 to 49 3′, exon 2 (accession no. M13347), and 5′ 650 to 640 3′, exon 1 (accession no. J02617); AGAGGAAATTCATCTCTCAGGCTGTA |
| Probe | 5′ 4838 to 4820 3′, exon 8, and 5′ 3961 to 3944 3′, exon 7 (accession no. X15666); CAAAGTCACAGTGTAAACCCTCGTCTCTGCTAATAAG | 5′ 636 to 617 3′; exon 1 (accession no. J02617); TGCCTGCATGCCGAACACCG |
(i) RNR transcripts.
For RNR-specific RT-PCR, probe RNR-M2-taq1 directed against the exon 7/8 splicing junction comprised nucleotides 5′ 4,838 to 4,820 on exon 8 and 5′ 3,961 to 3,944 on exon 7 (GenBank accession no. X15666). Oligonucleotide 5′ 3,900 to 3,917 served as the forward primer RNR_taq_forw1, and oligonucleotide 5′ 4,870 to 4,847 served as the reverse primer RNR_taq_rev1, yielding a 113-bp amplification product.
(ii) TS transcripts.
For TS-specific RT-PCR, probe TS_taq1 directed against exon 1 comprised nucleotides 5′ 636 to 617. Oligonucleotide 5′ 582 to 600 served as the forward primer TS_taq_for1, and oligonucleotides 5′ 63 to 49 on exon 2 (GenBank accession no. M13347) and 5′ 650-640 on exon 1 (GenBank accession no. J02617) served as the reverse primer TS_taq_rev1, yielding an 84-bp amplification product.
(iii) GAPDH transcripts.
For GAPDH-specific RT-PCR (59), probe GAPDH_Taq_RC comprised nucleotides 5′ 491 to 516. Oligonucleotide GAPDH_for served as the forward primer, comprising nucleotides 5′ 348 to 367. Oligonucleotide GAPDH_rev served as the reverse primer, comprising nucleotides 5′ 584 to 565 (GenBank accession no. NM_008084). This resulted in a 237-bp amplification product.
(iv) β-Actin transcripts.
For β-actin-specific RT-PCR (59), probe BetaActin_Taq_Probe1 comprised nucleotides 5′ 833 to 852 on exon 4 (GenBank accession no. NM_007393). Oligonucleotide 5′ 809 to 830 on exon 4 served as the forward primer BetaActin_Taq_for1, and oligonucleotide 5′ 896 to 873 directed against the exon 4/5 splicing junction served as the reverse primer BetaActin_Taq_rev1, yielding a 88-bp amplification product.
Reactions were performed in a total volume of 25 μl, containing 5 μl of 5× Qiagen OneStep RT-PCR buffer, 1 μl of Qiagen OneStep RT-PCR enzyme mixture, 668 μM of each dNTP, 0.6 μM of each primer, 0.26 μM probe, 1.5 mM additional MgCl2, and 0.132 μM ROX (5-carboxy-X-rhodamine) as a passive reference. Reverse transcription was performed at 50°C for 30 min. The cycle protocol for cDNA amplification started with an activation step at 95°C for 15 min, followed by 45 cycles of denaturation for 15 s at 94°C and a combined primer-annealing/extension step for 1 min at 58°C. The efficiencies of the RT-PCRs were >90% throughout. Relative quantifications were made with the comparative CT (cycle threshold) method as described by the manufacturer (Applied Biosystems) (2a). This method uses arithmetic formulas, where the target is first normalized to an endogenous reference (ΔCT = CT target − CT reference)—here GAPDH or β-actin—and then related to a calibrator (ΔΔCT = ΔCT − ΔCT calibrator sample), that is, samples from uninfected cells, for calculation of the relative amount of target according to the formula 2−ΔΔCT (50).
Mathematical calculations and statistical analysis. (i) Calculation of DTs.
Linear regression lines in a semilogarithmic plot [log N(t) = at + log N(0), where N(t) is the experimentally determined number of infected cells or of viral genomes at time t after infection, a is the slope of the regression line, and log N(0) its ordinate intercept] were calculated by using Mathematica Statistics LinearRegression software, version 5.1 (Wolfram Research, Inc., Champaign, IL). The doubling time (DT) of the number of infected cells was then calculated according to the formula log 2/a. Accordingly, the upper and lower 95% confidence limit values of slope a (determined from the ellipsoidal parameter confidence region) gave the 95% confidence interval of the DT.
(ii) Bioinformatics prediction of the proteasomal cleavage probability.
Scores for the probability of N-terminal proteasomal cleavage of IE1 after amino acid position 165 (position −3 relative to the N-terminal amino acid Tyr +1 of the immunodominant antigenic IE1 peptide) were calculated by using the software MHC-Pathway (86) and NetChop 3.0 (57) provided at the Web sites http://www.mhc-pathway.net and http://www.cbs.dtu.dk/services/NetChop/, respectively.
(iii) Significance analysis.
Two independent sets of data were compared by using distribution-free Wilcoxon-Mann-Whitney (rank sum) statistics. A calculator is provided on the website http://www.socr.ucla.edu/Applets.dir/WilcoxonRankSumTable.html (Ivo Dinov, Statistics Online Computational Resources, University of California—Los Angeles Statistics, Los Angeles, CA). Differences were regarded as marginally significant if the P value (two-tailed test) was <0.05 and as highly significant if the P value was <0.001.
RESULTS
Spontaneous mutation at the proteasomal cleavage site of the immunodominant IE1 peptide.
In previous work (76), we described a mutant virus in which the antigenicity of the IE1 peptide 168-YPHFMPTNL-176 of mCMV (64, 68) was selectively wiped out by point mutation L176A at the C-terminal anchor position that is critical for peptide binding to the peptide-presenting major histocompatibility complex (MHC) class I glycoprotein, to glycoprotein Ld in the specific case (5, 66, 76). In the process of generating revertant viruses for control, the Ala codon GCA was mutated back to the Leu codons CTA and CTT, thus either restoring the authentic sequence or leaving a single-nucleotide polymorphism in the wobble position as a molecular marker in viruses mCMV-IE1-A176L and mCMV-IE1-A176L*, respectively (76). During routine sequencing of the constructs to control mutational fidelity, we noticed in the wobble nucleotide-marked construct a spontaneous codon mutation, TGT, in place of TAT, leading to an amino acid point mutation, Y165C, replacing Tyr with Cys in position −3 relative to the N-terminal position +1 of the antigenic IE1 peptide (Fig. 1A). This example was an alert, showing that a mutation may occur in a revertant virus that is supposed to serve as a control for the mutant virus of interest.
FIG. 1.

Identification of the mutation Y165C at the N-terminal proteasomal cleavage site of the antigenic IE1 peptide precursor. (A) Sequencing of the IE1 peptide-coding region of BAC plasmids. Sequencing was originally undertaken with the intention to verify codon CTT specifying Leu at the C-terminal MHC class I anchor position of the antigenic IE1 peptide in single-nucleotide-tagged IE1-A176L* revertants of the epitope deletion mutant IE1-L176A (76). This revealed an unintended surplus mutation of codon TAT into codon TGT, replacing Tyr with Cys at amino acid position 165 of the IE1 protein, which is position −3 relative to the N terminus of the antigenic IE1 peptide 168-YPHFMPTNL-176. (Top) HindIII physical map of the mCMV-WT.Smith genome (20, 63) with the MIE region shown expanded. Exons (Ex) are numbered. MIEPE, MIE promoter and enhancer. (Bottom) Sequencing of the region ranging from nucleotide (n)181,067 to n181,020 within exon 4/m123 on the complementary strand (C strand), drawn in twisted orientation. Signals from nucleotides A, T, G, and C are shown in green, red, black, and blue, respectively. Mutated amino acid positions and viruses are highlighted by red lettering, and WT and revertant amino acid positions and viruses are highlighted by green lettering. Amino acids within the region of interest (position −3 to position 9 relative to the N terminus of the antigenic IE1 peptide) are given in one-letter code. (B) Presentation of the naturally processed IE1 peptide on MEF infected under conditions of selective and enhanced IE gene expression (MOI, 4; cycloheximide was replaced after 3 h by actinomycin D) with the viruses indicated. n.i., uninfected MEF. Shown are data from a gamma interferon secretion-based ELISPOT assay using IE1 epitope-specific CTL as effector cells and the IE-phase-arrested infected MEF as stimulator cells. The filled circles represent data (spot counts per 100 IE1-CTL seeded) of triplicate assay cultures, and the bars represent the median values. Green labeling and red labeling indicate absence and presence of the mutation, respectively. P values (two-sided), determined by the distribution-free (nonparametric) Wilcoxon-Mann-Whitney rank sum test, are indicated.
Although this surplus mutation was unintended, leading to a faulty virus, we became interested in it, as it was located precisely at the N-terminal proteasomal cleavage position for generating the antigenic IE1 peptide. As shown in previous work by Knuehl et al. (44) with an IE1 epitope-encompassing 25-mer substrate in an in vitro proteasome assay, both the constitutive proteasome and the immunoproteasome generate an 11-mer IE1 precursor peptide, 166-DMYPHFMPTNL-176, that is transported into the endoplasmic reticulum (ER), where N-terminal trimming leads to the mature IE1 peptide for MHC class I loading and transport of the complex to the cell surface (for a review, see reference 64).
We first employed the current bioinformatics algorithms MHC-Pathway (86) and NetChop 3.0 (57) to predict a possible influence of the mutation Y165C on the probability of proteasomal cleavage after amino acid position 165, also including theoretical mutations Y165A and Y165F in these calculations (Table 1). Both algorithms predicted cleavage to occur for all four amino acid 165X analogs (X = Y, C, A, or F) considered but also concurred in the prediction that the probability was lowest for cleavage after amino acid 165C. According to MHC-Pathway, this applied to both the constitutive proteasome and the immunoproteasome. To test experimentally if the mutation Y165C prevented or significantly reduced the proteasomal processing of the IE1 peptide, we used a polyclonal but IE1 epitope-monospecific CD8 T-cell line (5, 60) to detect Ld-presented IE1 peptide on MEF infected under conditions of IE-phase arrest with the mutant virus mCMV-IE1-Y165C, with the revertant virus mCMV-IE1-C165Y (which actually corresponds to the previously published virus mCMV-IE1-A176L* [76]), and with mCMV-WT.BAC (Fig. 1B). Whereas the numbers of responding cells were the same for stimulator cells infected with wild-type (WT) virus and the revertant virus (P = 0.7), it was slightly reduced, with marginal statistical significance (P = 0.048), after infection of stimulator cells with the mutant virus. Thus, in agreement with the bioinformatics predictions of cleavage probabilities, the mutation Y165C did not abolish IE1 peptide processing and presentation but caused some reduction.
TABLE 1.
Predictions of proteasomal cleavage
| Sequencea | Score
|
||
|---|---|---|---|
| MHC-Pathwayb
|
NetChop 3.0c | ||
| Constitutive proteasome | Immunoproteasome | ||
| GRLMY▾DMYPHFMPTNL | 1.04 | 1.43 | 0.9656 |
| GRLMC▾DMYPHFMPTNL | 0.94 | 1.14 | 0.8949 |
| GRLMA▾DMYPHFMPTNL | 1.01 | 1.2 | 0.9242 |
| GRLMF▾DMYPHFMPTNL | 0.98 | 1.41 | 0.9663 |
▾, cleavage site.
MHC-Pathway (http://www.mhc-pathway.net).
NetChop 3.0 Server (http://www.cbs.dtu.dk/services/NetChop/).
The mutation Y165C destroys the capacity of IE1 to transactivate cellular RNR and TS genes in quiescent fibroblasts.
As the IE1 protein, besides its role as an antigen, performs such an important regulatory function (see the introduction), our routine control program also included functional assessments, such as of the transactivation of cellular genes of dNTP synthesis. The respective DLR gene assays were performed upon transient cotransfection of growth-arrested NIH 3T3 fibroblasts with the appropriate donor and reporter plasmids, as well as with the Renilla luciferase-encoding control plasmid pRL-TK for standardization (Fig. 2). We also controlled for equivalent expression of IE1 protein from the donor plasmids pWT, pMut, and pRev (data not shown). To our surprise, the mutation Y165C, that is, the codon mutation TGT in place of TAT, in the donor plasmid pMut completely failed to transactivate RNR (Fig. 2A) (P < 0.001) and TS (Fig. 2B) (P < 0.001). The finding that the back mutation C165Y, that is, the reversion to codon TAT in the expression plasmid pRev, restored the transactivation of both cellular reporter genes to the respective levels found with expression plasmid pWT (P = 0.050 and P = 0.436 for RNR and TS, respectively) provided reasonable evidence to conclude that the mutation Y165C is a sufficient cause for the loss of transactivator function.
FIG. 2.

Impact of the mutation Y165C on the transactivation of cellular genes involved in dNTP biosynthesis. (A) Transactivation of the RNR promoter PR2. (B) Transactivation of the TS promoter PTS. DLR assays were performed with growth-arrested NIH 3T3 cells transiently cotransfected with an IE1-encoding donor plasmid, the respective firefly luciferase-encoding reporter plasmid, and the Renilla luciferase-encoding plasmid pRL-TK for standardization of the transfection efficacy. Vector pUC19 was used as an empty donor plasmid for control. Plasmid maps (not drawn to scale) are illustrated. The donor plasmids were pIE100/1 (here referred to as pWT), specifying the authentic IE1 protein; pMut, specifying the mutated protein IE1-Y165C; and pRev, specifying the rescued protein IE1-C165Y. MIEPE, MIE promoter and enhancer; Ex, exon. The arrows indicate the direction of transcription; the arrowheads mark the location of the mutation in exon 4. Correct expression of the IE1 protein was inspected for all donor plasmids by IE1-specific immunofluorescence (not depicted). Luminescence data are expressed as RLU and are normalized for transfection efficacy by forming the quotient of firefly luciferase activity (RLUFL) and Renilla luciferase activity (RLURL). The solid circles and gray-shaded bars represent data from triplicate transfection cultures and their median value, respectively, for each experiment, with a total of three independent experiments performed for each type of donor plasmid tested. P values (two-sided), determined by the distribution-free (nonparametric) Wilcoxon-Mann-Whitney rank sum test (n1 = n2 = 9), are indicated.
The mutation Y165C does not affect the capacity of IE1 to disrupt repressive nuclear domains ND10.
The mutation Y165C could possibly have interfered with all regulatory functions of IE1, for instance, due to a global protein refolding leading to an essentially different structure. This would have been like a deletion and thus not particularly attractive with regard to learning more about IE1 function. We therefore asked if other known functions of IE1 were also affected by the Y165C mutation (Fig. 3). As discussed above (see the introduction), IE1's capacity to disrupt repressive ND10 structures is a feature that is likely important for viral replicative fitness. In accordance with previous work by other groups (25, 84), as well as by our own group (76), CLSM analysis of infected MEF revealed a disruption of ND10 structures by mCMV-WT.BAC expressing the authentic IE1 protein and a failure in disrupting ND10 by the IE1 deletion mutant mCMV-ΔIE1 (Fig. 3A and B), indicating that disruption of ND10 requires IE1, at least early after infection (25). Notably, the mutant virus mCMV-IE1-Y165C and the revertant virus mCMV-IE1-C165Y both maintained the capacity to disrupt ND10 (Fig. 3A, CLSM images, and B, statistical analysis of ND10 counts). As side issues, the CLSM images revealed that the mutation Y165C in IE1 does not destroy the Croma 101 antibody epitope and does not interfere with the nuclear transport and localization of IE1 (Fig. 3A, e). In addition, in cells infected with the IE1 deletion mutant, some but not all ND10 are found to colocalize with nuclear inclusions formed by the E1 (M112-M113) protein (10, 17, 83) (Fig. 3A, d).
FIG. 3.

Functions of mutated and rescued IE1 proteins. (A) Dispersion of nuclear domains (ND10). Shown are CLSM images of representative triple-labeled individual cells (MEF) infected for 4 h (corresponding to the E phase) at an MOI of 4 (0.2 PFU/cell × 20; centrifugal enhancement of infectivity for synchronization) with the IE1 deletion mutant mCMV-ΔIE1 (images a to d) or the point mutant mCMV-IE1-Y165C (images e to h). (a and e) Red staining of intranuclear viral proteins E1 (M112-M113) and IE1 (m123), respectively. Note that green Alexa Fluor 488 fluorescence was electronically converted into red for better contrast. (c and g) Green staining of PML protein. Note that red Alexa Fluor 546 fluorescence was electronically converted into green for better contrast. The arrow points to an intranuclear PML body/ND10. (b and f) Visualization of cell nuclei by blue staining of DNA with Hoechst 33342 dye. (d and h) Merge of red E1 or IE1 protein, green PML, and blue Hoechst dye staining. The bar marker represents 10 μm. (B) Quantification of intranuclear PML bodies. MEF were infected (see above) with the viruses indicated. For statistical-significance analysis, intranuclear PML bodies were counted for 30 infected cell nuclei per group. The dots represent the numbers of PML bodies in individual cell nuclei. The median values are marked by horizontal bars. Two-sided P values (Wilcoxon-Mann-Whitney rank sum test; n = 30) are indicated for group comparisons of major interest. (C) Time course of IE- and E-phase protein expression represented by proteins IE1 (pp89/76) and m164 (gp36.5), respectively. Shown are Western blots of MEF total lysate proteins isolated at the indicated time points after infection with the viruses shown below for lanes 1 to 4. n.i., not infected; lysate proteins derived from uninfected MEF.
Mutation Y165C does not affect protein expression kinetics.
Another relevant issue was whether the mutation Y165C alters the kinetics of the expression of the IE1 protein itself or of downstream E-phase gene expression. Western blot analysis (Fig. 3C) did not reveal any notable difference in the expression kinetics between the mutated and the authentic IE1 proteins. The posttranslational modification of the pp89 form to the pp76 form of IE1 (42) was not affected either, which is important, considering that we do not know if functions ascribed to IE1 are performed differently by these two molecular species. As IE1 fulfills an accessory function in the transactivation of E-phase genes, it was important also to analyze the kinetics of the expression of E-phase proteins, here exemplified by the immunodominant protein m164/gp36.5. Again, no difference was found for mutant and revertant viruses. In accordance with the detection of the prototypic mCMV E-phase protein E1 (10, 17) in cells infected with the deletion mutant mCMV-ΔIE1 (Fig. 3A, a), expression of m164/gp36.5 was, however, not affected by complete absence of IE1, either.
Mutation Y165C does not affect viral replicative fitness in growth-arrested fibroblasts.
So far, the only phenotype seen for mutation Y164C was the failure of the mutated protein in the transactivation of RNR and TS gene promoters in quiescent cells upon transfection. Since we expected transactivation to be important for viral replication efficiency in quiescent cells, in which dNTPs are a limiting factor, rather than in proliferating cells, which have high intrinsic dNTP biosynthesis, we performed a multistep virus replication kinetics in quiescent MEF to assess the impact of the mutation on viral replicative fitness upon infection (Fig. 4). Opposite to what one would have expected, the mutant virus mCMV-IE1-Y165C replicated like mCMV-WT.BAC and the revertant virus mCMV-IE1-C165Y, regardless of whether virus replication was measured by quantitating viral DNA present within infected cells (Fig. 4A) or by quantitating infectivity released from infected cells into the cell culture supernatant (Fig. 4B). The most striking finding, however, was that the deletion mutant mCMV-ΔIE1 was capable of multistep growth and reached similar endpoint titers, with perhaps a somewhat lower release of infectivity after the first round of replication.
FIG. 4.

Viral replicative fitness in quiescent fibroblasts in cell culture. MEF were growth arrested in low-serum starvation medium and were centrifugally infected at an MOI of 0.4 (0.02 PFU/cell × 20; centrifugal enhancement of infectivity) with the viruses indicated. (A) Quantitation of viral genomes in infected cells as determined by gene M55(gB)-specific real-time PCR with normalization to the cellular gene pthrp. (B) Quantitation of infectious virus released into the cell culture supernatant as determined by virus plaque assay. The data refer to 106 cells. Shown are the multistep growth curves for a period of 72 h postinfection (p.i.) corresponding to three viral growth cycles. Time zero is defined as the end of the 30-min period of centrifugal infection revealing the inoculum viral genomes and infectivity. The dots represent data from three six-well cultures for each of the time points indicated. The median values are marked by horizontal lines. The productivity after the first viral growth cycle is highlighted by gray shading, indicating the onset of viral-DNA replication after 16 h and virus release after 24 h, which is in perfect accordance with previous findings (39).
The IE1 protein is not essential for RNR and TS gene expression in growth-arrested fibroblasts.
In view of the replication of the mutant viruses mCMV-IE1-Y165C and mCMV-ΔIE1 in quiescent cells, we wondered if activation of genes of dNTP synthesis is critical at all. We therefore established quantitative RT-PCRs (Table 2) to assess RNR and TS transcription after infection of quiescent MEF. Successful growth arrest was controlled for by immunofluorescence staining of Ki-67, a marker for proliferating cells (23) (Fig. 5A), and comparable rates of infection by the various viruses were confirmed by the quantitation of E1 transcripts (Fig. 5B). As opposed to low to absent RNR and TS transcription in uninfected cells, as well as in cells mock infected with UV light-inactivated mCMV-WT-BACUV, transcription was activated in infected cells regardless of whether IE1 was present or whether IE1 was mutated (Fig. 5C). For all four viruses tested, activation of RNR and TS transcription became apparent only beyond 6 h. These data suggest that activation of genes for dNTP synthesis may be required for efficient virus replication, but they also indicate that the transactivating function of the IE1 protein plays no essential role in this process.
The mutation Y165C differs from IE1 deletion with regard to the in vivo growth attenuation phenotype.
Previous work by Ghazal and colleagues (25; reviewed in reference 12) showed that the deletion mutant mCMV-ΔIE1 grows like mCMV-WT.BAC in proliferating MEF in cell culture but that growth is attenuated in vivo. For evaluating a possible contribution of IE1-transactivated dNTP synthesis to virus multiplication in host tissues, we compared the impact of IE1 deletion versus Y165C mutation in the BALB/c mouse model of the immunocompromised, total-body gamma-irradiated host (for the model, see references 32 and 34) (Fig. 6). Immunohistological images detecting the intranuclear proteins IE1 and MCP/M86 visualize a clear quantitative difference in the degrees of liver infection and histopathology. While in representative tissue section areas of livers infected with mCMV-ΔIE1 only few foci of infected IE1− MCP+ cells were seen, many plaque-like foci of IE1+ MCP+ cells, which are mainly hepatocytes, were seen after infection with mCMV-IE1-Y165C (Fig. 6A1).
FIG. 6.

Viral replicative fitness in host tissues. (A1) Immunocompromised BALB/c mice were infected with 105 PFU (corresponding to ∼5 × 107 viral genomes) of the deletion mutant mCMV-ΔIE1 (images a1 to b2) or of the point mutant mCMV-IE1-Y165C (images c1 to d2). On day 10 after infection, liver tissue sections were analyzed by IHC staining of intranuclear IE1 protein (IE1-IHC, black) and of intranuclear MCP (MCP-IHC, red). The index 1 images (a1 to d1) provide overviews of section areas of ∼0.5 mm2. The arrows point to sites of interest that are resolved to greater detail in the corresponding index 2 (a2 to d2) images. The bar markers represent 50 μm. Note the absence of IE1 protein in the intranuclear inclusion bodies of hepatocytes infected with the deletion mutant mCMV-ΔIE1 (image a2). (A2) Quantitation of liver infection in the time course of viral spread. Plotted are semilogarithmic growth curves for the indicated viruses based on the numbers of infected, MCP+ liver cells counted in representative 10-mm2 liver tissue section areas. The solid circles represent data from individual mice, with the median values marked by horizontal lines. The DTs and their 95% confidence intervals (given in parentheses) are calculated from the slopes a (95% confidence intervals of a) of the calculated log-linear regression lines according to the following formula: DT = log 2/a. (B) DTs of the viral-DNA load in spleen and lungs. The numbers of viral genomes per 106 tissue cells were quantified by real-time PCR specific for the viral gene M55(gB) normalized to the cellular gene pthrp. For further explanation, see the legend to panel A2. p.i., postinfection.
A small degree of tissue infection detected at a fixed time point does not necessarily indicate growth attenuation in cells within the tissue analyzed. Two fundamentally different mechanisms could lead to this result. (i) In the case of inhibition of virus dissemination from the portal of entry into the tissue, colonization of tissue occurs with delay, and as a consequence, virus replication in the tissue is retarded even though the growth rate may be like that of WT virus. (ii) Alternatively, virus dissemination is unaffected and colonization of tissue occurs with no delay, but the growth rate is reduced. One can distinguish between these two mechanisms by determining the time axis intercept and slope of log-linear growth curves. Of course, deficiency in dissemination from the portal of entry and deficiency in virus replication in distant target organs may also occur in combination. In the specific case considered here, all viruses under test had colonized the liver by day 6 and thus did not show a dissemination defect (Fig. 6A2). Whereas the DT of cells infected with the mutant virus mCMV-IE1-Y165C was like those of mCMV-WT.BAC and the revertant virus mCMV-IE1-C165Y, namely, ∼20 h, the DT was significantly prolonged for cells infected with the deletion mutant mCMV-ΔIE1, namely, ∼34 h, with no overlap of the 95% confidence intervals. Essentially the same result, albeit with different DTs, was obtained for the number of viral genomes in spleen and in lungs (Fig. 6B).
Together, these data confirm the attenuation of mCMV-ΔIE1 reported by Ghazal and colleagues (25) and add the information that this attenuation does not reflect a limitation in virus dissemination but results from growth retardation within the infected tissues. Interestingly, this is true for different organs constituted by different virus-producing cell types, including hepatocytes, endothelial-lining cells, and lung epithelial cells.
Infection induces RNR gene expression in the liver independently of the IE1 protein.
So far, the activation of genes for dNTP synthesis had been tested only in vitro. We therefore wondered if mCMV infection also induces RNR and TS transcription in host tissues. Notably, gene expression above the baseline defined by uninfected liver tissue, as well as by liver tissue of mice mock infected with UV-inactivated virus, was seen only for RNR (Fig. 7A). At first glance, one might conclude that RNR gene expression is induced by mCMV-WT.BAC and not by the deletion mutant mCMV-ΔIE1, suggesting a role for the IE1 protein in the in vivo induction of RNR. This, conclusion, however, is essentially wrong, as we have to consider the different viral burden due to the growth attenuation of mCMV-ΔIE1 (Fig. 6). Consequently, we normalized RNR gene expression to the number of E1 transcripts determined by quantitative RT-PCR, as well as to the number of infected cells determined by MCP-specific immunohistology (Fig. 7B). With this correction performed, no differences in RNR transcription were found between the four viruses tested, regardless of whether IE1 was present or whether IE1 was mutated.
FIG. 7.

In situ activation of cellular genes for dNTP synthesis during infection of the liver. Immunocompromised BALB/c mice were infected with 105 PFU of the viruses indicated. n. i. (not infected), uninfected but immunocompromised mice, showing that 6.5-Gy total-body gamma irradiation does not by itself stimulate the expression of genes involved in dNTP synthesis; UV, mice mock infected with UV light-inactivated mCMV-WT.BACUV in a dose corresponding to 105 PFU; WT, infectious mCMV-WT.BAC. The analysis was performed 10 days after infection. (A) Expression levels (ΔΔCT values) relative to GAPDH transcripts. The bars (solid bars, RNR; open bars, TS) represent the median values for three mice per experimental group; the error bars indicate the range. (B) Normalization of the relative expression levels of RNR to the numbers of E1 transcripts per 500 ng of total RNA (top) or to the numbers of infected MCP+ cells per defined liver tissue section area of 10 mm2 in order to take account of differences in tissue infection density. (C) Expression levels (ΔΔCT values) relative to β-actin in preparatively purified hepatocytes (Hc) and NPLCs derived from livers infected with mCMV-IE1-Y165C or from livers of a control group that was immunocompromised by total-body gamma irradiation but left uninfected. The section NPLC* shows the conservatively estimated contribution of contaminating NPLCs (10% in this experiment) to RNR and TS transcription in the hepatocyte fraction. The bars (solid bars, RNR; open bars, TS) represent the median values for three mice per experimental group; the error bars indicate the range. n. i., not infected.
Other organs, including the spleen, lungs, salivary glands, adrenal glands, kidneys, and heart, were tested with the qualitatively concordant result that the mutation Y165C had no consequences, although the induction of RNR gene expression was less significant due to a higher baseline expression and a generally lower infection density in these organs than in the liver (70), so that the influence of RNR expression in uninfected cells dominated the quantitation of transcripts (data not shown). It should be noted that baseline RNR expression also precluded a histological documentation of enhanced RNR expression in infected hepatocytes (J. Podlech, not shown). Thus, in our hands, transactivation of RNR expression in vivo could be detected only with the high sensitivity of RT-PCR.
Although the immunoablative treatment largely wiped out immune cells and thus prevented an inflammatory infiltration of the liver, which is also reflected in uncontrolled virus replication (Fig. 6), one can reasonably argue that some residual liver-resident nonparenchymal cells, such as Kupffer-type macrophages and hepatic natural killer cells, might become activated in response to the infection. In fact, recent work by Erlach et al. (21) has shown that CD49b+ NKG2D+ T-cell receptor β− hepatic natural killer cells expand relatively and absolutely in infected livers of immunocompromised BALB/c mice in the first 3 days after the gamma irradiation until the absolute numbers decline again. Such activation would also upregulate RNR expression in uninfected cells and could possibly account for the enhanced RNR transcription in the infected liver. Therefore, we purified hepatocytes and NPLCs to separately evaluate their contributions to RNR transcription in livers infected with the mutant virus mCMV-IE1-Y165C (Fig. 7C). The infection was found to enhance RNR transcription in purified hepatocytes, as well as in NPLCs, which is in accordance with the fact that hepatocytes, as well as Kupffer cells and liver endothelial-lining cells, are targets of mCMV (16, 70). As the contamination of the hepatocyte fraction with NPLCs was <10% in this experiment (C. K. Seckert, data not shown), proliferating NPLCs cannot account for the RNR transcription measured in the hepatocyte fraction. In accordance with the experiment shown in Fig. 7A, TS was not activated by the infection, either in hepatocytes or in NPLCs.
Together, these data show that mCMV induces RNR transcription in infected cells, not only in cell culture, but also in vivo. As in growth-arrested fibroblasts in vitro, however, this induction proved to be independent of IE1.
DISCUSSION
The IE proteins of CMVs are regarded as decisive regulators initiating the downstream viral transcriptional program for lytic replication (see the introduction). Whereas mCMV IE3 (M122), the homolog of hCMV IE2 (UL122), has been identified as an essential transactivator of E-phase gene expression that is critical for viral replication (2), mCMV IE1 (m123) has a pleiotropic function (see the introduction) that is not essential for virus replication in a variety of cell types proliferating in cell culture but determines viral replicative fitness in vivo. Accordingly, a deletion mutant, mCMV-ΔIE1 (Δexon 4 of the MIE transcription unit) showed growth attenuation in host organs, including the spleen, liver, and lungs (25; reviewed in reference 12). It was previously not possible to attribute different roles to the different functions of IE1, since deletion of IE1 abrogates all of its functions. A similar problem would likewise apply to small-interfering-RNA-based gene knockdown strategies. One noted function of IE1 is its capacity to transactivate cellular genes involved in dNTP synthesis, as specifically shown for RNR (48) and TS (28) genes in reporter gene assays. This function may relate directly to viral replicative fitness, since as far as is known, mCMV does not code for viral equivalents of nucleotide metabolism enzymes and thus depends on cellular dNTP synthesis for its DNA replication. As recently reviewed for hCMV (71), IE1 has the potential to contribute to cell cycle arrest in the S and G2/M phases and to delay cell cycle exit. Clearly, viral transactivation of cellular dNTP synthesis may not be essential in growing cells, in which the supply of dNTPs is not a limiting factor, but it may become important for virus growth in quiescent cells that are deprived of dNTPs. Cells constituting host tissues in typical target organs of productive CMV infection and disease are for the most part fully differentiated and contact inhibited, performing their physiological tasks in the G0 phase of the cell cycle. As an example, the hepatocyte, the major virus-producing cell type during mCMV infection (70), is metabolically highly active but is “resting” in terms of cellular DNA synthesis and cell division, unless tissue damage triggers regeneration. Histological analysis, however, has never provided any evidence for mCMV infection being restricted to regenerating tissue (J. Podlech, unpublished observations). The ability to trigger cellular dNTP synthesis in quiescent cells is thus likely to be relevant to virus multiplication in host tissues and an important pathogenicity factor. We therefore considered it attractive to hypothesize that transactivation of cellular genes involved in dNTP synthesis is a critical function of IE1 for virus replication in quiescent cells, which might explain the reported in vivo growth attenuation of the deletion mutant mCMV-ΔIE1.
Although we have to apologize for serendipity, we have identified here a spontaneous point mutation, Y165C, in the mCMV IE1 protein, a mutation that abolishes the transactivation of both RNR and TS in reporter gene assays but maintains IE1's capacity to disperse ND10, a feature that distinguishes the point mutation Y165C from IE1 deletion. It was an obvious side issue to ask if the loss of transactivator function is due to loss of tyrosine or to gain of cysteine. We should first recall the textbook knowledge that cysteines in cytosolic proteins are not usually involved in disulfide bonds, since protein disulfide isomerase is an ER protein catalyzing disulfide bond formation in the rough ER, but not in the cytosol. Furthermore, glutathione prevents the spontaneous formation of disulfide bonds in the cytosol, except for special enzymes catalyzing redox processes or under conditions of oxidative stress (18). We were therefore not particularly surprised to find that the IE1 protein with the designed mutation Y165A also failed to transactivate RNR and TS in reporter gene assays (data not shown). Further mutational analysis, in particular of mutant Y165F, may provide insight into the molecular basis of the loss of function, as replacement of tyrosine with phenylalanine removes a potential phosphorylation site. Our interest in the mutation Y165C was originally prompted by the fact that the proteasome cleaves the IE1 protein after Y165 to generate the precursor 166-DMYPHFMPTNL-176 of the immunodominant antigenic IE1 peptide 168-YPHFMPTNL-176 (for a review, see reference 64). Our finding that the mutation Y165C does not notably interfere with the presentation of the IE1 peptide to CTL is in accordance with bioinformatics cleavage predictions for both the constitutive proteasome and the immunoproteasome.
Ghazal and colleagues (25), who generated and first described the mutant virus mCMV-ΔIE1, had already shown for different cell types that deletion of IE1 does not have an impact on virus growth under regular cell culture conditions. Therefore, we did not expect an effect of mutation Y165C, either. The supply of dNTPs is not a limiting factor in dividing cells; however, we expected to see an effect with quiescent cells. Contrary to our expectations, the mutant virus mCMV-IE1-Y165C replicated in verified growth-arrested, Ki-67-negative MEF just like the parental virus, mCMV-WT.BAC, and the revertant virus mCMV-IE1-C165Y, even under the low-MOI infection conditions of a multistep growth curve analysis. This indicated for the first time that transactivation of dNTP biosynthesis by IE1 is not essential for the growth of mCMV in quiescent cells. Even more surprising, however, was the finding that the deletion mutant mCMV-ΔIE1 also replicated like the parental virus, indicating that none of the functions of IE1 is essential. That IE1's capacity to disperse ND10 is not crucial for progression to E-phase gene expression became apparent from the fact that the E1 protein was detected by CLSM analysis in the nuclei of cells infected with mCMV-ΔIE1 at a time when ND10 structures were still present in the very same nuclei. Likewise, in our immunohistological analysis, infection of hepatocytes with mCMV-ΔIE1 proceeded to the late-phase protein MCP visible in IE1-negative nuclei of infected cells. The failure of mCMV-ΔIE1 to disperse ND10, as well as the detection of MCP in IE1-negative cell nuclei, together with routine PCR control of virion DNA, proved the authenticity of the virus preparation. Thus, taking these data together, IE1 is dispensable for virus growth in quiescent cells.
Though IE1 proved to be dispensable, this does not necessarily mean that activation of dNTP biosynthesis in quiescent cells is also dispensable. We therefore considered the possibility that transactivation, precisely because it is so important for the virus, is guaranteed by redundancy, assuming that another viral factor, here operationally called “factor X,” might replace IE1 in this crucial role. Since viruses with mutations affecting IE1 were not available at the time when Gribaudo et al. and Lembo et al. discovered the transactivation of RNR and TS by reporter gene assays upon transient transfection of quiescent cells (28, 48), these authors had no opportunity to analyze the specific role of IE1 in the activation of dNTP biosynthesis upon infection, although pharmacological inhibition of RNR and TS indicated that both enzymes are, in principle, critically implicated in virus replication in quiescent cells. Our data now show that both RNR and TS are indeed activated after the infection of quiescent cells. Notably, however, this activation was independent of IE1. We thus propose a model in which factor X can step in for IE1 after infection with the deletion mutant mCMV-ΔIE1 or with the specific point mutant mCMV-IE1-Y165C (Fig. 8 shows a summarizing model). The fact that enhanced RNR transcription in infected cells became apparent only after 6 h, which is not an immediate-early kinetics, might tempt us to conclude that factor X is not an IE-phase protein. This is not logical, however, since delayed RNR transcription was also observed after infection with mCMV-WT.BAC expressing the transactivator protein IE1.
FIG. 8.

Summarizing model of viral-DNA replication in quiescent cells. Activation of the TS and RNR gene promoters, PTS and PRNR, is a redundant function accomplished by the IE1 protein (green) and at least one unknown “factor X” (yellow), which could be a virus-encoded transactivator or a cellular transactivator triggered by the infection. The mutation Y165C (red) destroys the transactivator function of IE1 but does not affect the alternative transactivator, factor X. Activation of dNTP biosynthesis promotes viral replication in quiescent cells in cell culture and in host tissues. The cylindrical symbols represent promoters and structural genes, the wavy symbols represent mRNAs, and the oval symbols represent proteins.
We do not know if factor X is an alternative virally encoded transactivator or if it describes a viral function that triggers a cellular signaling pathway for the activation of dNTP biosynthesis. In this context, it is important to emphasize that mCMV's main transactivator, the MIE protein IE3, the homolog of hCMV IE2, failed to transactivate RNR and TS (28, 48), suggesting that IE3 is not factor X. Since the UV-inactivated parental virus mCMV-WT.BACUV failed to stimulate RNR and TS expression, factor X does not seem to be a virion protein. Global alteration of the cellular transcriptome ascribed to multipathway signaling triggered by gB binding and the virion entry process does not seem to be involved either, even though numerous cellular transcription factors are activated upon mere virus-cell contact (for review articles, see references 6 and 75). It thus appears that viral-gene expression is required for generating factor X.
It is an advantage of animal models that the consequences of viral mutations can be tested in the pathophysiological context of host organs that are relevant to viral disease. Although the deletion mutant mCMV-ΔIE1 is not growth attenuated in quiescent fibroblasts, previous work by Ghazal and colleagues showed in vivo growth attenuation of the mutant (25). We therefore wondered if factor X is operative in vivo or if a failure in activating dNTP synthesis, specifically in host tissues, might explain the in vivo growth attenuation. We first endeavored to reproduce the reported in vivo attenuation of mCMV-ΔIE1 and to test if the point mutant mCMV-IE1-Y165C would differ in this respect. The quantitative analysis of log-linear growth curves in host tissues indeed revealed a reduced growth rate for the mutant mCMV-ΔIE1 compared to the parental virus, mCMV-WT.BAC, albeit with subtle differences between host organs. Specifically, attenuation appeared to be less pronounced in the lungs, in particular if one considered the confidence intervals. The fact that the slopes of the regression lines differed shows that the attenuation does not result from an inhibition of virus dissemination from the portal of entry to the distant target organs but results from a reduced rate of replication within the organotypic tissue cells. As opposed to the deletion mutant mCMV-ΔIE1, the point mutant mCMV-IE1-Y165C replicated like revertant virus and the parental virus in all three organs tested. Thus, the mutation Y165C has no phenotype, which implies that the transactivator function of IE1 is not essential for in vivo virus replication. As shown for the liver, attenuation of mCMV-ΔIE1 is not paralleled by reduced RNR transcription. Instead, both mutants, as well as the C165Y revertant, induced RNR comparably to the parental virus, mCMV-WT.BAC, which implies that factor X is operative in the liver. Our liver cell separation experiment performed for livers infected with the mutant virus mCMV-IE1-Y165C suggested that RNR transactivation by factor X could occur in hepatocytes. We have no immediate explanation for the failure to detect activation of TS expression in vivo, but a shortage of TS cannot explain the attenuation of mCMV-ΔIE1, since mCMV-WT.BAC did not differ in this respect.
In conclusion, the activation of genes involved in dNTP metabolism in quiescent cells and host tissues is a redundant function of mCMV that does not depend exclusively on IE1. The in vivo attenuation seen after deletion of the complete IE1 protein is apparently not related to IE1's transactivator function, which maps to position 165Y, but must be caused by another function of IE1. A crucial role for adaptive immunity is unlikely in immunocompromised-host models, and in any case, deletion of IE1 should rather favor virus growth in BALB/c mice due to the elimination of the immunodominant IE1 peptide. Since the in vivo phenotypes of mCMV-ΔIE1 were comparable in the SCID mouse model (25), in which innate immunity is intact, and in the model of hematoablative treatment (this report), in which all immune functions are affected, attenuation of mCMV-ΔIE1 due to a putatively higher susceptibility to the innate immune response is unlikely. In addition, the log-linear virus growth within various host organs argues against any growth limitation by immune control.
One feature distinguishing mCMV-IE1-Y165C and mCMV-ΔIE1 is the ability and lack of ability, respectively, to disperse ND10. Therefore, this remains a candidate mechanism for in vivo attenuation of mCMV-ΔIE1, although it is difficult to conceive why dispersal of ND10 should be critical for virus growth in vivo but not in quiescent cells in cell culture. Certainly, yet another function of IE1 might eventually prove to be critical for in vivo replication. In this context, it is interesting that Busche and colleagues (12) noted that mCMV IE1, unlike hCMV IE1 (61), enhances rather than inhibits the antiviral type I interferon response, so this property of mCMV IE1 cannot explain the attenuation of mCMV-ΔIE1. It must be mentioned that deletion of hCMV IE1, unlike the deletion of mCMV IE1, attenuates virus growth even in regular cell culture under conditions of low MOI (22, 27). Whether this difference is due to differences in the respective IE1 proteins (for a recent review, see reference 52) or whether another mCMV protein compensates for a loss of mCMV IE1 function will be an issue for future research. In any case, our data have helped to reject the hypothesis that transactivation of genes involved in dNTP biosynthesis is the critical function of mCMV IE1 in virus growth in quiescent cells in cell culture, as well as in host tissues.
Acknowledgments
We thank Martin Messerle (Hannover, Germany), Ana Angulo (Barcelona, Spain), and Peter Ghazal (Edinburgh, United Kingdom), for the IE1 deletion mutant of mCMV and Martin Messerle for helpful discussions and advice. Reporter gene plasmids for the dual-luciferase assays were kindly provided by Santo Landolfo, Turin, Italy, with permission from Lars Thelander, Umeå, Sweden, and Lee Johnson, Columbus, OH. MAbs Croma 101 and Croma 103 were generously supplied by Stipan Jonjic, Rijeka, Croatia. We thank the Confocal Laser Scanning Microscopy Core Facility of the Immunology Cluster of Excellence for assistance with image collection and the Central Laboratory Animal Facility (CLAF) team for animal care.
Extramural support was provided by the Deutsche Forschungsgemeinschaft, SFB 490, individual projects E2 (V.W., C.O.S., A.R., C.K.S., M.J.R., and N.K.A.G.), E3 (V.B.), and E4 (T.D., S.E., D.S., and M.J.R.), as well as SFB 432, individual project A10 (J.P. and M.J.R.), and Clinical Research Group KFO 183 (N.A.W.L. and M.J.R.). Intramural support was provided by the young investigators program MAIFOR of the Medical Faculty of the Johannes Gutenberg-University (V.W. and N.K.A.G.). Special thanks go to the Dr. Gerhard and Martha Röttger-Stiftung and to the Dr. Hans-Joachim and Ilse Brede-Stiftung for generous donations.
Footnotes
Published ahead of print on 6 August 2008.
REFERENCES
- 1.Adachi, N., and M. R. Lieber. 2002. Bidirectional gene organization: a common architectural feature of the human genome. Cell 109807-809. [DOI] [PubMed] [Google Scholar]
- 2.Angulo, A., P. Ghazal, and M. Messerle. 2000. The major immediate-early gene ie3 of mouse cytomegalovirus is essential for viral growth. J. Virol. 7411129-11136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2a.Applied Biosystems. 2001. User bulletin no. 2: ABI Prism 7700 Sequence Detection System (manual no. 10/2001). Applied Biosystems, Foster City, CA.
- 3.Ash, J., W.-C. Liao, Y. Ke, and L. F. Johnson. 1995. Regulation of mouse thymidylate synthase gene expression in growth-stimulated cells: upstream S phase control elements are indistinguishable from essential promoter elements. Nucleic Acids Res. 234649-4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bain, M., M. Reeves, and J. Sinclair. 2006. Regulation of human cytomegalovirus gene expression by chromatin remodeling, p. 167-183. In M. J. Reddehase (ed.), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Wymondham, Norfolk, United Kingdom.
- 5.Böhm, V., J. Podlech, D. Thomas, P. Deegen, M. F. Pahl-Seibert, N. A. Lemmermann, N. K. Grzimek, S. A. Oehrlein-Karpi, M. J. Reddehase, and R. Holtappels. 2008. Epitope-specific in vivo protection against cytomegalovirus disease by CD8 T cells in the murine model of preemptive immunotherapy. Med. Microbiol. Immunol. 197135-144. [DOI] [PubMed] [Google Scholar]
- 6.Böhme, K. W., and T. Compton. 2006. Virus entry and activation of innate immunity, p. 111-130. In M. J. Reddehase (ed.), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Wymondham, Norfolk, United Kingdom.
- 7.Borst, E. M., G. Hahn, U. H. Koszinowski, and M. Messerle. 1999. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: a new approach for construction of HCMV mutants. J. Virol. 738320-8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borst, E. M., G. Posfai, F. Pogoda, and M. Messerle. 2004. Mutagenesis of herpesvirus BACs by allele replacement. Methods Mol. Biol. 256269-279. [DOI] [PubMed] [Google Scholar]
- 9.Borysiewicz, L. K., J. K. Hickling, S. Graham, J. Sinclair, M. P. Cranage, G. L. Smith, and J. G. Sissons. 1988. Human cytomegalovirus-specific cytotoxic T cells. Relative frequency of stage-specific CTL recognizing the 72-kD immediate early protein and glycoprotein B expressed by recombinant vaccinia virus. J. Exp. Med. 168919-931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bühler, B., G. M. Keil, F. Weiland, and U. H. Koszinowski. 1990. Characterization of the murine cytomegalovirus early transcription unit e1 that is induced by immediate-early proteins. J. Virol. 641907-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bunde, T., A. Kirchner, B. Hoffmeister, D. Habedank, R. Hetzer, G. Cherepnev, S. Prösch, P. Reinke, H. D. Volk, H. Lehmkuhl, and F. Kern. 2005. Protection from cytomegalovirus after transplantation is correlated with immediate early 1-specific CD8 T cells. J. Exp. Med. 2011031-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busche, A., A. Angulo, P. Kay-Jackson, P. Ghazal, and M. Messerle. 2008. Phenotypes of major immediate-early gene mutants of mouse cytomegalovirus. Med. Microbiol. Immunol. 197233-240. [DOI] [PubMed] [Google Scholar]
- 13.Cardin, R. D., G. B. Abenes, C. A. Stoddard, and E. S. Mocarski. 1995. Murine cytomegalovirus IE2, an activator of gene expression, is dispensable for growth and latency in mice. Virology 209236-241. [DOI] [PubMed] [Google Scholar]
- 14.Chabes, A., and L. Thelander. 2000. Controlled protein degradation regulates ribonucleotide reductase activity in proliferating mammalian cells during the normal cell cycle and in response to DNA damage and replication blocks. J. Biol. Chem. 27517747-17753. [DOI] [PubMed] [Google Scholar]
- 15.Chatellard, P., R. Pankiewicz, E. Meier, L. Durrer, C. Sauvage, and M. O. Imhof. 2007. The IE2 promoter/enhancer region from mouse CMV provides high levels of therapeutic protein expression in mammalian cells. Biotechnol. Bioeng. 96106-117. [DOI] [PubMed] [Google Scholar]
- 16.Cicin-Sain, L., Z. Ruzsics, J. Podlech, I. Bubic, C. Menard, S. Jonjic, M. J. Reddehase, and U. H. Koszinowski. 2008. Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J. Virol. 822056-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciocco-Schmitt, G. M., Z. Karabekian, E. W. Godfrey, R. M. Stenberg, A. E. Campbell, and J. A. Kerry. 2002. Identification and characterization of novel murine cytomegalovirus M112-113 (e1) gene products. Virology 294199-208. [DOI] [PubMed] [Google Scholar]
- 18.Cumming, R. C., N. L. Andon, P. A. Haynes, M. Park, W. H. Fischer, and D. Schubert. 2004. Protein disulfide bond formation in the cytoplasm during oxidative stress. J. Biol. Chem. 27921749-21758. [DOI] [PubMed] [Google Scholar]
- 19.Dorsch-Häsler, K., G. M. Keil, F. Weber, M. Jasin, W. Schaffner, and U. H. Koszinowski. 1985. A long and complex enhancer activates transcription of the gene coding for the highly abundant immediate early mRNA in murine cytomegalovirus. Proc. Natl. Acad. Sci. USA 828325-8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebeling, A., G. M. Keil, E. Knust, and U. H. Koszinowski. 1983. Molecular cloning and physical mapping of murine cytomegalovirus DNA. J. Virol. 47421-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erlach, K. C., V. Böhm, M. Knabe, P. Deegen, M. J. Reddehase, and J. Podlech. 2008. Activation of hepatic natural killer cells and control of liver-adapted lymphoma in the murine model of cytomegalovirus infection. Med. Microbiol. Immunol. 197167-178. [DOI] [PubMed] [Google Scholar]
- 22.Gawn, J. M., and R. F. Greaves. 2002. Absence of IE1 p72 protein function during low-multiplicity infection by human cytomegalovirus results in a broad block to viral delayed-early gene expression. J. Virol. 764441-4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerdes, J., L. Li, C. Schlueter, M. Duchrow, C. Wohlenberg, C. Gerlach, I. Stahmer, S. Kloth, E. Brandt, and H. D. Flad. 1991. Immunobiochemical and molecular biologic characterization of the cell proliferation-associated nuclear antigen that is defined by monoclonal antibody Ki-67. Am. J. Pathol. 138867-873. [PMC free article] [PubMed] [Google Scholar]
- 24.Ghazal, P., M. Messerle, K. Osborn, and A. Angulo. 2003. An essential role of the enhancer for murine cytomegalovirus in vivo growth and pathogenesis. J. Virol. 773217-3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghazal, P., A. E. Visser, M. Gustems, R. Garcia, E. M. Borst, K. Sullivan, M. Messerle, and A. Angulo. 2005. Elimination of ie1 significantly attenuates murine cytomegalovirus virulence but does not alter replicative capacity in cell culture. J. Virol. 797182-7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibson, L., G. Piccinini, D. Lilleri, M. G. Revello, Z. Wang, S. Markel, D. J. Diamond, and K. Luzuriaga. 2004. Human cytomegalovirus proteins pp65 and immediate early protein 1 are common targets for CD8+ T cell responses in children with congenital or postnatal human cytomegalovirus infection. J. Immunol. 1722256-2264. [DOI] [PubMed] [Google Scholar]
- 27.Greaves, R. F., and E. S. Mocarski. 1998. Defective growth correlates with reduced accumulation of a viral DNA replication protein after low-multiplicity infection by a human cytomegalovirus ie1 mutant. J. Virol. 72366-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gribaudo, G., L. Riera, D. Lembo, M. De Andrea, M. Gariglio, T. L. Rudge, L. F. Johnson, and S. Landolfo. 2000. Murine cytomegalovirus stimulates cellular thymidylate synthase gene expression in quiescent cells and requires the enzyme for replication. J. Virol. 744979-4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grzimek, N. K. A., D. Dreis, S. Schmalz, and M. J. Reddehase. 2001. Random, asynchronous, and asymmetric transcriptional activity of enhancer-flanking major immediate-early genes ie1/3 and ie2 during murine cytomegalovirus latency in the lungs. J. Virol. 752692-2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grzimek, N. K. A., J. Podlech, H.-P. Steffens, R. Holtappels, S. Schmalz, and M. J. Reddehase. 1999. In vivo replication of recombinant murine cytomegalovirus driven by the paralogous major immediate-early promoter-enhancer of human cytomegalovirus. J. Virol. 735043-5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harlow, E., and D. Lane. 1988. Antibodies: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 32.Holtappels, R., V. Böhm, J. Podlech, and M. J. Reddehase. 2008. CD8 T-cell-based immunotherapy of cytomegalovirus infection: “proof of concept” provided by the murine model. Med. Microbiol. Immunol. 197125-134. [DOI] [PubMed] [Google Scholar]
- 33.Holtappels, R., D. Gillert-Marien, D. Thomas, J. Podlech, P. Deegen, S. Herter, S. A. Oehrlein-Karpi, D. Strand, M. Wagner, and M. J. Reddehase. 2006. Cytomegalovirus encodes a positive regulator of antigen presentation. J. Virol. 807613-7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holtappels, R., M. W. Munks, J. Podlech, and M. J. Reddehase. 2006. CD8 T-cell-based immunotherapy of cytomegalovirus disease in the mouse model of the immunocompromised bone marrow transplantation recipient, p. 383-418. In M. J. Reddehase (ed.), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Wymondham, Norfolk, United Kingdom.
- 35.Holtappels, R., M.-F. Pahl-Seibert, D. Thomas, and M. J. Reddehase. 2000. Enrichment of immediate-early 1 (m123/pp89) peptide-specific CD8 T cells in a pulmonary CD62Llo memory-effector cell pool during latent murine cytomegalovirus infection of the lungs. J. Virol. 7411495-11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holtappels, R., C. O. Simon, M. W. Munks, D. Thomas, P. Deegen, B. Kühnapfel, T. Däubner, S. F. Emde, J. Podlech, N. K. Grzimek, S. A. Oehrlein-Karpi, A. B. Hill, and M. J. Reddehase. 2008. Subdominant CD8 T-cell epitopes account for protection against cytomegalovirus independent of immunodomination. J. Virol. 825781-5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holtappels, R., D. Thomas, J. Podlech, and M. J. Reddehase. 2002. Two antigenic peptides from genes m123 and m164 of murine cytomegalovirus quantitatively dominate CD8 T-cell memory in the H-2d haplotype. J. Virol. 76151-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karrer, U., S. Sierro, M. Wagner, A. Oxenius, H. Hengel, U. H. Koszinowski, R. E. Philipps, and P. Klenerman. 2003. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J. Immunol. 1702022-2029. (Erratum, 170:3895.) [DOI] [PubMed] [Google Scholar]
- 39.Keil, G. M., A. Ebeling-Keil, and U. H. Koszinowski. 1984. Temporal regulation of murine cytomegalovirus transcription and mapping of viral RNA synthesized at immediate early times after infection. J. Virol. 50784-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keil, G. M., A. Ebeling-Keil, and U. H. Koszinowski. 1987. Immediate-early genes of murine cytomegalovirus: location, transcripts, and translation products. J. Virol. 61526-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keil, G. M., A. Ebeling-Keil, and U. H. Koszinowski. 1987. Sequence and structural organization of murine cytomegalovirus immediate-early gene 1. J. Virol. 611901-1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keil, G. M., M. R. Fibi, and U. H. Koszinowski. 1985. Characterization of the major immediate-early polypeptides encoded by murine cytomegalovirus. J. Virol. 54422-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kern, F., I. P. Surel, N. Faulhaber, C. Frömmel, J. Schneider-Mergener, C. Schönemann, P. Reinke, and H. D. Volk. 1999. Target structures of the CD8+-T-cell response to human cytomegalovirus: the 72-kilodalton major immediate-early protein revisited. J. Virol. 738179-8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knuehl, C., P. Spee, T. Ruppert, U. Kuckelkorn, P. Henklein, J. Neefjes, and P. M. Kloetzel. 2001. The murine cytomegalovirus pp89 immunodominant H-2Ld epitope is generated and translocated into the endoplasmic reticulum as an 11-mer precursor peptide. J. Immunol. 1671515-1521. [DOI] [PubMed] [Google Scholar]
- 45.Kurz, S. K., M. Rapp, H.-P. Steffens, N. K. A. Grzimek, S. Schmalz, and M. J. Reddehase. 1999. Focal transcriptional activity of murine cytomegalovirus during latency in the lungs. J. Virol. 73482-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurz, S. K., H.-P. Steffens, A. Mayer, J. R. Harris, and M. J. Reddehase. 1997. Latency versus persistence or intermittent recurrences: evidence for a latent state of murine cytomegalovirus in the lungs. J. Virol. 712980-2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lembo, D., M. Donalisio, A. Hofer, M. Cornaglia, W. Brune, U. H. Koszinowski, L. Thelander, and S. Landolfo. 2004. The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J. Virol. 784278-4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lembo, D., G. Gribaudo, A. Hofer, L. Riera, M. Cornaglia, A. Mondo, A. Angeretti, M. Gariglio, L. Thelander, and S. Landolfo. 2000. Expression of an altered ribonucleotide reductase activity associated with the replication of murine cytomegalovirus in quiescent fibroblasts. J. Virol. 7411557-11565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao, W. C., J. Ash, and L. F. Johnson. 1994. Bidirectional promoter of the mouse thymidylate synthase gene. Nucleic Acids Res. 224044-4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25402-408. [DOI] [PubMed] [Google Scholar]
- 51.Maul, G. G. 2008. Initiation of cytomegalovirus infection at ND10. Curr. Top. Microbiol. Immunol. 325117-132. [DOI] [PubMed] [Google Scholar]
- 52.Maul, G. G., and D. Negorev. 2008. Differences between mouse and human cytomegalovirus interactions with their respective hosts at immediate early times of the replication cycle. Med. Microbiol. Immunol. 197241-249. [DOI] [PubMed] [Google Scholar]
- 53.Meier, J. L., and M. F. Stinski. 2006. Major immediate-early enhancer and its gene products, p. 151-166. In M. J. Reddehase (ed.), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Wymondham, Norfolk, United Kingdom.
- 54.Messerle, M., B. Bühler, G. M. Keil, and U. H. Koszinowski. 1992. Structural organization, expression, and functional characterization of the murine cytomegalovirus immediate-early gene 3. J. Virol. 6627-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Messerle, M., I. Crnkovic, W. Hammerschmidt, H. Ziegler, and U. H. Koszinowski. 1997. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 9414759-14763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Messerle, M., G. M. Keil, and U. H. Koszinowski. 1991. Structure and expression of murine cytomegalovirus immediate-early gene 2. J. Virol. 651638-1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nielsen, M., C. Lundegaard, O. Lund, and C. Keçsmir. 2005. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 5733-41. [DOI] [PubMed] [Google Scholar]
- 58.O'Connor, M., M. Peifer, and W. Bender. 1989. Construction of large DNA segments in Escherichia coli. Science 2441307-1312. [DOI] [PubMed] [Google Scholar]
- 59.Overbergh, L., D. Valckx, M. Waer, and C. Mathieu. 1999. Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine 11305-312. [DOI] [PubMed] [Google Scholar]
- 60.Pahl-Seibert, M.-F., M. Juelch, J. Podlech, D. Thomas, P. Deegen, M. J. Reddehase, and R. Holtappels. 2005. Highly protective in vivo function of cytomegalovirus IE1 epitope-specific memory CD8 T cells purified by T-cell receptor-based cell sorting. J. Virol. 795400-5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paulus, C., S. Krauss, and M. Nevels. 2006. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc. Natl. Acad. Sci. USA 1033840-3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Podlech, J., R. Holtappels, N. K. A. Grzimek, and M. J. Reddehase. 2002. Animal models: murine cytomegalovirus, p. 493-525. In S. H. E. Kaufmann and D. Kabelitz (ed.), Methods in microbiology, immunology of infection, 2nd ed., vol. 32. Academic Press, San Diego, CA. [Google Scholar]
- 62a.Qiagen. 2006. DNeasy blood and tissue handbook (manual no. 07/2006). Qiagen, Hilden, Germany.
- 62b.Qiagen. 2007. RNeasy micro handbook (manual no. 12/2007). Qiagen, Hilden, Germany.
- 62c.Qiagen. 2006. RNeasy mini handbook (manual no. 04/2006). Qiagen, Hilden, Germany.
- 63.Rawlinson, W. D., H. E. Farrell, and B. G. Barrell. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 708833-8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reddehase, M. J. 2002. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2831-844. [DOI] [PubMed] [Google Scholar]
- 65.Reddehase, M. J., and U. H. Koszinowski. 1984. Significance of herpesvirus immediate early gene expression in cellular immunity to cytomegalovirus infection. Nature 312369-371. [DOI] [PubMed] [Google Scholar]
- 66.Reddehase, M. J., and U. H. Koszinowski. 1991. Redistribution of critical major histocompatibility complex and T cell receptor-binding functions of residues in an antigenic sequence after biterminal substitution. Eur. J. Immunol. 211697-1701. [DOI] [PubMed] [Google Scholar]
- 67.Reddehase, M. J., W. Mutter, K. Münch, H. J. Bühring, and U. H. Koszinowski. 1987. CD8-positive T lymphocytes specific for murine cytomegalovirus immediate-early antigens mediate protective immunity. J. Virol. 613102-3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reddehase, M. J., J. B. Rothbard, and U. H. Koszinowski. 1989. A pentapeptide as minimal antigenic determinant for MHC class I-restricted T lymphocytes. Nature 337651-653. [DOI] [PubMed] [Google Scholar]
- 69.Reddehase, M. J., C. O. Simon, C. K. Seckert, N. Lemmermann, and N. K. A. Grzimek. 2008. Murine model of cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol., vol 325315-332. [DOI] [PubMed] [Google Scholar]
- 70.Sacher, T., J. Podlech, C. A. Mohr, S. Jordan, Z. Ruzsics, M. J. Reddehase, and U. H. Koszinowski. 2008. The major virus-producing cell type during murine cytomegalovirus infection, the hepatocyte, is not the source of virus dissemination in the host. Cell Host Microbe 3263-272. [DOI] [PubMed] [Google Scholar]
- 71.Sanchez, V., and D. Spector. 2006. Exploitation of host cell cycle regulatory pathways by HCMV, p. 205-230. In M. J. Reddehase (ed.), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Wymondham, Norfolk, United Kingdom.
- 72.Seckert, C. K., A. Renzaho, M. J. Reddehase, and N. K. Grzimek. 2008. Hematopoietic stem cell transplantation with latently infected donors does not transmit virus to immunocompromised recipients in the murine model of cytomegalovirus infection. Med. Microbiol. Immunol. 197251-259. [DOI] [PubMed] [Google Scholar]
- 73.Seglen, P. O. 1972. Preparation of rat liver cells. I. Effect of Ca2+ on enzymatic dispersion of isolated, perfused liver. Exp. Cell Res. 74450-454. [DOI] [PubMed] [Google Scholar]
- 74.Seglen, P. O. 1973. Preparation of rat liver cells. III. Enzymatic requirements for tissue dispersion. Exp. Cell Res. 82391-398. [DOI] [PubMed] [Google Scholar]
- 75.Shenk, T. 2006. Human cytomegalovirus genomics, p. 49-62. In M. J. Reddehase (ed.), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Wymondham, Norfolk, United Kingdom.
- 76.Simon, C. O., R. Holtappels, H.-M. Tervo, V. Böhm, T. Däubner, S. A. Oehrlein-Karpi, B. Kühnapfel, A. Renzaho, D. Strand, J. Podlech, M. J. Reddehase, and N. K. A. Grzimek. 2006. CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J. Virol. 8010436-10456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simon, C. O., B. Kühnapfel, M. J. Reddehase, and N. K. Grzimek. 2007. Murine cytomegalovirus major immediate-early enhancer region operating as a genetic switch in bidirectional gene pair transcription. J. Virol. 817805-7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Simon, C. O., C. K. Seckert, D. Dreis, M. J. Reddehase, and N. K. Grzimek. 2005. Role for tumor necrosis factor alpha in murine cytomegalovirus transcriptional reactivation in latently infected lungs. J. Virol. 79326-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Simon, C. O., C. K. Seckert, N. K. A. Grzimek, and M. J. Reddehase. 2006. Murine model of cytomegalovirus latency and reactivation: the silencing/desilencing and immune sensing hypothesis, p. 483-500. In M. J. Reddehase (ed.), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Wymondham, Norfolk, United Kingdom.
- 80.Stinski, M. F., and H. Isomura. 2008. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med. Microbiol. Immunol. 197223-231. [DOI] [PubMed] [Google Scholar]
- 81.Stinski, M. F., and D. T. Petrik. 2008. Functional roles of the human cytomegalovirus essential IE86 protein. Curr. Top Microbiol. Immunol. 325133-152. [DOI] [PubMed] [Google Scholar]
- 82.Sylwester, A. W., B. L. Mitchell, J. B. Edgar, C. Taormina, C. Pelte, F. Ruchti, P. R. Sleath, K. H. Grabstein, N. A. Hosken, F. Kern, J. A. Nelson, and L. J. Picker. 2005. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 202673-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tang, Q., L. Li, and G. G. Maul. 2005. Mouse cytomegalovirus early M112/113 proteins control the repressive effect of IE3 on the major immediate-early promoter. J. Virol. 79257-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tang, Q., and G. G. Maul. 2003. Mouse cytomegalovirus immediate-early protein 1 binds with host cell repressors to relieve suppressive effects on viral transcription and replication during lytic infection. J. Virol. 771357-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang, Q., and G. G. Maul. 2006. Immediate-early interactions and epigenetic defense mechanisms, p. 131-149. In M. J. Reddehase (ed.), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Wymondham, Norfolk, United Kingdom.
- 86.Tenzer, S., B. Peters, S. Bulik, O. Schoor, C. Lemmel, M. M. Schatz, P. M. Kloetzel, H. G. Rammensee, H. Schild, and H. G. Holzhütter. 2005. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol. Life Sci. 621025-1037. [DOI] [PubMed] [Google Scholar]
- 87.Trinklein, N. D., S. F. Aldred, S. J. Hartman, D. I. Schroeder, R. P. Otillar, and R. M. Myers. 2004. An abundance of bidirectional promoters in the human genome. Genome Res. 1462-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Voigt, S., G. R. Sandford, G. S. Hayward, and W. H. Burns. 2005. The English strain of rat cytomegalovirus (CMV) contains a novel captured CD200 (vOX2) gene and a spliced CC chemokine upstream from the major immediate-early region: further evidence for a separate evolutionary lineage from that of rat CMV Maastricht. J. Gen. Virol. 86263-274. [DOI] [PubMed] [Google Scholar]
- 89.Wagner, M., S. Jonjic, U. H. Koszinowski, and M. Messerle. 1999. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 737056-7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu, Y., S. A. Cei, A. Rodriguez Huete, K. S. Celletti, and G. S. Pari. 2004. Human cytomegalovirus DNA replication requires transcriptional activation via an IE2- and UL84-responsive bidirectional promoter element within oriLyt. J. Virol. 7811664-11677. [DOI] [PMC free article] [PubMed] [Google Scholar]