

Abstract
A novel frontier in the treatment of tumors that are difficult to treat is oncolytic virotherapy, in which a replication-competent virus selectively infects and destroys tumor cells. Herpes simplex virus (HSV) represents a particularly attractive system. Effective retargeting to tumor-specific receptors has been achieved by insertion in gD of heterologous ligands. Previously, our laboratory generated an HSV retargeted to human epidermal growth factor receptor 2 (HER2), a receptor overexpressed in about one-third of mammary tumors and in some ovarian tumors. HER2 overexpression correlates with increased metastaticity and poor prognosis. Because HER2 has no natural ligand, the inserted ligand was a single-chain antibody to HER2. The objective of this work was to genetically engineer an HSV that selectively targets the HER2-expressing tumor cells and that has lost the ability to enter cells through the natural gD receptors, HVEM and nectin1. Detargeting from nectin1 was attempted by two different strategies, point mutations and insertion of the single-chain antibody at a site in gD different from previously described sites of insertion. We report that point mutations at gD amino acids 34, 215, 222, and 223 failed to generate a nectin1-detargeted HSV. An HSV simultaneously detargeted from nectin1 and HVEM and retargeted to HER2 was successfully engineered by moving the site of single-chain antibody insertion at residue 39, i.e., in front of the nectin1-interacting surface and not lateral to it, and by deleting amino acid residues 6 to 38. The resulting recombinant, R-LM113, entered cells and spread from cell to cell solely via HER2.
A novel frontier in the treatment of tumors that are difficult to treat with surgery, radiotherapy, and chemotherapy is oncolytic virotherapy, in which a replication-competent virus infects and destroys tumor cells (3, 47). Herpes simplex virus (HSV) represents a particularly attractive system for a number of reasons (2, 4, 5, 30, 33, 57). Genetic engineering is readily feasible, space is available for the insertion of heterologous genes, and if necessary, HSV replication in humans can be controlled by acyclovir treatment. In numerous preclinical and phase 1 clinical studies, some recombinants proved safe and exerted beneficial effects (20, 22, 26, 31, 44, 45). The best-studied tumor system has been glioblastoma stereotactically injected with HSV recombinants exhibiting conditional replication in dividing tumor cells (32). In general, safety has been obtained at the expense of attenuation (23, 39). More robust viruses are needed that selectively destroy the tumor cells and spare neighboring cells and tissues.
The entry apparatus of HSV is fairly well known. HSV enters cells by fusion of its envelope with plasma or endocytic cell membranes. Fusion is executed by a trio of glycoproteins conserved across the family Herpesviridae, gB and the gH/gL heterodimer (8, 51). What varies in the different herpesvirus species is the virion glycoprotein deputed to recognize specific receptors and, consequently, the cells targeted by each virus. For HSV, this glycoprotein is gD, which is capable of binding three alternative receptors, nectin1, HVEM, and 3-O-sulfated sites on heparan sulfate (3-OS HS) (12, 17, 40, 48). The first two receptors, particularly nectin1, are expressed in most of the tissues targeted by HSV, including cells of nerve tissues (12, 17, 18, 34, 53). The presence of 3-O-modified HS in human tissues and the importance of this interaction in natural infection are less well documented; infection of cultured human corneal cells appears to be mediated by modified HS (55). Structurally, the gD ectodomain is made of an immunoglobulin-folded core (amino acids [aa] 56 to 184) bracketed by N- and C-terminal extensions (10, 28). The HVEM-binding site on gD was clearly identified in the cocrystal of gD and HVEM; it maps at the very N terminus (aa 7 to 32), a segment that is unstructured in gD alone and forms a hairpin in HVEM-bound gD (10). The nectin1-binding site is less well known, is discontinuous, and requires approximately aa 32 to 250. The critical residues identified by mutagenesis are aa 34, 38, 215, 222, and 223 (13, 29, 63). Mutations in the N terminus of HSV type 1 gD that abolish binding to HVEM, located near a positively charged pocket, also abrogate functional interactions with 3-OS HS (59, 60), suggesting a partial overlap between the binding sites for HVEM and 3-OS HS.
The ability of gD to accept foreign sequences and retarget the virus tropism to heterologous receptors is of utmost importance in the design of oncolytic HSVs. Zhou et al. and Kamiyama et al. provided proof of principle that insertion of heterologous ligands in gD yields recombinants that use the ligands' receptors as portals of entry (25, 65). Ideally, the targeted receptors should be selectively expressed or overexpressed in tumor cells and absent or little expressed in normal cells. Virions carrying a gD-interleukin 13 (IL-13) chimera enter glioblastoma cells through the IL-13 receptor (IL-13R) (62, 65). Similarly, virions carrying a gD-urokinase plasminogen activator (uPA) chimera target the uPA receptor (uPAR) (25). In our laboratory, we chose to target HER2 (human epidermal growth factor receptor 2) (36), a receptor overexpressed in about 30% of mammary tumors, as well as in some ovarian tumors, and little, if at all, expressed in other tissues (21, 41, 50). Importantly, HER2 overexpression correlates with increased metastaticity and poor prognosis of these cancers (21, 24). Because of these properties, HER2 is the target of therapy with the humanized monoclonal antibody (MAb) Herceptin (43). An obstacle to the genetic engineering of HER2-retargeted viruses stems from the fact that it is an orphan receptor, i.e., no natural ligand is known. To obviate this limit, we inserted in gD a single-chain antibody (scFv) to HER2 (scHER2), a molecule comparable in size to the gD ectodomain itself (247 versus 310 aa). To our surprise, gD tolerated this large insert, and viruses carrying the gD-scHER2 chimera were able to use HER2 as a receptor (36). The insertion at aa 24 interrupted the HVEM-binding site, and consequently, the recombinant virus was no longer able to infect through HVEM but retained the ability to infect via nectin1.
The strategy employed so far to generate HSV recombinants retargeted to IL-13Rα2, uPAR, and HER2 yielded viruses that are capable of entering through the novel receptors. While detargeting from HVEM has been readily achieved by deletion of or insertion N-terminal sequences (60, 63), detargeting from nectin1 has been a much more demanding task. Some of the difficulties arise from the fact that gD residues critical to the interaction with nectin1 have not been clearly defined. Residues 38, 215, 222, and 223 were implicated by site-directed mutagenesis as being important (13, 29, 56). However, whether they simply affect the gD structure or are responsible for the physical interaction is not known. Furthermore, recombinants carrying these substitutions were not generated; hence, the impact of these residues is still unclear. In one example, the V34S substitution introduced in the IL-13-gD chimera yielded the only nectin1-detargeted virus described so far (63).
The purpose of this study was to generate an oncolytic HSV prototype retargeted to HER2 and detargeted from both HVEM and nectin1. We took two approaches. The first was to introduce the V34S mutation reported to affect gD binding to nectin1 or substitutions at aa 215, 222, and 223. These viruses, named here as second-generation HER2-retargeted recombinants, were not hampered in the ability to use nectin1 as a receptor. The second approach relied on a structure-based prediction (third-generation HER2-retargeted HSV) and consisted of the insertion of scHER2 at a different site in gD so that it would lie in front of the nectin1-interacting surface. At the same time, the N terminus was deleted up to amino acid residue 38. This approach yielded an HSV recombinant that was simultaneously detargeted from HVEM and nectin1 and retargeted to HER2.
MATERIALS AND METHODS
Cells and viruses.
Cells were grown in Dulbecco's modified Eagle medium supplemented with 5% fetal calf serum. The J cell line, a derivative of baby hamster kidney (BHK)-tk− cells that lacks gD receptors, has been described previously (12). J-hNectin1, J-mNectin1, J-HER2, J-HVEM, and R6, a gD-complementing cell line, were described previously (12, 36, 37, 61). TT12.E2 is a mouse cell line expressing rat HER2 (16). The SKOV3 (a human ovary adenocarcinoma cell line expressing HER2 at high levels) and MCF7 (a breast adenocarcinoma cell line expressing HER2 at intermediate or low levels) cell lines were grown in RPMI+Glutamax-I medium (Gibco) supplemented with 10% fetal calf serum. RH4 cells are HER2-negative human rhabdomyosarcoma cells (46). Viruses were grown in RS (rabbit skin), BHK, or J-HER2 cells and titrated by plaque assay in Vero or SKOV3 cells.
Construction of gD-minus HSV-BAC.
The first step in the generation of recombinant HSVs through bacterial artificial chromosome (BAC) technology was the generation of a gD-minus HSV-BAC genome by means of the “ET-cloning” procedure in bacteria. A kanamycin (Kan) resistance cassette flanked by two FLP recombination target (FRT) sites was PCR amplified from the plasmid pFRT-2 with primers that contained at their 5′ ends 60 nucleotides of sequence flanking the gD open reading frame: gDup_Kan_f (TGT TCG GTC ATA AGC TTC AGC GCG AAC GAC CAA CTA CCC CGA TCA TCA GTT ATC CTT AAG CCA GTG AAT TCG AGC TCG GTA C) and gDdown_Kan_r (ACT TAT CGA CTG TCC ACC TTT CCC CCC TTC CAG ACT CGC TTT ATA TGG AGT TAA GGT CCC GAC CAT GAT TAC GCC AAG CTC C). pFRT-2 was constructed by insertion of the Kan resistance cassette derived from pCP15 into the NsiI sites of pCP16 (11), replacing the tetracycline resistance gene. The PCR product was electroporated into Escherichia coli DH10B harboring YEbac102 HSV-BAC (54) and transiently expressing lambda phage Red-β and Red-γ recombinases from the pKD46 plasmid (15). Recombinant clones were selected on plates containing 25 μg/ml Kan (the marker contained in the PCR product) and 20 μg/ml chloramphenicol (Cam) (the marker contained in HSV-BAC sequences) to ensure replacement of the gD coding sequence by the Kan resistance cassette. To remove the Kan cassette, the positive clones were electroporated with pCP20 (11), a plasmid expressing Saccharomyces cerevisiae FLP recombinase, which targets FRT sequences. Finally, the colonies were assayed for loss of the Kan marker and for Cam resistance. The resulting gD-minus HSV-BAC genome, designated 102gD-FRT, was checked by PCR and sequencing of relevant insertions and for the ability to form plaques only in R6 and not in other cell lines.
Construction of gD-minus HSV-BAC backbones containing EGFP or LacZ reporter genes.
The second step in the engineering of HSV-BAC recombinants was the insertion of a reporter gene, for either enhanced green fluorescent protein (EGFP) or LacZ, which resulted in the generation of gD-minus-EGFP-HSV-BAC, or gD-minus-LacZ-HSV-BAC, respectively. We chose as the site of the reporter gene insertion the pBeloBAC sequences themselves, so that the marker gene could be deleted together with the BAC sequences by Cre recombinase if required. The coding sequence of EGFP followed by the polyadenylation signal from bovine growth hormone was PCR amplified from pCMS-EGFP (Clontech) with primers EGFP_BamHI_f (5′-CAA CCC GGG ATC CAC CGG TCG CCA CCA TGG TGA GC-3′) and EGFP+pA_BamHI_r (5′-CCC CTT GGG ATC CTG CCC CAC CCC ACC CCC CAG AAT AG-3′) and cloned downstream of the HSV α27 promoter. The α27-EGFP cassette was inserted between two 700-bp sequences PCR amplified from the plasmid pBeloBac11 (GenBank accession no. U51113). The amplimers were designated pBeloBac11-up (primers Sal_pBelo_1209_f [5′-TTG CCA GTC GAC ATT CCG GAT GAG CAT TCA TCA GGC GGG CA-3′] and pBelo_1897_Xho_r [5′-GCA AAA ACT CGA GTG TAG ACT TCC GTT GAA CTG ATG GAC-3′]) and pBeloBac11-down (primers Mun_pBelo_1898_f [5′-GGA AGT CAA TTG GAA GGT TTT TGC GCT GGA TGT GGC TGC CC-3′] and pBelo_2586_Eco_r [5′-CAC ACT GAA TTC GCA ATT TGT CAC AAC ACC TTC TCT AGA AC-3′]). In the resulting construct, the α27-EGFP cassette was inserted between nucleotides 1897 and 1898 (original coordinates) of pBeloBac11. The α27-EGFP cassette plus the pBeloBac11 flanking sequences were subcloned into the shuttle vector pST76KSR (1) for homologous recombination in bacteria. For LacZ insertion, we followed the same strategy, cloning pBeloBac11-up and -down sequences into a plasmid already containing the α27-LacZ cassette. The relevant insert and adjacent regions were sequenced for accuracy in all plasmids.
Construction of shuttle vectors for insertion of chimeric gD into gD-minus BACs.
The gD shuttle vector named pS31 carries scHER2L (scFv anti-HER2 plus a 9-aa serine glycine linker) inserted between amino acid residues 24 and 25 of gD, plus the V34S substitution (Fig. 1B, b). It was constructed as follows. First, the V34S substitution was introduced by site-directed mutagenesis in pLM13, a construct carrying scHER2L inserted between amino acid residues 24 and 25 of gD. Mutagenesis was performed by means of the Stratagene Quickchange II kit with primers gD_34S_StuI (5′-TCC TCC GGG GAG CCG GCG CGT GTA CCA CAT CCA GGC AGG CCT ACC GG-3′) and its reverse. The primers contained the indicated silent restriction sites for ease of mutant-clone screening. Next, the cassette containing the mutagenized gD-scHER2L plus gD genomic upstream and downstream flanking sequences (about 500 bp each) was transferred to the pST76KSR shuttle vector to enable homologous recombination in E. coli (see the supplemental material).
FIG. 1.

(A) Schematic representation of the recombinant HSV-BACs generated in this study. The backbone of gD-minus-EGFP-HSV-BAC is shown as an example. The HSV-BACs are derived from pYEbac102 (54), which carries pBeloBAC11 sequences inserted between UL3 and UL4. In gD-minus-EGFP-HSV-BAC, the reporter cassette (α27-EGFP) is inserted in the BAC sequences. gD-minus-LacZ-HSV-BAC has the same structure but carries LacZ in place of EGFP. (B) Schematic representations of wt gD (a) and the gD-scHER2 chimeric proteins. gD of recombinant R-LM31 carries a substitution at amino acid residue 34 (b); gD of recombinant R-LM39 carries mutations at amino acid residues 34, 215, 222, and 223 (c); and gD of recombinant R-LM113 carries scHER2 in place of amino acid residues 6 to 38 (d). The boldface numbers indicate the length in amino acid residues of each fragment. The lightface numbers refer to amino acid residues according to wt gD coordinates. Mutated residues are indicated by ovals (Table 1). For each virus expressing wt or chimeric gD, the pattern of receptor recognition is summarized in the right-hand columns as +, −, or not determined (nd). TM, transmembrane domain. VH and VL, heavy- and light-chain variable domains of the anti-HER2 antibody 4D5; Δ, deletion. In panel B, the bars are drawn to scale.
To construct pS39, the D215G, R222N, and F223I substitutions were added to gD cloned in pS31 by means of the primer gD_215G-222N-223I_PvuI (5′-AGG GGG TGA CGG TGG GCT CGA TCG GGA TGC TGC CCA ACA TCA TCC CCG AGA ACC-3′) and its reverse (Fig. 1B, c).
pS113 is a shuttle vector containing gD, in which amino acid residues 6 to 38 were deleted and replaced with scHER2L (Fig. 1B, d). To generate this construct, two restriction sites were sequentially introduced into the gD open reading frame, namely, EcoRI at the sequence coding for aa 6 to 8 and BamHI at the sequence coding for aa 37 to 39, by means of the mutagenic primers gD_6/8_EcoRI_f (5′-CAA ATA TGC CTT GGC GGA GAA TTC TCT CAA GAT GGC CG-3′) and gD_37/38_BamHI_f (5′-CGG GGG TCC GGC GCG GAT CCC ACA TCC AGG CGG G-3′), respectively. The insertion of the EcoRI site introduced the substitutions D6E and A7N. scHER2L was amplified from pS2019a (49) with primers scFv_x6_Eco_f (5′-GCA AAG GAA TTC CGA TAT CCA GAT GAC CCA GTC CCC G-3′) and scFv_SG_x37_BamH (5′-CGG AGG ATC CAC CGG AAC CAG AGC CAC CGC CAC TCG AGG-3′). The final shuttle plasmid, pS113, was constructed by subcloning the engineered gD, along with genomic flanking sequences, into pST76KSR. The relevant insert and adjacent regions were sequenced for accuracy in all plasmids.
Generation of recombinant genomes by two-step replacement in bacteria.
The procedure applied to generate recombinant genomes in E. coli was essentially as described previously, with slight modifications (6, 38, 42). Briefly, electrocompetent E. coli DH10B harboring the relevant gD-minus HSV-BAC genomes was electroporated with the shuttle vector in 0.2-cm electroporation cuvettes (Bio-Rad) at 200 Ω, 25 μF, and 2.5 kV; plated on LB agar containing 25 μg/ml Kan (the shuttle vector's marker) and 20 μg/ml Cam (the BAC's marker); and incubated at 30°C overnight to allow the expression of RecA from the shuttle vector. The clones were replated onto LB plus Kan plus Cam at 43°C to allow the identification of those harboring the cointegrates (visible as large colonies compared to the temperature-sensitive “small-colony” phenotype determined by nonintegrated shuttle vectors). Subsequently, the cointegrates were allowed to resolve by plating the clones onto LB plus Cam at 30°C, and clones containing the resolved HSV-BAC were selected on LB-plus-Cam plates supplemented with 10% sucrose. Finally, the clones were checked for loss of Kan resistance and for the presence of the desired insert by colony PCR.
gD-minus-EGFP-HSV-BAC and gD-minus-LacZ-HSV-BAC.
Recombination between the 102gD-FRT HSV-BAC and the appropriate shuttle vectors generated gD-minus-EGFP-HSV-BAC or gD-minus-LacZ-HSV-BAC DNAs, which contained the α27 promoter-EGFP (or α27 promoter-LacZ) cassette inserted into the BAC sequences (Fig. 1A). The viruses were reconstituted by transfection of the BAC DNA into the gD-complementing R6 cells.
The gD-minus HSV-BACs were used as recipients for the generation of recombinants containing the engineered gD (Fig. 1 and Table 1; see Fig. S1 in the supplemental material). The recombinant genomes were checked by PCR and sequencing of relevant insertions. The viruses were reconstituted by transfection of the BAC DNAs into R6 cells, followed by a single passage in BHK cells and subsequent growth in J-HER2 cells. The virus stocks were grown in J-HER2 cells and serially passaged for 11 passages. The virus titer was determined in SKOV3 or J-HER2 cells; the figures obtained in the two cell lines were very similar.
TABLE 1.
Summary of recombinant viruses generated in this study
| Virus | Reporter gene | scHER2L insert between aa: | Changes in gD aimed at modifying receptor binding | Technology |
|---|---|---|---|---|
| R-LM5 | EGFP | None | None | Homologous recombination in mammalian cells |
| R-LM13 | EGFP | 24-25 | None | Homologous recombination in mammalian cells |
| R-LM31 | LacZ | 24-25 | V34S | BAC |
| R-LM39 | EGFP | 24-25 | V34S, D215G, R222N, F223I | BAC |
| R-LM113 | EGFP | 5-39 | Δ6-38 | BAC |
Generation of recombinant viruses R-LM5 and R-LM13 by homologous recombination in mammalian cells.
R-LM5, a recombinant virus carrying wild-type (wt) gD and an EGFP reporter in HSV BAC, was generated by homologous recombination in mammalian cells. RS cells were cotransfected with DNA from gD-minus-EGFP-HSV-BAC and pLM5, a plasmid carrying the coding sequences of wt gD flanked by 500 bp of genomic upstream and downstream flanking sequences. R-LM13 was generated in a similar fashion, by means of the pLM13 plasmid, which carries scHER2L inserted in gD between aa 24 and 25.
Infection assays.
Cells were infected at an increasing multiplicity of infection (MOI) in 96-well plates: after 90 min of absorption, the viral inoculum was removed. Infection of EGFP-expressing viruses was monitored by means of a Zeiss Axioplan fluorescence microscope and measured as fluorescence 24 h later by means of a Synergy HTTR-I fluorometer (Bio-Tek).
Inhibition of virus infection by antibodies.
SKOV3 cells grown in 96-well plates were incubated for 2 h on ice with increasing concentrations of antibodies (R1.302 against nectin1, Herceptin against HER2, or mouse immunoglobulins) diluted in Dulbecco's modified Eagle medium without serum and then with the viral inoculum at an MOI of 2 PFU/cell (as titrated in SKOV3 cells) for a further 90 min on ice. Following virus adsorption, the unattached virus was removed and the cells were washed twice with ice-cold RPMI+Glutamax supplemented with 2.5% fetal bovine serum. The cells were overlaid with medium containing the same concentration of antibodies or immunoglobulin G (IgG), rapidly shifted to 37°C, and incubated for 16 h. Infection was quantified as the EGFP fluorescence intensity by means of a Victor plate reader (Perkin Elmer). The 100% value represented data obtained with cells infected with virus in the absence of antibodies.
Virus replication assay.
J, J-hNectin1, J-HVEM, J-HER2, SKOV3, I-143 tk−, and HEp-2 cells grown in 12-well plates were infected with the indicated viruses at 1 PFU (as determined in SKOV3 cells)/cell for 90 min at 37°C. Following virus adsorption, the inoculum was removed and the unpenetrated virus was inactivated by means of an acid wash (40 mM citric acid, 10 mM KCl, 135 mM NaCl [pH 3]) (7). Replicate cultures were frozen at the indicated times (3, 24, and 48 h) after infection. The progeny virus (intracellular plus extracellular) was titrated in SKOV3 cells.
Inhibition of plaque formation by Herceptin.
SKOV3 cells were infected with serial dilutions of the indicated viruses and overlaid with medium containing 1% SeaPlaque agarose, with or without the addition of 10 μg/ml Herceptin (MAb to HER2). Fluorescent plaques were monitored with a Zeiss fluorescence microscope, and pictures were taken 48 h after infection. The areas of the plaques were measured with the Photoshop histogram tool.
RESULTS
Second-generation HER2-retargeted recombinants carrying single or multiple substitutions maintain tropism for nectin1.
The first-generation recombinants R-LM11 and R-LM11L carried scHER2 inserted between amino acid residues 24 and 25 of gD (36). The insertion altered the N terminus so that entry through HVEM was hampered while entry through nectin1 was maintained (36). The first attempt to generate a nectin1-detargeted recombinant consisted of the insertion of the V34S mutation into gD-scHER2 (Fig. 1B and Table 1). When introduced into the IL-13-gD chimera, the V34S substitution strongly decreased entry via nectin1 (63). The recombinant LM31-BAC DNA was generated by homologous recombination in E. coli (Table 1) (see Materials and Methods and the supplemental material). The recipient genome was gD-minus-LacZ-HSV-BAC. The R-LM31 recombinant virus was obtained by transfection of the LM31-BAC DNA into the gD-complementing R6 cells. R-LM31 tropism was assayed in J cells expressing human or murine nectin1 or human HER2 and monitored as β-galactosidase activity. As shown in Fig. 2, the R-LM31 recombinant infected J-nectin1 cells (via either the human or murine receptor); hence, it was not detargeted from nectin1. This result indicates that the effect of the V34S substitution varies depending on the insert present in gD.
FIG. 2.
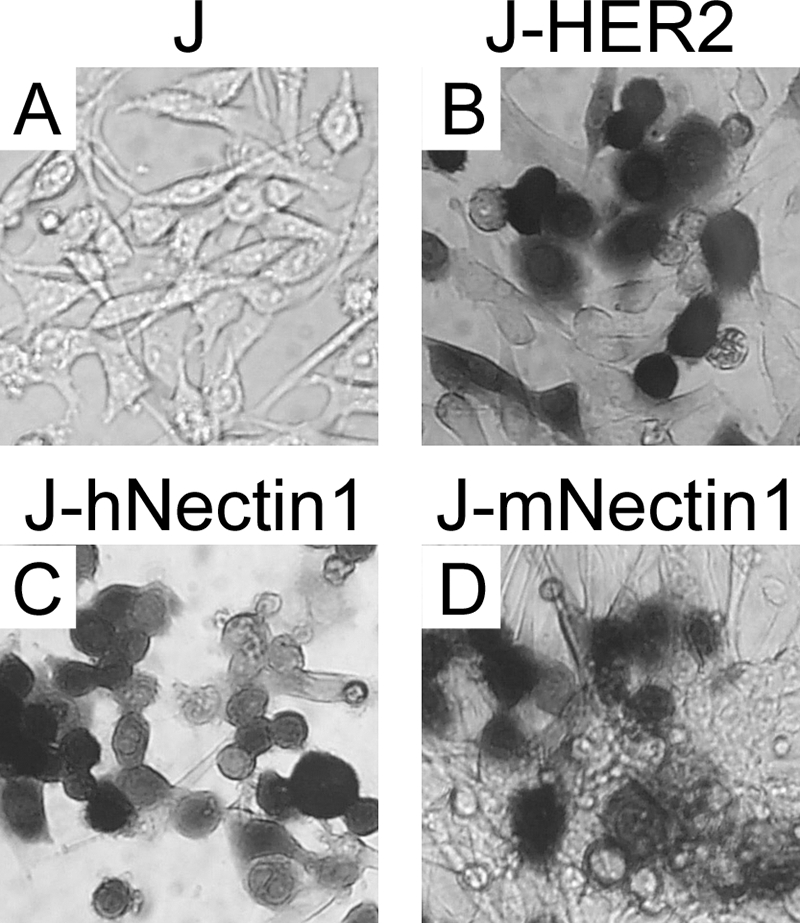
The recombinant virus R-LM31 is not detargeted from the nectin1 receptor. Shown are micrographs of receptor-negative J cells (A) and J-HER2 (B), J-hNectin1 (C), and J-mNectin1 (D) expressing human HER2 and human or murine nectin1, respectively, exposed to R-LM31 at 10 PFU/cell. Infection was monitored as β-galactosidase activity by in situ X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) staining 16 h after infection.
To enable detection of virus growth in live cells, all subsequent recombinants carried EGFP, and not LacZ, as the reporter gene (Table 1). The prototypic virus in this series carried wt gD and was named R-LM5. R-LM13 carried the gD-scHER2L chimera without mutations. Next, the D215G, R222N, F223I, and V34S substitutions (29, 56) were inserted into the chimeric gD from R-LM13, thus generating R-LM39. The electrophoretic mobility in denaturing gels of R-LM39 gD was similar to that of R-LM13 and slower than that of R-LM5 gD, as expected (Fig. 3). R-LM39 was characterized with respect to replication in J cells expressing individual receptors and in human cell lines expressing or not expressing HER2 and with respect to its ability to be blocked by a MAb against nectin1 (MAb R1.302) or against HER2 (Herceptin). The main results were as follows (Fig. 4A to G). R-LM39 was unable to grow in J-HVEM cells but replicated in J-HER2 and J-nectin1 cells, implying that it could use both HER2 and nectin1 as receptors (Fig. 4B, C, and D). Accordingly, it replicated in the human cell line SKOV3, which expresses both nectin1 and HER2, as well as in I-143 tk− and HEp-2, which express nectin1 (Fig. 4E, F, and G). Receptor usage was more directly assessed in experiments in which virus entry was blocked by Herceptin (directed against HER2), MAb R1.302 (directed against nectin1), or a mixture of the two antibodies (Fig. 5). Replicate aliquots of virions were added to cells preincubated with decreasing concentrations of the antibodies and then allowed to infect SKOV3 cells, which express both HER2 and nectin1 receptors. The results in Fig. 5 show that R-LM39 was not blocked by Herceptin alone or R1.302 alone, but only by the two antibodies in combination (Fig. 5A). The results imply that R-LM39 can use nectin1 or, alternatively, HER2 as a receptor, further documenting the lack of detargeting from nectin1. Thus, unexpectedly, the mutations V34S, D215G, R222N, and F223I were unable to detarget HSV tropism from nectin1. We ascertained that no mutations other than those introduced on purpose were present in R-LM39 gD. Previously, the effects of the D215G, R222N, and F223I mutations were assayed by infectivity complementation assay or soluble-gD-mediated infection (29, 56), and the two assays may have underestimated the residual ability to enter via nectin1. These results underscore the need to assay the effects of the mutations in the context of the viral genome.
FIG. 3.
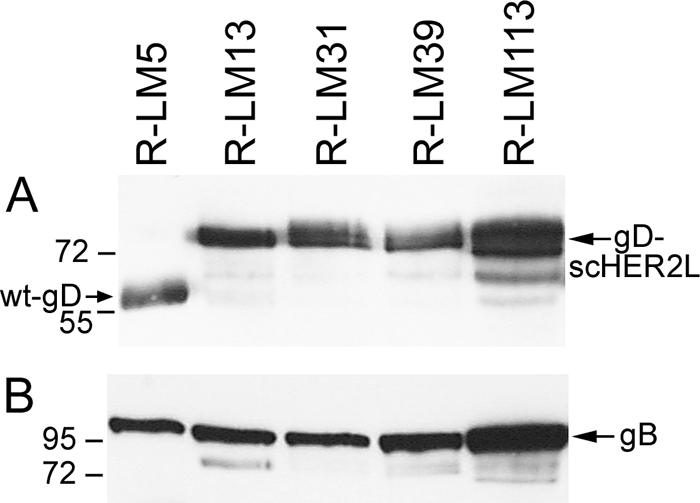
Electrophoretic mobility of chimeric scHER2-gDs (A) or gB (B) expressed in SKOV3 cells infected with R-LM5, R-LM13, R-LM39, or R-LM113. The infected cell lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and visualized by Western blotting with MAb BD80 against the C-terminal portion of the gD ectodomain (A) or MAb H1817 against gB (B), followed by peroxidase-conjugated anti-mouse IgG and enhanced chemiluminescence. In the recombinants, the insertion of scHER2L in gD resulted in slower electrophoretic mobility. The numbers on the left represent the migration positions of molecular mass markers (in kilodaltons).
FIG. 4.

Growth of R-LM39 and R-LM113 recombinants and of control viruses R-LM5 and R-LM13. (A to G) Replicate cultures of J (A), J-Nectin1 (B), J-HVEM (C), J-HER2 (D), SKOV3 (E), I-143 tk− (F), and HEp-2 (G) cells were infected with R-LM5, R-LM13, R-LM39, or R-LM113 at 1 PFU/cell. Progeny virus was harvested at 3, 24, and 48 h after infection and titrated in SKOV3 cells. (H) Viral growth values at 48 h. (I) Infection of an array of cell lines with R-LM113. Monolayers were infected at 5 PFU/cell. Infection was quantified at 24 h as EGFP expression by means of a fluorometer. R.F.U., relative fluorescence units.
FIG. 5.

Blocking of infection of SKOV3 cells with R-LM39 (A) or R-LM113 (B) by antibodies to HER2 (Herceptin) or nectin1 (R1.302). SKOV3 cells were preincubated with the indicated concentrations of purified IgG from Herceptin, R1.302, the combination of Herceptin plus R1.302 or irrelevant mouse IgGs for 2 h at 4°C. For the combination of the two antibodies, the figures on the x axis indicate the concentration of each antibody. Virus was added to the antibody-containing medium and allowed to adsorb to the cells for 90 min at 4°C. Infection was monitored 16 h later as EGFP expression. One hundred percent represents the EGFP readings in untreated virus-infected cultures.
The third-generation recombinant carrying scHER2 in place of aa 6 to 38 of gD is detargeted from nectin1.
The design of the third-generation recombinant was structure based. Inspection of the wt gD structure showed that scHER2 insertion at residue 39 would locate the insert in front of the nectin1-reacting surface (identified by residues 34 and 38 and 215, 222, and 223) (29) and not lateral to it, as happens when scHER2 is inserted at residue 24. Given that scHER2 is comparable in overall size to the gD ectodomain (247 versus 310 aa), we hypothesized that the insert might render the nectin1-interacting surface inaccessible. Furthermore, amino acid residues 6 to 38 were deleted in order to eliminate entry through HVEM (9, 10) and, at the same time, to eliminate V34 and Y38, two residues reported to play some role in the interaction with nectin1 (13, 63). The possibility that steric hindrance might be sufficient to preclude receptor binding stemmed from the finding that scHER2 insertion between aa 24 and 25 abrogated entry via HVEM in R-LM11 and R-LM11L, even though the HVEM-reacting sequences were still present (36). For virus stock production, R-LM113 was grown in J-HER2 cells; the virus has been stable after 11 serial passages. We analyzed the electrophoretic mobility of R-LM113 gD in denaturing gels and observed that it was slower than that of wt gD and indistinguishable from that of R-LM39 gD (Fig. 3), as expected. However, R-LM113 gD accumulated in smaller amounts than wt gD or its other, chimeric forms; thus, in Fig. 3, the lane marked R-LM113 was loaded with a 10-fold-greater amount of lysate than the other lanes. The amounts of gD strictly mirror the amounts of gB in the same virus stocks (Fig. 3B). The smaller amounts of gD and gB in R-LM113 likely reflect the lesser extent of replication (Fig. 4).
Assay of R-LM113 tropism showed that the virus maintained the retargeting to HER2 and was detargeted from both nectin1 and HVEM, as follows. First, we determined R-LM113's ability to infect and replicate in cell lines expressing a single (J-Nectin1, J-HVEM, or J-HER2) or multiple (SKOV3) receptors or commonly used human cell lines. We compared R-LM113 to the recombinant viruses R-LM5 and R-LM13 carrying wt gD or the gD-scHER2 chimera, respectively (Fig. 4A to G). For every cell line, the input MOI was 1 PFU (as determined in SKOV3 cells)/cell. Of note, the ratios of the genome copy numbers to the numbers of PFU were similar for all viruses, irrespective of the cells in which the viruses were grown (see the supplementary material); thus, measurement of virus particles through two different assays resulted in essentially similar figures. The main results were as follows. (i) R-LM113 grew efficiently in J-HER2 cells; the virus titer was comparable to that of R-LM13 in the same cells (Fig. 4D) and about 1 order of magnitude lower than that of R-LM13 in J-nectin1 cells (compare Fig. 4B and D). In SKOV3 cells, R-LM113 replicated to titers only 1 to 1.5 orders of magnitude lower than those of the control viruses R-LM5 and R-LM13 and of R-LM39 (Fig. 4E). (ii) R-LM113 was detargeted from both nectin1 and HVEM, as assessed by its inability to grow in J-nectin1 and J-HVEM cells, as well as in the human I-143 tk− and HEp-2 cells, to titers higher than 103 to 104 PFU/ml (Fig. 4B, C, F, and G). The comparative yields of progeny virus/cell for the different viruses are summarized in Fig. 4H, which underscores the inability of R-LM113 to grow in cells that express nectin1 or HVEM as sole receptors and, at the same time, the ability to replicate in the HER2-expressing SKOV3 cells. Because replication in different cell lines reflects not only the different receptor usage, but also the intrinsic abilities of different cells to support virus growth, as a more direct measure of receptor usage we monitored R-LM113 EGFP expression in a number of cell lines of human and rodent origin. Extensive literature on HSV entry supports the contention that expression of a reporter gene (EGFP) placed under an immediate-early promoter closely mirrors the ability to enter, and thus the receptor usage in the cell under examination. The results in Fig. 4I show that R-LM113 infected J-HER2 cells, but not J cells expressing human nectin1 or murine nectin1. The detargeting from murine nectin1 was confirmed by failure to infect L and NIH 3T3 cells. Human cells were susceptible to R-LM113, provided that they expressed HER2 at a high level (SKOV3 cells). The MCF7 cells, which express HER2 at low to intermediate levels (14), could not be infected. For nectin1 and HVEM, it has been shown that cells that express receptors at low density can be infected only at very high MOIs (27). Thus, the lack of MCF7 infection likely reflects the very low density of HER2 (14). Human HER2-negative cells, e.g., HEp-2, I-143 tk−, and RH4 cells, were infected at a low or negligible level. Interestingly, R-LM113 was specific for human HER2, as it failed to infect the TT12.E2 mouse cell line expressing the rat ortholog of HER2 (neu-NT) (16).
Receptor usage was confirmed in virus-blocking experiments with Herceptin, MAb R1.302, or a mixture of the two antibodies, as detailed above. The results in Fig. 5 show that R-LM113 was blocked by Herceptin. The combination of Herceptin plus MAb R1.302 exerted the same inhibition as Herceptin alone; MAb R1.302 had no effect (Fig. 5B). The results with R-LM113 are in sharp contrast to those obtained with R-LM39, which is capable of infecting J-nectin1 cells. R-LM39 infection could be inhibited only by the mixture of Herceptin plus MAb R1.302 (Fig. 5A). R-LM113 infection was inhibited by Herceptin alone, while MAb R1.302 alone had no effect. We conclude that R-LM113 can enter cells only through the HER2 receptor, in accordance with the results shown in Fig. 4.
Finally, we asked whether R-LM113 used HER2, not only for virus infection, but also for cell-to-cell spread. As shown in Fig. 6, exposure of R-LM13-, R-LM39-, and R-LM113-infected SKOV3 monolayers to Herceptin reduced the plaque size. The plaque size of R-LM5 was not reduced by Herceptin.
FIG. 6.

(A) Inhibition of cell-to-cell spread by Herceptin. SKOV3 cells infected with serial dilutions of the indicated viruses were overlaid with medium containing 1% SeaPlaque agarose ± 10 μg/ml Herceptin. Individual plaques were photographed at 48 h, and the plaque areas were measured by means of the Photoshop histogram tool and expressed as pixels. The histograms represent averages; the error bars represent standard deviations. (B) Representative plaques.
DISCUSSION
The ideal virus for selective destruction of cancer cells is one that infects only cancer cells by virtue of altered receptor usage and is unable to infect the cells naturally targeted by the virus. The host range of HSVs can be altered by genetic manipulation of the receptor-binding glycoprotein gD, which is able to interact with the natural receptors nectin1 and HVEM. Chimeric forms of gD carrying heterologous ligands to the receptor of choice redirect the virus to the ligands' receptors. By this strategy, HSV retargeted to IL-13Rα2 and uPAR was generated (25, 62, 65). An inherent difficulty arises with receptors like HER2, for which no natural ligand is available. In this instance, the selected ligand was a single-chain antibody. Surprisingly, gD tolerated the insertion and could mediate virus entry into cells that expressed HER2 as the receptor (36). What has been more difficult to achieve is detargeting from natural receptors, a feature common to other retargeted viruses, adenovirus, adeno-associated virus, and measles virus (19, 35, 52, 58). Here, we have reported the successful construction of a candidate oncolytic HSV retargeted to the HER2 receptor and detargeted from both HVEM and nectin1.
Detargeting from HVEM has been readily achieved in the past (36, 63, 64). Two features have contributed to the ease of detargeting. The HVEM-binding site on gD is well defined. Residues involved in the physical interaction are localized to a small, continuous N-terminal region (aa 7 to 32) that can be deleted from gD without impairing binding to nectin1 and the profusion activity of gD (10, 59, 60). By contrast, detargeting from nectin1 has not been achieved so far, except in one case (63). The reasons for that are twofold; the nectin1-binding site is not known in detail and appears to be a discontinuous rather than a continuous region. Furthermore, residues critical for the nectin1-binding site, identified by mutagenesis, have not been mutated in virions; rather, the effect of their replacement has been assayed in infectivity complementation assays or in soluble-gD-mediated infection (13, 29, 56). The only virus generated so far that was debilitated in entry via nectin1 is the IL-13-retargeted R5141 carrying the V34S substitution (63). As reported here, when the same substitution was engineered in the HER2-retargeted R-LM31, it did not decrease entry through nectin1. Thus, the effect of the V34S substitution is not universal but appears to be dependent on the resulting conformation of gD. It is likely that the presence of the IL-13 insert confers upon gD a structure that is further modified by the V34S substitution so that the nectin1-binding site is either deformed or not readily accessible. Insertion of the triple substitutions at aa 215, 222, and 223 also did not decrease entry through nectin1. As mentioned above, recombinants carrying these substitutions were not generated previously; hence, the effects of these substitutions in wt gD viruses are unknown.
The rationale for the design of R-LM113 was to rotate the position of the scHER2 insert so that it would be located in front of, and not lateral to, the nectin1-interacting surface and, at the same time, to delete the N-terminal sequences up to aa 38, thus removing aa 34 and 38, which have been described as potentially important for nectin1 binding. This strategy indeed resulted in the generation of a nectin1-detargeted HSV. Because of the Δ6-38 deletion, R-LM113 was also simultaneously detargeted from HVEM. Given that mutations that hamper HVEM binding also hamper interaction with 3-OS HS and the partial overlap between the binding sites for HVEM and 3-OS HS (59, 60), R-LM113 is expected to also be detargeted from 3-OS HS. In any case, the importance of the latter receptor to the infection of human tissues remains to be determined. Importantly, the insertion at position 38 did not affect the ability of scHER2 to interact with HER2, the ability of gD to respond to heterologous receptor binding by adopting a conformation suitable for the subsequent steps in virus entry, or the ability of gD to recruit/activate the downstream glycoproteins gB and gH/gL, responsible for fusion execution. Although we did not formally prove it, given that the strategy adopted here to generate a nectin1-detargeted HSV was designed on the wt gD structure, it seems likely that it is generally applicable. Furthermore, the growth curves reported in Fig. 4 indicate that the modifications in R-LM113 allow replication at titers just 1 order of magnitude lower than those attained with wt gD viruses. Cumulatively, the tumor-restricted tropism coupled to relatively high virus yields makes R-LM113 a good candidate oncolytic HSV. Furthermore, taking into consideration that retargeting to HER2 has been obtained through insertion of a single-chain antibody, the effective retargeting to a heterologous receptor, including an orphan receptor with no natural ligand, and the simultaneous detargeting from HVEM and nectin1 described here appear to be a generally applicable strategy in the design of oncolytic HSVs.
Supplementary Material
Acknowledgments
We thank Genentech (San Francisco, CA) for the gift of Herceptin and cDNA encoding the scFv to HER2; Gary Cohen and Roselyn Eisenberg (University of Pennsylvania, Philadelphia) for antibodies; Pier-Luigi Lollini, Patrizia Nanni, and Carla De Giovanni (University of Bologna) for cell lines; and Yasushi Kawaguchi for pYEbac102 HSV-BAC. We are indebted to Elisabetta Romagnoli for invaluable help with cell cultures. We thank Albert Zimmermann (University of Düsseldorf) for help and discussions. Real-time PCR was carried out at CRBA (Centro unificato di Ricerca Biomedica Applicata, S. Orsola-Malpighi Hospital, Bologna, Italy).
This work was supported by a TargetHerpes EU grant (LSHG-CT-2006-037517) to G.C.-F. and H.H., by PRIN-MIUR, by the University of Bologna (RFO), by Fondo Pallotti and Alma Medicina grants, and by a FEMS-ESCMID Fellowship 2006-1 to L.M.
Footnotes
Published ahead of print on 6 August 2008.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Adler, H., M. Messerle, M. Wagner, and U. H. Koszinowski. 2000. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J. Virol. 746964-6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Advani, S. J., R. R. Weichselbaum, R. J. Whitley, and B. Roizman. 2002. Friendly fire: redirecting herpes simplex virus-1 for therapeutic applications. Clin. Microbiol. Infect. 8551-563. [DOI] [PubMed] [Google Scholar]
- 3.Aghi, M., and R. L. Martuza. 2005. Oncolytic viral therapies—the clinical experience. Oncogene 247802-7816. [DOI] [PubMed] [Google Scholar]
- 4.Andreansky, S., L. Soroceanu, E. R. Flotte, J. Chou, J. M. Markert, G. Y. Gillespie, B. Roizman, and R. J. Whitley. 1997. Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human malignant brain tumors. Cancer Res. 571502-1509. [PubMed] [Google Scholar]
- 5.Andreansky, S. S., B. He, G. Y. Gillespie, L. Soroceanu, J. Markert, J. Chou, B. Roizman, and R. J. Whitley. 1996. The application of genetically engineered herpes simplex viruses to the treatment of experimental brain tumors. Proc. Natl. Acad. Sci. USA 9311313-11318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borst, E. M., G. Hahn, U. H. Koszinowski, and M. Messerle. 1999. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: a new approach for construction of HCMV mutants. J. Virol. 738320-8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunetti, C. R., R. L. Burke, B. Hoflack, T. Ludwig, K. S. Dingwell, and D. C. Johnson. 1995. Role of mannose-6-phosphate receptors in herpes simplex virus entry into cells and cell-to-cell transmission. J. Virol. 693517-3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campadelli-Fiume, G., M. Amasio, E. Avitabile, A. Cerretani, C. Forghieri, T. Gianni, and L. Menotti. 2007. The multipartite system that mediates entry of herpes simplex virus into the cell. Rev. Med. Virol. 17313-326. [DOI] [PubMed] [Google Scholar]
- 9.Carfi, A., H. Gong, H. Lou, S. H. Willis, G. H. Cohen, R. J. Eisenberg, and D. C. Wiley. 2002. Crystallization and preliminary diffraction studies of the ectodomain of the envelope glycoprotein D from herpes simplex virus 1 alone and in complex with the ectodomain of the human receptor HveA. Acta Crystallogr. D 58836-838. [DOI] [PubMed] [Google Scholar]
- 10.Carfi, A., S. H. Willis, J. C. Whitbeck, C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and D. C. Wiley. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8169-179. [DOI] [PubMed] [Google Scholar]
- 11.Cherepanov, P. P., and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1589-14. [DOI] [PubMed] [Google Scholar]
- 12.Cocchi, F., L. Menotti, P. Mirandola, M. Lopez, and G. Campadelli-Fiume. 1998. The ectodomain of a novel member of the immunoglobulin superfamily related to the poliovirus receptor has the attributes of a bonafide receptor for herpes simplex viruses 1 and 2 in human cells. J. Virol. 729992-10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connolly, S. A., D. J. Landsburg, A. Carfi, C. J. Whitbeck, Y. Zuo, D. C. Wiley, G. H. Cohen, and R. J. Eisenberg. 2005. Potential nectin-1 binding site on herpes simplex virus glycoportein D. J. Virol. 791282-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuello, M., S. A. Ettenberg, A. S. Clark, M. M. Keane, R. H. Posner, M. M. Nau, P. A. Dennis, and S. Lipkowitz. 2001. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res. 614892-4900. [PubMed] [Google Scholar]
- 15.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 976640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Giovanni, C., G. Nicoletti, L. Landuzzi, A. Astolfi, S. Croci, A. Comes, S. Ferrini, R. Meazza, M. Iezzi, E. Di Carlo, P. Musiani, F. Cavallo, P. Nanni, and P. L. Lollini. 2004. Immunoprevention of HER-2/neu transgenic mammary carcinoma through an interleukin 12-engineered allogeneic cell vaccine. Cancer Res. 644001-4009. [DOI] [PubMed] [Google Scholar]
- 17.Geraghty, R. J., C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and P. G. Spear. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 2801618-1620. [DOI] [PubMed] [Google Scholar]
- 18.Haarr, L., D. Shukla, E. Rodahl, M. C. Dal Canto, and P. G. Spear. 2001. Transcription from the gene encoding the herpesvirus entry receptor nectin-1 (HveC) in nervous tissue of adult mouse. Virology 287301-309. [DOI] [PubMed] [Google Scholar]
- 19.Hadac, E. M., K. W. Peng, T. Nakamura, and S. J. Russell. 2004. Reengineering paramyxovirus tropism. Virology 329217-225. [DOI] [PubMed] [Google Scholar]
- 20.Harrow, S., V. Papanastassiou, J. Harland, R. Mabbs, R. Petty, M. Fraser, D. Hadley, J. Patterson, S. M. Brown, and R. Rampling. 2004. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. 111648-1658. [DOI] [PubMed] [Google Scholar]
- 21.Holbro, T., and N. E. Hynes. 2004. ErbB receptors: directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 44195-217. [DOI] [PubMed] [Google Scholar]
- 22.Hu, J. C., R. S. Coffin, C. J. Davis, N. J. Graham, N. Groves, P. J. Guest, K. J. Harrington, N. D. James, C. A. Love, I. McNeish, L. C. Medley, A. Michael, C. M. Nutting, H. S. Pandha, C. A. Shorrock, J. Simpson, J. Steiner, N. M. Steven, D. Wright, and R. C. Coombes. 2006. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 126737-6747. [DOI] [PubMed] [Google Scholar]
- 23.Hunter, W. D., R. L. Martuza, F. Feigenbaum, T. Todo, T. Mineta, T. Yazaki, M. Toda, J. T. Newsome, R. C. Platenberg, H. J. Manz, and S. D. Rabkin. 1999. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primates. J. Virol. 736319-6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hynes, N. E., and H. A. Lane. 2005. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer 5341-354. [DOI] [PubMed] [Google Scholar]
- 25.Kamiyama, H., G. Zhou, and B. Roizman. 2005. Herpes simplex virus 1 recombinant virions exhibiting the amino terminal fragment of urokinase-type plasminogen activator can enter cells via the cognate receptor. Gene Ther. 13621-629. [DOI] [PubMed] [Google Scholar]
- 26.Kemeny, N., K. Brown, A. Covey, T. Kim, A. Bhargava, L. Brody, B. Guilfoyle, N. P. Haag, M. Karrasch, B. Glasschroeder, A. Knoll, G. Getrajdman, K. J. Kowal, W. R. Jarnagin, and Y. Fong. 2006. Phase I, open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum. Gene Ther. 171214-1224. [DOI] [PubMed] [Google Scholar]
- 27.Krummenacher, C., F. Baribaud, M. Ponce De Leon, I. Baribaud, J. C. Whitbeck, R. Xu, G. H. Cohen, and R. J. Eisenberg. 2004. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology 322286-299. [DOI] [PubMed] [Google Scholar]
- 28.Krummenacher, C., V. M. Supekar, J. C. Whitbeck, E. Lazear, S. A. Connolly, R. J. Eisenberg, G. H. Cohen, D. C. Wiley, and A. Carfi. 2005. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 244144-4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manoj, S., C. R. Jogger, D. Myscofski, M. Yoon, and P. G. Spear. 2004. Mutations in herpes simplex virus glycoprotein D that prevent cell entry via nectins and alter cell tropism. Proc. Natl. Acad. Sci. USA 10112414-12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markert, J. M., G. Y. Gillespie, R. R. Weichselbaum, B. Roizman, and R. J. Whitley. 2000. Genetically engineered HSV in the treatment of glioma: a review. Rev. Med. Virol. 1017-30. [DOI] [PubMed] [Google Scholar]
- 31.Markert, J. M., M. D. Medlock, S. D. Rabkin, G. Y. Gillespie, T. Todo, W. D. Hunter, C. A. Palmer, F. Feigenbaum, C. Tornatore, F. Tufaro, and R. L. Martuza. 2000. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 7867-874. [DOI] [PubMed] [Google Scholar]
- 32.Markert, J. M., J. N. Parker, D. J. Buchsbaum, W. E. Grizzle, G. Y. Gillespie, and R. J. Whitley. 2006. Oncolytic HSV-1 for the treatment of brain tumours. Herpes 1366-71. [PubMed] [Google Scholar]
- 33.Martuza, R. L., A. Malick, J. M. Markert, K. L. Ruffner, and D. M. Coen. 1991. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 252854-856. [DOI] [PubMed] [Google Scholar]
- 34.Mata, M., M. Zhang, X. Hu, and D. J. Fink. 2001. HveC (nectin-1) is expressed at high levels in sensory neurons, but not in motor neurons, of the rat peripheral nervous system. J. Neurovirol. 7476-480. [DOI] [PubMed] [Google Scholar]
- 35.Mathis, J. M., M. A. Stoff-Khalili, and D. T. Curiel. 2005. Oncolytic adenoviruses—selective retargeting to tumor cells. Oncogene 247775-7791. [DOI] [PubMed] [Google Scholar]
- 36.Menotti, L., A. Cerretani, and G. Campadelli-Fiume. 2006. A herpes simplex virus recombinant that exhibits a single-chain antibody to HER2/neu enters cells through the mammary tumor receptor, independently of the gD receptors. J. Virol. 805531-5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menotti, L., M. Lopez, E. Avitabile, A. Stefan, F. Cocchi, J. Adelaide, E. Lecocq, P. Dubreuil, and G. Campadelli Fiume. 2000. The murine homolog of human-Nectin1δ serves as a species non-specific mediator for entry of human and animal αherpesviruses in a pathway independent of a detectable binding to gD. Proc. Natl. Acad. Sci. USA 974867-4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Messerle, M., I. Crnkovic, W. Hammerschmidt, H. Ziegler, and U. H. Koszinowski. 1997. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 9414759-14763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mineta, T., S. D. Rabkin, T. Yazaki, W. D. Hunter, and R. L. Martuza. 1995. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1938-943. [DOI] [PubMed] [Google Scholar]
- 40.Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87427-436. [DOI] [PubMed] [Google Scholar]
- 41.Natali, P. G., M. R. Nicotra, A. Bigotti, I. Venturo, D. J. Slamon, B. M. Fendly, and A. Ullrich. 1990. Expression of the p185 encoded by HER2 oncogene in normal and transformed human tissues. Int. J. Cancer 45457-461. [DOI] [PubMed] [Google Scholar]
- 42.O'Connor, M., M. Peifer, and W. Bender. 1989. Construction of large DNA segments in Escherichia coli. Science 2441307-1312. [DOI] [PubMed] [Google Scholar]
- 43.Pal, S. K., and M. Pegram. 2007. HER2 targeted therapy in breast cancer: beyond Herceptin. Rev. Endocr. Metab. Disord. 8269-277. [DOI] [PubMed] [Google Scholar]
- 44.Papanastassiou, V., R. Rampling, M. Fraser, R. Petty, D. Hadley, J. Nicoll, J. Harland, R. Mabbs, and M. Brown. 2002. The potential for efficacy of the modified (ICP 34.5(−)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 9398-406. [DOI] [PubMed] [Google Scholar]
- 45.Rampling, R., G. Cruickshank, V. Papanastassiou, J. Nicoll, D. Hadley, D. Brennan, R. Petty, A. MacLean, J. Harland, E. McKie, R. Mabbs, and M. Brown. 2000. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 7859-866. [DOI] [PubMed] [Google Scholar]
- 46.Ricci, C., L. Landuzzi, I. Rossi, C. De Giovanni, G. Nicoletti, A. Astolfi, S. Pupa, S. Menard, K. Scotlandi, P. Nanni, and P. L. Lollini. 2000. Expression of HER/erbB family of receptor tyrosine kinases and induction of differentiation by glial growth factor 2 in human rhabdomyosarcoma cells. Int. J. Cancer 8729-36. [PubMed] [Google Scholar]
- 47.Russell, S. J., and K. W. Peng. 2007. Viruses as anticancer drugs. Trends Pharmacol. Sci. 28326-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shukla, D., J. Liu, P. Blaiklock, N. W. Shworak, X. Bai, J. D. Esko, G. H. Cohen, R. J. Eisenberg, R. D. Rosenberg, and P. G. Spear. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 9913-22. [DOI] [PubMed] [Google Scholar]
- 49.Sidhu, S. S., B. Li, Y. Chen, F. A. Fellouse, C. Eigenbrot, and G. Fuh. 2004. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J. Mol. Biol. 338299-310. [DOI] [PubMed] [Google Scholar]
- 50.Slamon, D. J., G. M. Clark, S. G. Wong, W. J. Levin, A. Ullrich, and W. L. McGuire. 1987. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235177-182. [DOI] [PubMed] [Google Scholar]
- 51.Spear, P. G., and R. Longnecker. 2003. Herpesvirus entry: an update. J. Virol. 7710179-10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Springfeld, C., V. von Messling, M. Frenzke, G. Ungerechts, C. J. Buchholz, and R. Cattaneo. 2006. Oncolytic efficacy and enhanced safety of measles virus activated by tumor-secreted matrix metalloproteinases. Cancer Res. 667694-7700. [DOI] [PubMed] [Google Scholar]
- 53.Takai, Y., and H. Nakanishi. 2003. Nectin and afadin: novel organizers of intercellular junctions. J. Cell Sci. 11617-27. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka, M., H. Kagawa, Y. Yamanashi, T. Sata, and Y. Kawaguchi. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 771382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tiwari, V., C. Clement, D. Xu, T. Valyi-Nagy, B. Y. Yue, J. Liu, and D. Shukla. 2006. Role for 3-O-sulfated heparan sulfate as the receptor for herpes simplex virus type 1 entry into primary human corneal fibroblasts. J. Virol. 808970-8980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsvitov, M., A. R. Frampton, Jr., W. A. Shah, S. K. Wendell, A. Ozuer, Z. Kapacee, W. F. Goins, J. B. Cohen, and J. C. Glorioso. 2007. Characterization of soluble glycoprotein D-mediated herpes simplex virus type 1 infection. Virology 360477-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Varghese, S., and S. D. Rabkin. 2002. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther. 9967-978. [DOI] [PubMed] [Google Scholar]
- 58.Waehler, R., S. J. Russell, and D. T. Curiel. 2007. Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 8573-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoon, M., and P. G. Spear. 2004. Random mutagenesis of the gene encoding a viral ligand for multiple cell entry receptors to obtain viral mutants altered for receptor usage. Proc. Natl. Acad. Sci. USA 10117252-17257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoon, M., A. Zago, D. Shukla, and P. G. Spear. 2003. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J. Virol. 779221-9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou, G., V. Galvan, G. Campadelli-Fiume, and B. Roizman. 2000. Glycoprotein D or J delivered in trans blocks apoptosis in SK-N-SH cells induced by a herpes simplex virus 1 mutant lacking intact genes expressing both glycoproteins. J. Virol. 7411782-11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou, G., and B. Roizman. 2005. Characterization of a recombinant herpes simplex virus 1 designed to enter cells via the IL13Rα2 receptor of malignant glioma cells. J. Virol. 795272-5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou, G., and B. Roizman. 2006. Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13α2 receptor. Proc. Natl. Acad. Sci. USA 1035508-5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou, G., and B. Roizman. 2007. Separation of receptor binding and pro-fusogenic domains of glycoprotein D of herpes simplex virus 1 into distinct interacting proteins. Proc. Natl. Acad. Sci. USA 1044142-4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou, G., G. J. Ye, W. Debinski, and B. Roizman. 2002. Engineered herpes simplex virus 1 is dependent on IL13Rα2 receptor for cell entry and independent of glycoprotein D receptor interaction. Proc. Natl. Acad. Sci. USA 9915124-15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.