

Abstract
The Pentapeptide Repeat Protein (PRP) family has over 500 members in the prokaryotic and eukaryotic kingdoms. These proteins are composed of, or contain domains composed of, tandemly repeated amino acid sequences with a consensus sequence of [S,T,A,V][D,N][L,F]-[S,T,R][G]. The biochemical function of the vast majority of PRP family members is unknown. The three-dimensional structure of the first member of the PRP family was determined for the fluoroquinolone resistance protein (MfpA) from Mycobacterium tuberculosis. The structure revealed that the pentapeptide repeats encode the folding of a novel right-handed quadrilateral β-helix. MfpA binds to DNA gyrase and inhibits its activity. The rod-shaped, dimeric protein exhibits remarkable size, shape and electrostatic similarity to DNA.
Discovery of the Pentapeptide Repeat Family
The first protein identified with what is now recognized as the pentapeptide repeat motif was the hglK-encoded protein from Anabena sp. Strain PCC 7120 (1). This filamentous, cyanobacterium is known to form heterocysts, specialized cells capable of fixing nitrogen, when reduced forms of nitrogen are unavailable. Chemical mutagenesis of the PCC 7120 strain allowed mutants to be identified that were incapable of forming the thick glycolipid outer cell component characteristic of heterocysts. Complementation analysis revealed that the hglK gene could reverse the mutant phenotype and that, in the mutant strain, a mutation had introduced a stop codon at amino acid position 496 of the 727 residue long protein. Starting at position 501, a series of 36 uninterrupted, tandem repeats of a pentapeptide with the consensus sequence ADLSG was observed. The amino terminus contained four possible membrane spanning domains, suggesting that these might anchor the protein into the membrane, and that glycolipid transport or assembly into the heterocyst might be the function of the pentapeptide repeat.
Bioinformatic Approaches to Identifying Pentapeptide Repeat Proteins
The first bioinformatic approach to the genome-wide identification of pentapeptide repeat proteins was reported in 1998 by Bateman et al., (2) who searched the Synechocystis sp. PCC 6803 genome using a single pentapeptide repeat sequence (gene: SLR1819). This putative 331 amino acid protein contained 60 uninterrupted pentapeptide repeats representing 91% of the total sequence. Using this query sequence and the Blast program (3), 15 additional Synechocystis sp. PCC 6803 proteins were identified that contained between 13 and 44 tandem pentapeptide repeats. Several additional sequences were identified as being members of the pentapeptide repeat family, including the McbG gene product, known to confer resistance to the antibacterial Microcin B17 (4). A more robust approach to the identification of proteins that contain the pentapeptide repeat motif has used a Hidden Markov Model (HMM; 5) and a motif that contains eight consecutive pentapeptide repeats. The Pfam (6) database (www.sanger.ac.uk/cgi-bin/Pfam) currently lists 1061 pentapeptide repeat-containing proteins (Pfam00805). Included in the database are all PRP's identified by Bateman et al. While the vast majority of these are found in prokaryotes, there are examples of proteins containing pentapeptide repeat domains in Plasmodium falciparum, Anopheles gambia, Arabidopsis, zebrafish, mouse and human. Essentially all (19 of 20) eukaryotic PRP's contain 32 uninterrupted tandem pentapeptide repeats at the C-terminus of 300-390 residue long proteins whose N-terminus is the cytoplasmic tetramerization domain of the voltage-gated K+ channel. PRP's have also been identified in bacteriophages as well as mycobacteriophages. Given this broad phylogenetic distribution, it is surprising that no PRP's have been identified in any sequenced yeast genome.
Phylogenetic categorization and polydomain PRP's
Our bioinformatic studies indicate a wide phylogenetic distribution of PRP's. Using the PFAM Hidden Markov Model (HMM) definitions of these motifs, we used the HMMER program to scan the Non-Redundant (NR) sequence database (7) that currently contains approximately 2.3 million entries, and identified 1702 sequence domains that contain pentapeptide motifs. PRP motifs are highly repetitive in nature and HMMER often locates PRP motifs multiple times in the same protein. Once redundancy is removed from the hits, we arrived at 525 currently known, unique proteins with Pentapeptide motifs. These numbers can further be divided into PRP's in prokaryotic (484) and eukaryotic (41) species.
While many bacteria contain one or few PRP's (see below), some microorganisms, especially the photosynthetic cyanobacteria such as Anabena, have numerous chromosomally-encoded PRP's. Synechocystis sp. Strain 6803 has 16 PRP's. We can identify 40 PRP's in Nostoc punctiforme which range in size from 75 amino acids to >400 residues. As noted above in the case of the Anabena HglK protein and the human voltage-gated potassium channel tetramerization protein, these often contain multiple domains, with the PRP domain generally found at the C-terminus of these poly-domain proteins.
In order to explore the types and occurrence of additional domains in all 525 of the identified PRP proteins, we mapped all the currently known 7868 Pfam (6) domain definitions using the HMMER program. All data are available online at http://andes.aecom.yu.edu/prp/. Of the 484 prokaryotic proteins containing pentapeptide motifs, 172 had at least one additional domain. The associated domains fall into 20 different types (Table 1) with the most common being WD40 domains (β-transducin repeat; coordination of multiprotein complex formation in signal transduction, transcriptional regulation, cell cycle control and apoptosis), Pkinase domains (Ser/Thr protein kinases), TPR_1 and TPR_2 domains (tetratricopeptide repeat, role in protein-protein interactions), DnaJ domains (J-domains; associated with hsp70 heat shock system and mediating protein-protein interactions), Acetyltranf_1 domains (N-acetyltransferase; GNAT family), RDD domains (membrane localization) and 11 other domain types with 2 or fewer representatives. These characteristic observations also hold true for the eukaryotic PRPs. These 20 extra domains are of two distinct types, the K_tetra (voltage-gated potassium channel tetramerization domain) and GnRH (Gonadotropin-releasing hormone; role in reproduction) domains.
Table 1.
Domains fused to Pentapeptide Repeat Proteins
| Pentapeptide | Pentapeptide_2 |
|---|---|
| Prokaryotic (172 total domains) | |
| WD40 β-transducin repeat (56) | PPE domain (54) |
| Ser/Thr protein kinase (11) | Fibronectin type III (3) |
| Tetratricopeptide_1 repeat (11) | |
| DNAJ domain (7) | |
| Tetratricopeptide_2 repeat (7) | |
| GNAT acetyltransferase (6) | |
| RDD membrane spanning (4) | |
| Eukaryotic (21 total domains) | |
| K+ tetramerization (19) | Serine proteinase inhibitor (1) |
| Gonadotropin-releasing hormone (1) |
In addition to the pentapeptide repeat motif originally described by Bateman et al. (2), a second pentapeptide repeat family has been identified in the Pfam database and termed the “Pentapeptide_2” family with 896 sequence domains listed in Pfam libraries. The pentapeptide_2 repeat proteins contain tandemly repeated sequences with the consensus sequence [N][T,L,I][G][S,N][G]. After removing redundancy, 61 examples of Pentapeptide_2 proteins occur in prokaryotes, with only two identified in eukaryotes. Like the original pentapeptide repeat proteins, the prokaryotic pentapeptide_2 proteins are associated with additional domains, including 54 examples of N-terminal PPE domains in mycobacterial species (unknown function; but implicated in antigenic variation in M. tuberculosis) and three fn3 domains (fibronectin type III domain; putative role in structural complexes). A single eukaryotic pentapeptide_2 protein with an associated Kazal_2 (Kazal-type serine proteinase inhibitor) domain was identified. There is not a single instance where the two different PRP motifs occur in the same protein, nor is there any overlap of the types of additional domains associated with the two types of PRP's; the additional domains are uniquely associated with one, or the other, pentapeptide repeat domains. Since it is unlikely that these proteins share the same three-dimensional fold, no further discussion of the pentapeptide_2 repeat proteins will be presented.
PRP's and fluoroquinolone resistance
Fluoroquinolones are synthetic derivatives of nalidixic acid and exhibit broad-spectrum and powerful antibacterial activity (Scheme 1). Prior to 1998, all mechanisms of bacterial fluoroquinolone resistance had been shown to be either due to a) mutations in the type II topoisomerases, DNA gyrase and topoisomerase IV, that are the targets of these drugs or b) increased expression of efflux pumps that reduce the intracellular concentration of the drug. Neither of these resistance mechanisms has been shown to be transmissible between organisms. Therefore, the discovery of a plasmid-mediated, transmissible form of fluoroquinolone resistance in 1998 heralded a significant clinical concern. The 218 residue long QnrA protein was originally found on a conjugative plasmid isolated from a fluoroquinolone resistant strain of Klebsiella pneumoniae in Alabama (8). Since then it has been found on transferable plasmids from fluoroquinolone resistant strains of K. pneumoniae isolated in 6 different states in the United States (9). Outside of the United States, qnr genes have been found on plasmids in fluoroquinolone resistant isolates of Enterobacteriae from China (10), Hong Kong (11), Korea (12), France (13), Germany (14) and Egypt (15). In some cases, the qnr genes were adjacent to genes that confer resistance to other antibiotics, such as sulfamethoxazole and the extended spectrum β-lactam antibiotics within Type I integrons that were apparently mobile, as they were found on an assortment of conjugative plasmids (16, 17). Thus, although several studies have found qnr genes in a small percentage of fluoroquinolone resistant strains in the United States (8 of 72; 9) and Europe, it appears that the qnrA gene is present worldwide on integrons that can integrate into a series of promiscuous plasmids within many species of Enterobacteriaceae.
Scheme 1.

The Qnr protein is a pentapeptide repeat protein (see below for complete description). Qnr has been biochemically characterized by Jacoby et al. (18) who showed that, in vitro, the Qnr protein protects both E. coli DNA gyrase and the E. coli topoisomerase IV (19) from the inhibitory effects of fluoroquinolones. More recently, these investigators have shown that Qnr competes with DNA for binding to the DNA gyrase (20), suggesting that this 218 residue long PRP, consisting of 40 uninterrupted pentapeptide repeats (92% of the sequence), binds to DNA gyrase (see below). The original source of the qnr gene was unclear, since in all cases the G+C content (52%) of the Qnr gene is much higher than the chromosomal G+C content of the clinical Enterobacteriae isolates in which it has so far been found. The origin of QnrA was recently discovered by PCR screening of 48 Gram-negative bacterial species for the presence of a qnrA gene (21). In Shewanella algae, an environmental species from marine and fresh water, four QnrA-like proteins were found. These four chromosomally encoded proteins (QnrA2-5) differ from each other, and from the original QnrA (now termed QnrA1), by only 1-4 amino acids. It is interesting that a qnr allele found on four different, transferable plasmids from four fluoroquinolone-resistant strains of Salmonella enterica serotype Enteriditidis in Hong Kong (11) are 100% identical to QnrA3.
Another qnr homologue, QnrS, that is only 59% identical to QnrA, was found on a plasmid from a broadly drug resistant strain of Shigella flexneri isolated in Japan (22). Transfer of the conjugative plasmid also conferred resistance to β-lactam antibiotics, but qnrS was not located in an integron, but rather within a putative transposon (Tn3-like) sequence, together with a TEM-1 β-lactamase gene. Another protein belonging to the Qnr family, QnrB, is only 40% identical to QnrA1, and has been found in various Enterobacteriaciae in both the United States and India (23). Genes encoding proteins similar to Qnr were recently identified in the chromosomes of three species of the Vibrionaceae family (58-66% identical to QnrA, 52-64% identical to QnrS), so it is likely that the origin of QnrS and other putative Qnr proteins may be found in the chromosomes of other Gram-negative bacteria. Interestingly, when one of the qnr-like genes from Vibrio parahaemolyticus was cloned onto a plasmid, it did not confer fluoroquinolone resistance unless cysteine 115 was mutated to a tyrosine, perhaps allowing the mutant form to bind to the gyrase (24). A recent review summarizes the rapidly changing field of Qnr function (23).
A genetic selection for fluoroquinolone resistance in the fast-growing M. smegmatis identified a gene expressed on a multi-copy plasmid responsible for a 2-8 fold increase in the MIC (Minimum Inhibitory Concentration) values for ciprofloxacin and sparfloxacin (Scheme 1, 25). The gene, termed mfpA for mycobacterial fluoroquinolone resistance protein, encodes a 192 amino acid protein whose nearly complete open reading frame consisted of 32 uninterrupted pentapeptide repeats. The role of MfpA in fluoroquinolone resistance was confirmed by deletion of the gene and demonstrating that the knockout strain was 2-4 fold more sensitive to ciprofloxacin and sparfloxacin. The 183 residue long M. tuberculosis Rv3361 gene is 67% identical to the M. smegmatis MfpA protein, and contains a similar pentapeptide repeat motif.
The Pentapeptide Repeat Fold
The expression and purification of the M. tuberculosis MfpA protein (MtMfpA), and its crystallization in four distinct forms, were recently reported (26). The three-dimensional structure of MtMfpA demonstrated that the pentapeptide repeat sequence codes for a right-handed quadrilateral β-helix (RHQBH; Figure 1). Each pentapeptide forms one side of a nearly square repeating unit, which we have termed a coil. Each coil is composed of four pentapeptides, with twenty residues per coil, and with a rise in the helical axis of 4.8 Å per revolution. In general, the quadrilateral coils stack directly atop one another although there is a slight left-handed twist to the helical axis. Due to this stacking and the quadrilateral nature of the coils, the protein can be designated to have four faces, termed F1-F4 (Figure 2). MtMfpA has eight complete coils, the first seven of which are composed exclusively of pentapeptide repeats. The eighth coil has three strands, as strand F3 is replaced by a two-turn α-helix (α1). What would be the ninth coil has a single strand on F1, followed by a three-turn α-helix (α2) and a β-strand that is not part of the regular coiled structure.
Figure 1.
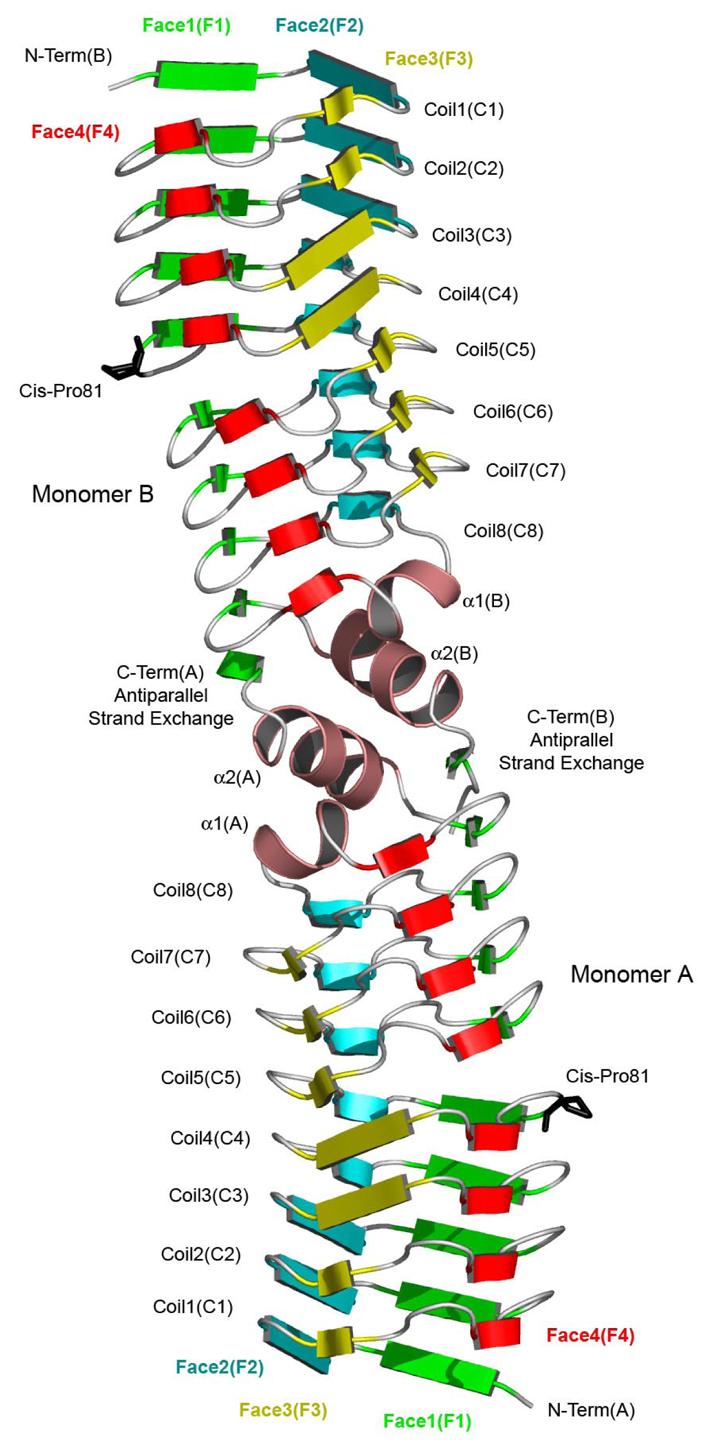
Ribbon diagram of the Mycobacterium tuberculosis MfpA dimer. The four faced of the quadrilateral β-helix are colored green (face 1), blue (face 2), yellow (face 3) and red (face 4).
Figure 2.

Structure-based sequence alignment of MfpA. Regions comprising α1 and α2 are shaded salmon.
MtMfpA was found to exist as a dimer in solution by ultracentrifugation (unpublished results) and in all four crystal forms a consistent dimer interface, located at the C-terminal end of the β-helix, was found. At this interface the C-terminal α-helices form an alternating perpendicular α-helical stack (α1-α2-α2'-α1') with a hydrophobic core at the molecular two-fold axis. The final β-strand crosses over to the two-fold related β-helix adding a final β-strand in an antiparallel fashion to F1. The two β-helices are coaxial, producing a highly asymmetric dimer with an axial length of 100 Å. While the mainchain atoms exhibit a quadrilateral cross section, the addition of surface side chains yields a molecule with a more cylindrical appearance when viewed down the helical axis, whose diameter varies from 27 to 18 Å from the N-terminus to the dimer interface.
The individual residues of the pentapeptide are designated as follows: the conserved hydrophobic residue (Leu or Phe) at the center of pentapeptide alignments is the ‘i’ residue, while the residues N-terminal to the ‘i’ residue are the i−1 and i−2 residues and the residues C-terminal to the ‘i’ residue are the i+1 and i+2 residues. The side chains of the i and i−2 residues pack in the interior of the β-helix, whereas the side chains of the remaining residues point outwards and generate the surface of the protein (Figure 3). As suggested by the repeating nature of the primary sequence, the pentapeptide residues adopt similar conformations throughout the β-helix. There are two predominant mainchain phi-psi dihedral orientations encoded by the pentapeptide repeat which differ only in the orientation of a single peptide bond (Figure 4). In general, the pentapeptide secondary structure can be viewed as type II turns (Figure 5) composed of the i, i+1, i+2 and i−2 residues [(−120, 20), (−60, 120), (60, 20) and (−80, 160), respectively] connected by isolated β-bridges involving the i−1 residue [(−120, 120)]. In this conformation the carbonyl of the ith residue is hydrogen bonded to the i−2 amide of the following pentapeptide and only the i−1 residue contributes both backbone amide nitrogens and carbonyls to intercoil hydrogen bonding. This is illustrated in the ribbon diagram in Figure 1 where the isolated β-bridges are shown as short β-strands. This phi-psi composition occurs in 19 of the 30 turns in MtMfpA (Figure 4). In the other cases, the peptide bond between the i and i+1 is rotated 90 degrees such that both the i and i+1 mainchain atoms participate fully in intercoil hydrogen bonding (Type IV turns). These pentapeptides are illustrated in the ribbon diagram in Figure 1 as the longer β-strands and have phi-psi angles for the i−2, i−1, i, i+1 and i+2 residues of [(−80, 160), (−120, 120), (−120, 120), (−120, 120) and (60, 20)], respectively. The choice between the type II and type IV turn appears to be related to the identities of the internal sidechains (i−2 and i). The type IV turn of the pentapeptide repeat lacks the intracoil hydrogen bond of the type II turn, and yields a pentapeptide with a more extended conformation (∼0.5-0.75 Å). The type IV turns, which frequently occur in the N-terminal coils, have predominately phenylalanine at the i position, and this larger sidechain is accommodated by extended pentapeptides forming a larger core. In contrast the C-terminal coils have only leucines, and all exhibit the type II turn.
Figure 3.

Electron density and molecular model of MfpA. Shown is 2Fo-Fc electron density contoured at 1σ and residues comprising the second coil.
Figure 4.
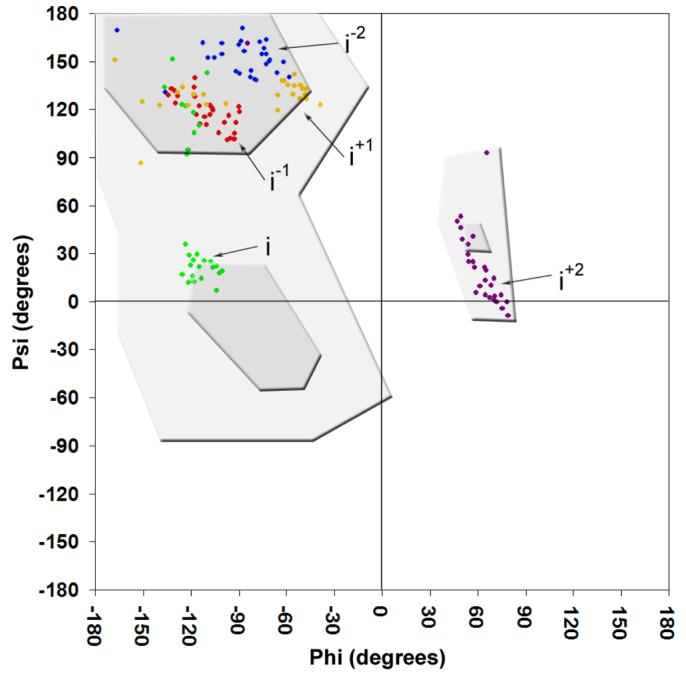
Ramachandran plot of the first 165 residues that make up the right-handed quadrilateral β-helix domain of MfpA. Pentapeptide repeat positions are colored blue (i−2), red (i−1), green (i), yellow (i+1) and purple (i+2). The most favored and additionally allowed phi-psi orientations are shaded dark and light grey, respectively.
Figure 5.

Side chain and main chain interactions of the pentapeptide repeat type II turns.
Each of the residues of an individual pentapeptide exhibit a propensity towards a restricted number of amino acids, with a structure-based consensus sequence of [S,T,A,V][D,N][L,F][S,T,R][G]. The central i residue is the main component of the hydrophobic core of the RHQBH and is predominately either a phenylalanine or a leucine. Typically, the central hydrophobic side chain is in van der Waals contact with the i residue in neighboring coils above and below both on its own face and on neighboring faces and also the i−2 residue of its own face and the i−2 residue of the next consecutive pentapeptide. Due to the equivalent nature of each of the quadrilateral sides and the distances from Cα to Cα of opposite faces, the i residue never interacts with any of the internal residues of the opposite face. Stacks of phenylalanine residues exist internally on F2 (Phe9, 29 and 49) and F3 (Phe14, 34, 54 and 74) with similar conformations of their sidechain torsion angles (χ1∼-70, χ2∼-70) (Figure 6). The aromatic ring conformation and the left handed twist to the coil stacking is used to minimize the negative interaction between their π-electron clouds and maximize their interaction with the somewhat positive ring hydrogens in an interaction that has been described for the interaction of phenylalanines in general, and phenylalanine stacking in β-helices (27). Stacks of leucine residues of at least three leucines occur on F1 (Leu84, 104,124, 144) and F4 (Leu19, 39,59, 79, 99, and 119) (Figure 7). The stacks of leucine sidechains have a much wider conformational space when compared to the phenylalanine stacks and tend to assume conformations that maximize van der Waals contacts with nearby residues.
Figure 6.

The interior phenylalanine stack on face 3 of MfpA.
Figure 7.
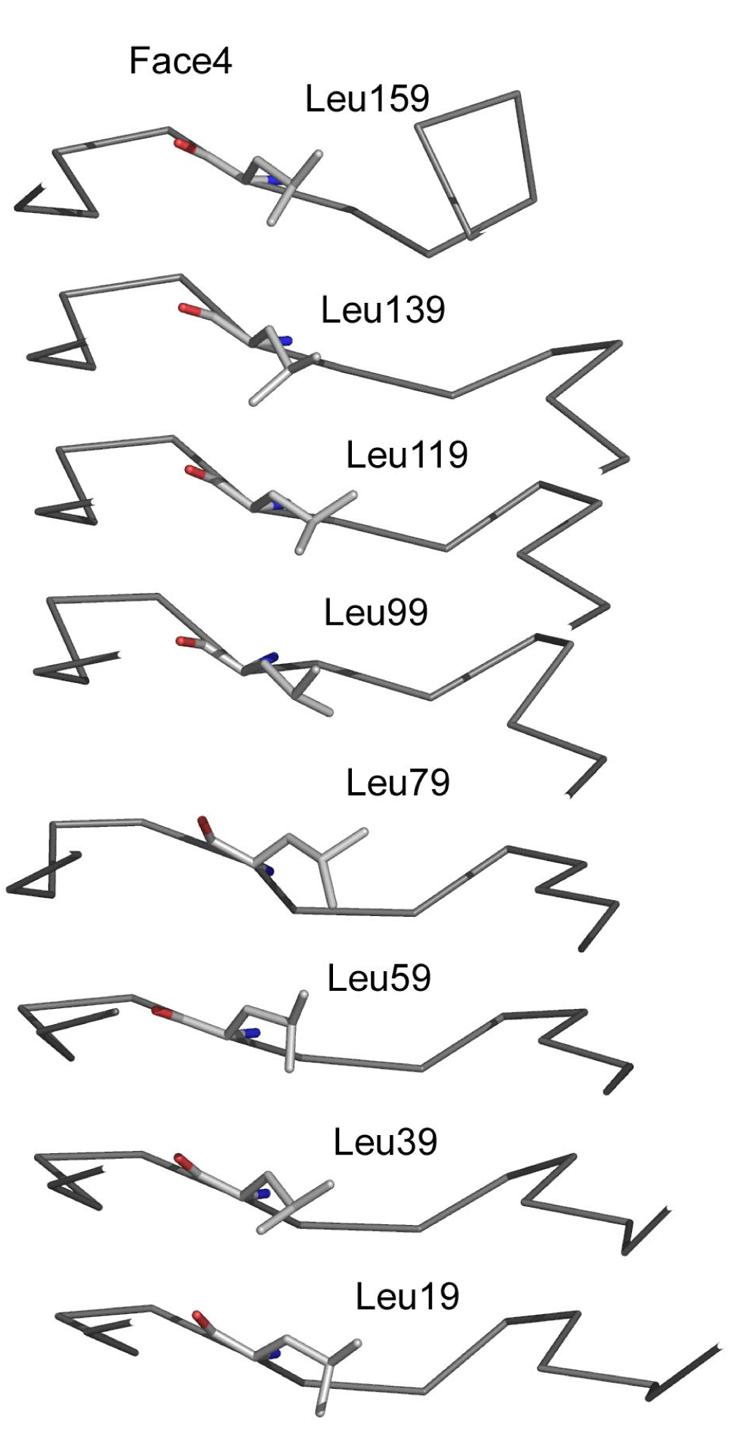
The interior leucine stack on face 4 of MfpA
The i−2 residues typically are alanine, serine, threonine, or valine (18/33). The side chains of the i−2 residues project inward, participate in hydrogen bonding with mainchain atoms and form part of the hydrophobic core. In most cases, the hydroxyl groups of serine and threonine residues in the i−2 position hydrogen bond to the amide backbone of the i−2 or the carbonyl of the i residue in the turn directly below. MtMfpA is unique among pentapeptide repeat proteins in that the i−2 residue is often a cysteine (7/33). The cysteine residues are in conformations similar to the serine residues at this position.
The i+2 residue adopts a left-handed α-helical conformation (αL) and is typically either a glycine (11/32) or a polar residue (15/32). Glycine is the most frequent amino acid found in type II turns at this position, where a bulkier amino acid would have limited conformational flexibility due to peptide clashes with Cβ. Finally, the i−1 residue is often an asparagine or aspartate while the i+1 residue is often a serine, threonine or arginine. The structure reveals that the propensities for specific amino acids at these two positions are linked. When the carbonyl of the i residue participates in a type II turn, the amide group of the i+1 residue projects outward, but is shielded from possible solvation by the side chain of the i−1 residue. The sidechains of asparagine and aspartate at the i−1 position are of the correct length to form a hydrogen bond with this i+1 amide group (Figure 4). In addition, to further stabilize this arrangement the asparagine or aspartate residues at the i−1 position will also form a hydrogen bond with the i+1 side chain when it is a serine, or threonine or a salt link when it is an arginine.
There is a 12° kink in the helical axis mid-way through the β-helix. This results in the disruption of intercoil hydrogen bonding between coil 4 and 5 on F4 and coil 5 and 6 on F1. This disruption is due to a cis-proline (Pro 81) in the turn adjoining coils 4 and 5. The exposed mainchain atoms in this interface are bridged by several water molecules, and the larger intercoil space is partially filled by bulkier residues at the i−2 position (Leu82, Leu102, Asn97). This intercoil disruption affords MfpA a degree of flexibility. The structure of MtMfpA was determined from crystal forms that had three, two, two and one molecules per asymmetric unit, respectively, yielding eight MfpA monomer structures. The largest deviation between monomers was an RMSD of 0.77 Å over 180 common C α atoms. The largest component of the deviation is a difference in the intercoil distance near the cis-proline, yielding minor differences in the kink angle between the two helical axes. A second component of the deviation is flexibility at the C-termini, probably owing to the nature of the hydrophobic interaction between the C-terminal α-helices (unpublished data).
Because the i residues of opposing pentapeptides do not interact there is unoccupied space at the core of the structure along the helical axis. Indeed, in the original analysis of pentapeptide repeat proteins by Bateman et. al. (2), models of pentapeptide repeat proteins with four sides per coil were rejected due to poor packing of the internal sidechains. This is why a model similar to the left-handed β-helix observed for hexapeptide repeat proteins with a triangular cross-section (28) was proposed for pentapeptide repeat proteins. The largest continuous space is within and between coils 4, 5 and 6 with a volume of ∼100 Å3, whose size is larger than the unoccupied space of other coils due to the kink in the helical axis at this point. Other smaller ‘cavities’ are in the 15- 30 Å3 size range. It is unclear if these internal cavities have functional consequences. The highest resolution structure was refined to 2.0 Å, and all of the refined waters are located on the protein surface, and no water molecules are observed within these protein cavities.
Modeling the Interaction of MfpA and DNA Gyrase
DNA gyrase is involved in the negative supercoiling of DNA. The proposed mechanism of DNA gyrase involves coordinated binding of two sections of DNA, termed the T and G segments, cleavage of both strands of the G-segment, the passage of the T-segment through the cleavage point, and religation of the cleaved strands (29). Fluoroquinolones act by stabilizing the tyrosyl122-DNA-Gyrase cleavage complex preventing religation of the DNA. This results in the hydrolysis of the phospho-phenolic linkage and release of cleaved DNA. The fragmentation of the DNA is the bactericidal consequence of fluoroquinolone treatment. DNA gyrase is an α2β2 heterotetramer. The structure of the entire (αβ)2 complex has not been amenable to structure determination to date, but the structure of various subdomains of the DNA gyrase have been structurally characterized. The α-subunit is the site at which fluroquinolones bind to form the tyrosyl-DNA-Gyrase adduct. The structure of an N-terminal construct composed of residues 2-523 of the α subunit (GyrA59) has been determined (30). GyrA59 is a dimer in the crystal structure, whose conformation is proposed to be consistent with the α2 dimer binding the G-segment DNA. A large electropositive “patch” at the dimer interface contains the catalytic tyrosine residues and residues whose mutation are known to confer resistance to fluoroquinolones. This is the proposed binding site for the G-segment of DNA and where the tyrosyl-DNA-Gyrase adduct is generated. Modeling of DNA into this electropositive region indicates that approximately 30-35 bp of B-DNA would occupy the entire length of the dimer. Interestingly the length of 30-35 bp of B-DNA is similar to the axial length of the MtMfpA dimer. MtMfpA has a preponderance of electronegative charge over its entire surface (net charge = −10) and molecular modeling indicated that MfpA would also fit comfortably in this groove with the dimer axis of MfpA aligning with the dimer axis of GyrA59 (26). The negative charge of MtMfpA is projected more prevalently from face 1 and 2, so MfpA was positioned so that these faces were in closer contact with the electropositive groove of GyrA59 (Figure 8). The binding of MfpA to this surface would provide a compelling hypothesis as to how MfpA can provide an organism with resistance to fluoroquinolones. Overexpression and binding of MfpA to the GyrA subunit of DNA-gyrase would inhibit the binding of DNA, and block the formation of the target of fluoroquinolones: the DNA-gyrase binary complex. This would give the population of bacteria time to achieve resistance through alternative means such as mutation of GyrA or expression of drug efflux pumps. However, caution must be applied to such a model, since there are likely to be several surfaces on DNA gyrase that are involved in the capture, cleavage, transfer, religation, and release of DNA and this involves many complex structural rearrangements. However, the general interaction of pentapeptide repeat proteins with GyrA is supported by the biochemical data. Increasing concentrations of MfpA inhibit the topoisomerase activity of DNA-gyrase (26). Utilizing surface plasmon resonance, MfpA was found to interact with DNA-gyrase with a kon and koff values of ∼103 M−1 sec−1 and ∼10−4 sec−1, respectively, allowing a Kd of 450 nM to be calculated. In addition, the QnrA protein was found to promote the release of DNA from a DNA-gyrase complex (20).
Figure 8.
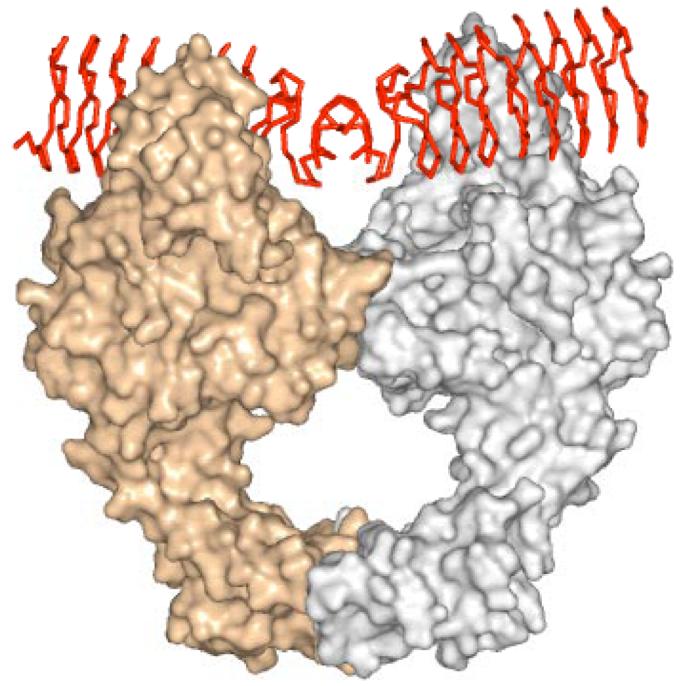
Model for the interaction of MfpA with DNA gyrase. DNA gyrase monomers are presented as silver and gold surface renderings, while the MfpA dimer is shown as a red Cα trace.
The Genomic Environment and Potential Regulation
The role of MtMfpA in the biology of mycobacteria is unknown, but there are some indications that it could play an important role. First, the gene is present and very similar in all mycobacterial genomes (M. tuberculosis, M. smegmatis, M. bovis, M. ulcerans, M. avium.), but appears to be present as a pseudogene in M. leprae. Second, just upstream of mfpA, and apparently expressed as part of the same transcriptional unit, are four genes that are highly conserved in the mycobacteria. This four gene cluster (Rv3362c-3365c; Figure 9) is found together only in the Actinomycetes, the family of bacteria to which the mycobacteria belong. It is presumed to be a regulatory team because the four encoded proteins are similar to other regulatory proteins. Blast searching with Rv3362c finds approx. 30 putative GTPases (31) that are all at least 50% identical to Rv3362c, and these are found only in the Actinomycetes family of bacteria, and are all located within conservons (see below). Rv3363c encodes a possible protein of 122 amino acids that is most similar to the DUF472 family (pfam05331.4) of proteins whose function is unknown. Rv3364c encodes a possible protein of 130 amino acids that is most similar to the Roadblock/LC7 (pfam03259.10) family of proteins, which in eukaryotes are associated with dynein, and in Myxococcus xanthus, with gliding motility. Rv3365c is similar to bacterial histidine kinases, and is likely membrane bound and may serve as a sensor. However, the presumptive histidine kinases in the conservon units, such as Rv3365c, appear to form a subgroup that is distinct from most of those in two-component systems (32). The histidine residue phosphorylated in the classic histidine kinase proteins appears to be conserved, but the surrounding amino acids in the “H-box” are different than those well conserved in the histidine kinases that are part of two component systems.
Figure 9.

Genomic environment of M. tuberculosis MfpA. The four upstream genes colored in red are termed the “conservon” in other Actinomyces species.
In the chromosome of Streptomyces coelicolor A3(2), another actinomycete, this four-gene group is present 13 times and has been labeled a “conservon” (33, 34). The four-gene unit is also found in some other actinomyces, with ten representatives in Streptomyces avermitilis, three in different Frankia species, five in Nocardia farcinica, five in Thermobifida fusca, and two in Kineococcus radiotolerans. Others examples may appear as more actinomycetes genomes are sequenced. There is only one conservon in mycobacteria, and this is the only one associated with a pentapeptide protein.
Biological Functions?
A very small fraction of the PRP's are associated with any defined biochemical function. We will first discuss those PRP's whose expression leads to drug resistance to compounds that inhibit DNA gyrase. While it is clear that MfpA inhibits DNA gyrase activity by binding to the enzyme, possibly in the same region to which the G-segment of DNA binds based on our modeling studies and the phenotype of fluoroquinolone resistance, the physiological role of the chromosomally-encoded protein remains unclear. M. tuberculosis does not contain a second type II topoisomerase, and thus it's DNA gyrase must fulfill the roles of DNA supercoil introduction, relaxation and decatanation. Given the long doubling time of the organism, one possible function is to prevent the relaxation of the supercoiled DNA and maintain a condensed chromosome during periods of replicative senescence. This would require that the protein's expression be coordinated with the cell cycle. Alternatively, if it's expression were constitutive, then the regulatory team of proteins found upstream of the gene, and presumably co-transcribed with the gene, could function to modulate the activity of MfpA. Whether this physiological role is shared with the functionally homologous family of Qnr proteins that are responsible for transmissible, clinical resistance to fluoroquinolones is unclear at present. The mcbG gene is located in an operon that includes many of the Microcin B17 biosynthetic genes, and was initially identified as an immunity factor (35). Based on the results with MfpA, it seems very likely that the McbG protein will adopt a similar fold and act via a similar mechanism. Like fluoroquinolones, Microcin B17 induces double-strand breaks in chromosomal DNA (36), however, resistance to Microcin B17 occurs via mutations in the gyrB-encoded subunit (37). This argues that Microcin B17 binds to DNA gyrase (38), or a DNA gyrase-DNA complex (39), somewhere in the GyrB subunit, and that the McbG protein prevents its binding, possibly by mimicking a DNA gyrase-DNA complex.
A fourth member of the PRP family involved in resistance is the oxrA-encoded oxetanocin A resistance factor. Bacillus megaterium contains an operon that includes biosynthetic genes for oxetanocin A as well as the oxrA resistance locus (40). Oxetanocin A is an unusual nucleoside whose nucleotide triphosphate form powerfully inhibits viral DNA polymerases (41), human telomerase (42) and HIV reverse transcriptase (43). OxrA has been shown to confer resistance to oxetanocin A, in a manner similar to that discussed above for MicrocinB17 and the McbG resistance protein. OxrA contains only 9 pentapeptide repeats in a 185 amino acid protein, suggesting an alternative mechanism of resistance.
The HglK protein was the first PRP discovered (1), and has been ascribed potential functions in glycolipid transport and/or localization. The four N-terminal membrane-spanning domains suggest that the protein is membrane-associated, but the function of the pentapeptide repeat domain is unclear. The small, discontinuous cavities in the MfpA structure would require a decrease in the van der Waals volume of the i residues, or a small expansion of the exterior of the fold to allow for the insertion of unbranched lipids into the core. A second potential transport function for a pentapeptide repeat domain was reported for a gene in the photosynthetic cyanobacterium, Synechocystis sp. Strain 6803, termed the rfrA gene, that bears pronounced similarity to HglK in overall domain organization, containing both four membrane-spanning sequences at its N-terminus and an uninterrupted tandem repeat of 12 pentapeptides. The RfrA gene was shown to be involved in the regulation of manganese uptake (44), although the authors did not suggest that RfrA itself transported manganese, but rather regulated a high affinity transport system by an unknown mechanism. Whether the extremely high genome density of PRP's in the photosyntheric cyanobacteria, including Synechocystis (16 PRP's), Gloeobacter (18), Anabena (31) and Nostoc (40), is related to multiple functional roles in these organisms, not associated with DNA mimicry, is unclear at the present time.
Finally, many PRP's are polydomain proteins with additional associated motifs, some of which are homologous to catalytic domains. The best studied is the Synechocystis sp. Strain 6803 SpkB protein (45). Its N-terminal domain is homologous to mammalian protein Ser/Thr kinase domains. The SpkB protein is one of thirteen proteins encoded in the genome of this organism with putative Ser/Thr protein kinase activity, but the only one that contains an additional associated pentapeptide repeat domain. The SpkB protein catalyzes its autophosphorylation, as well as the phosphorylation of bovine myelin basic protein, casein and calf thymus histones. The ability of this bacterial protein to phosphorylate mammalian proteins is highly reminiscent of bacterial aminoglycoside phosphotransferases that have been shown to phosphorylate both aminoglycoside antibiotics, conferring high-level resistance to these compounds, as well as mammalian proteins (46). A second example of a polydomain PRP with an N-terminal catalytic domain is the Bacillus anthracis PRP. In this case, the protein has an N-terminal Gcn5-related N-acetyltransferase (GNAT) domain similar to those found in eucaryotic histone acetyltransferases. Although this protein has not been functionally characterized to date, it is again highly reminiscent of other bacterial aminoglycoside N-acetyltransferases that acetylate mammalian histone proteins (47). While the role of the catalytic domains is clear in these systems, the role of the pentapeptide repeat domain is cryptic. One possibility is that the pentapeptide domain, by potentially mimicking DNA in these proteins, might target the catalytic domains to DNA-binding proteins, thus controlling the selectivity of the catalytic domains that post-translationally modify the normal bacterial proteins.
Conclusions and Perspectives
The first PRP was identified only ten years ago, but we now know that they are broadly present in bacteria and multi-cellular eukaryotes. In only a handful of cases, have any biochemical functions been ascribed to these proteins, however, their role in clinical resistance to fluoroquinolones and other DNA gyrase inhibitors is now clear. Whether they serve additional transport and post-translational modification functions is unclear at the present time. As additional structural information is obtained about how the PRP fold can accommodate peptide excursions, and how additional domains are appended, and biochemical information that define functions and specificities of these domains becomes available, we will start to have a more complete picture of this large family of proteins.
Acknowledgments
This work was supported by NIH grants AI336996 (to JSB), T32 AI07501 (to MWV) and the Seaver Foundation (to AF)
Abbreviations
- PRP
Pentapeptide Repeat Proteins
- MfpA
Mycobacterial Fluoroquinolone Resistance Protein
- MtMfpA
Mycobacterium tuberculosis Mycobacterial Fluoroquinolone Resistance Protein
- MHM
Hidden Markov Model
- NR
Non-redundant
- Qnr
Quinolone Resistance Protein
- MIC
Minimum Inhibitory Concentration
- RHQBH
Right-handed Quadrilateral β-Helix
- GNAT
Gcn5-related N-acetyltransferases
References
- 1.Black K, Buikema WJ, Haselkorn R. The hglK Gene is required for Localization of Heterocyst-Specific Glycolipids in the Cyanobacterium Anabena sp. Strain PCC 7120. J. Bact. 1995;177:6440–6448. doi: 10.1128/jb.177.22.6440-6448.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman A, Murzin AG, Teichman SA. Structure and Distribution of Pentapeptide Repeats in Bacteria. Prot. Sci. 1998;7:1477–1480. doi: 10.1002/pro.5560070625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic Alignment Search Tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 4.Kolter R, Moreno F. Genetics of Ribosomally Synthesized Peptide Antibiotics. Ann. Rev. Microbiol. 1992;46:141–163. doi: 10.1146/annurev.mi.46.100192.001041. [DOI] [PubMed] [Google Scholar]
- 5.Krogh A, Brown M, Mian IS, Sjolander K, Haussler D. Hidden Markov Models in Computational Biology. J. Mol. Biol. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- 6.Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O'Donovan C, Phan I, Pilbout S, Schneider M. The SWISS-PROT Protein Knowledgebase and Its Supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez-Martinez L, Pascual A, Jacoby GA. Quinolone resistance from a transferable plasmid. Lancet. 1998;351:797–9. doi: 10.1016/S0140-6736(97)07322-4. [DOI] [PubMed] [Google Scholar]
- 9.Wang M, Sahm DF, Jacoby GA, Hooper DC. Emerging Plasmid-mediated Quinolone Resistance Associated with the qnr Gene in Klebsiella pneumoniae Clinical Isolates in the United States. Antimicrob. Agents Chemother. 2004;49:1295–1299. doi: 10.1128/AAC.48.4.1295-1299.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M, Tran JH, Jacoby GA, Zhang Y, Wang F, Hooper DC. Plasmid-mediated Quinolone Resistance in Clinical Isolates of Escherichia coli from Shanghai, China. Antimicrob. Agents Chemother. 2003;47:2242–2248. doi: 10.1128/AAC.47.7.2242-2248.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung TK, Chu YW, Chu MY, Ma CH, Yung RW, Kam KM. Plasmid-mediated resistance to ciprofloxacin and cefotaxime in clinicial isolates of Salmonella enterica serotype Enteritidis in Hong Kong. J. Antimicrob Chemother. 2005;56:586–9. doi: 10.1093/jac/dki250. [DOI] [PubMed] [Google Scholar]
- 12.Jeong J-Y, Yoon HJ, Kim ES, Lee Y, Choi S-H, Kim NJ, Woo JH, Kim YS. Detection of qnr in Clinical Isolates of Escherichia coli from Korea. Antimicrob. Agents Chemother. 2005;49:2522–24. doi: 10.1128/AAC.49.6.2522-2524.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mammeri H, Van de Loo M, Poirel L, Martinez-Martinez L, Nordmann P. Emergence of Plasmid-mediated Quinolone Resistance in Escherichia coli in Europe. Antimicrob. Agents Chemother. 2005;49:71–76. doi: 10.1128/AAC.49.1.71-76.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jonas D, Biehler K, Hartung D, Spitzmuller B, Daschner FD. Plasmid-mediated Quinlone Resistance in Isolates Obtained in German Intensive Care Units. Antimicrob. Agents Chemother. 2005;49:773–775. doi: 10.1128/AAC.49.2.773-775.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiegand I, Khalaf N, Al-Agamy MHM, et al. First detection of the transferable Quinolone Resistance Determinant in Clinical Providencia stuartii strains in Egypt. Clin. Microbiol. Infect. 2004;10:64. [Google Scholar]
- 16.Poirel L, De Loo MV, Mammeri H, Nordmann P. Association of Plasmid-Mediated Quinolone Resistance with Extended-Spectrum β-Lactamase VEB-1. Antimicrob. Agents Chemother. 2005;49:3091–3094. doi: 10.1128/AAC.49.7.3091-3094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodriguez-Martinez JM, Pascual A, Garcia I, et al. Detection of the plasmid-mediated quinolone resistance determinant qnr among clinical isolates of Klebsiella pneumoniae producing AmpC-type β-lactamase. J. Antimicrob. Chemother. 2003;52:703–6. doi: 10.1093/jac/dkg388. [DOI] [PubMed] [Google Scholar]
- 18.Tran JH, Jacoby GA. Mechanism of Plasmid-mediated Quinolone Resistance. Proc. Nat'l Acad. Sci. 2002;99:5638–5642. doi: 10.1073/pnas.082092899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran JH, Jacoby GA, Hooper DC. Interaction of the plasmid-encoded Quinolone Resistance Protein QnrA with Escherichia coli Topoisomerase IV. Antimicrob. Agents Chemother. 2005;49:3050–3052. doi: 10.1128/AAC.49.7.3050-3052.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran JH, Jacoby GA, Hooper DC. Interaction of the Plasmid-encoded Quinlone Resistance Protein Qnr with Escherichia coli DNA gyrase. Antimicrob. Agents Chemother. 2005;49:118–125. doi: 10.1128/AAC.49.1.118-125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poirel L, Rodriguez-Martinez J-M, Mammeri H, Liard A, Nordmann P. Origin of Plasmid-Mediated Quinolone Resistance Determinant QnrA. Antimicrob. Agents Chemotherap. 2005;49:3523–3525. doi: 10.1128/AAC.49.8.3523-3525.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hata M, Suzuki M, Matsumoto M, Takahashi M, Sato K, Ibe S, Sakae K. Cloning of a Novel Gene for Quinlone Resistance from a Transferable Plasmid in Shigella flexneri 2b. Antimicrob. Agents Chemother. 2005;49:801–803. doi: 10.1128/AAC.49.2.801-803.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nordmann P, Poirel L. Emergence of plasmid-mediated resistance to quinolones in Enterobacteriaceae. J. Antimicrob. Chemother. 2005;56:463–9. doi: 10.1093/jac/dki245. [DOI] [PubMed] [Google Scholar]
- 24.Saga T, Kaku M, Onodera Y, Yamachika S, Sato K, Takase H. Vibrio parahaemolyticus Chromosomal qnr Homologue VPA0095: Demonstration by Transformation with a Mutated Gene of Its Potential To Reduce Quinolone Susceptibility in Escherichia coli. Antimicrob. Agents Chemother. 2005;49:2144–5. doi: 10.1128/AAC.49.5.2144-2145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montero C, Mateu G, Rodriguez R, Takiff H. Intrinsic Resistance of Mycobacterium smegmatis to Fluoroquinolones may be Influenced by New Pentapeptide Protein MfpA. Antimicrob. Agents Chemother. 2001;45:3387–3392. doi: 10.1128/AAC.45.12.3387-3392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hegde SS, Vetting MW, Roderick SL, Mitchenall LA, Maxwell A, Takiff HE, Blanchard JS. A Fluoroquinolone Resistance Protein from Mycobacterium tuberculosis that Mimics DNA. Science. 2005;308:1480–1483. doi: 10.1126/science.1110699. [DOI] [PubMed] [Google Scholar]
- 27.Jenkins J, Pickersgill R. The Architecture of Parallel beta-helices and Related Folds. Prog. Biophys. Mol. Bio. 2001;77:111–175. doi: 10.1016/s0079-6107(01)00013-x. [DOI] [PubMed] [Google Scholar]
- 28.Raetz CHR, Roderick SL. A Left-handed Parallel β Helix in the Structure of UDP-N-acetylglucosamine Acyltransferase. Science. 1995;270:997–1000. doi: 10.1126/science.270.5238.997. [DOI] [PubMed] [Google Scholar]
- 29.Drlica K, Malik M. Fluoroquinolones: Action and Resistance. Curr. Topics Med. Chem. 2003;3:249–282. doi: 10.2174/1568026033452537. [DOI] [PubMed] [Google Scholar]
- 30.Morais Cabral JH, Jackson AP, Smith CV, Shikotra N, Maxwell A, Liddington RC. Crystal Structure of the Breakage-reunion domain of DNA Gyrase. Nature. 1999;388:903–906. doi: 10.1038/42294. [DOI] [PubMed] [Google Scholar]
- 31.Caldon CE, March PE. Function of the universally conserved bacterial GTPases. Curr. Opin. Microbiol. 2003;6:135–139. doi: 10.1016/s1369-5274(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 32.Rison SCG, Kendall SL, Movahedzadeh F, Stoker NG. The Mycobacterial Two-component Regulatory Systems. In: Parish T, editor. Mycobacterium Molecular Microbiology. Horizon Bioscience; Wymondham, Norfolk: 2005. pp. 29–69. [Google Scholar]
- 33.Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2) Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 34.Komatsu M, Kuwahara Y, Hiroishi A, Hosono K, Beppu T, Ueda K. Cloning of the conserved regulatory operon by its aerial mycelium-inducing activity in an amfR mutant of Streptomyces griseus. Gene. 2003;306:79–89. doi: 10.1016/s0378-1119(03)00405-0. [DOI] [PubMed] [Google Scholar]
- 35.San Milan JL, Hernandez-Chico C, Pereda P, Moreno F. Cloning and mapping of the genetic determinants for Microcin B17 production and immunity. J. Bacteriol. 1985;163:275–281. doi: 10.1128/jb.163.1.275-281.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vizan JL, Hernandez-Chico C, del Castillo I, Moreno F. The peptide antibiotic Microcin B17 induces double-strand cleavage of DNA mediated by E. coli DNA gyrase. EMBO J. 1991;10:467–476. doi: 10.1002/j.1460-2075.1991.tb07969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.del Castillo FJ, del Castillo I, Moreno F. Construction and characterization of mutations at codon 751 of the Escherichia coli gyrB gene that confer resistance to the antimicrobial peptide Microcin B17 and alter the activity of DNA gyrase. J. Bacteriol. 2001;183:2137–2140. doi: 10.1128/JB.183.6.2137-2140.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heddle JG, Blance SJ, Zamble D/B, Hollfelder F, Miller DA, Wentzell LM, Walsh CT, Maxwell A. The antibiotic Microcin B17 is a DNA gyrase poison: characterization of the mode of inhibition. J. Mol. Biol. 2001;307:1223–1234. doi: 10.1006/jmbi.2001.4562. [DOI] [PubMed] [Google Scholar]
- 39.Pierrat OA, Maxwell A. Evidence for the role of strand passage in the mechanism of action of Microcin B17 on DNA gyrase. Biochemistry. 2005;44:4202–4215. doi: 10.1021/bi0478751. [DOI] [PubMed] [Google Scholar]
- 40.Morita M, Tomita K, Ishizawa M, Tagaki K, Kawamura F, Takahashi H, Morino T. Cloning of oxetanocin A biosynthetic and resistance genes that reside on a plasmid of Bacillus megaterium strain NK84-0128. Biosci Biotechnol Biochem. 1999;63:563–566. doi: 10.1271/bbb.63.563. [DOI] [PubMed] [Google Scholar]
- 41.Izuta S, Shimada N, Kitigawa M, Suzuki M, Kojima K, Yoshida S. Inhibitory effects of triphosphate derivatives of oxetanocin G and related compounds on eukaryotic and viral DNA polymerases and human immunodeficiency virus reverse transcriptase. J. Biochem. 1992;112:81–87. doi: 10.1093/oxfordjournals.jbchem.a123870. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi H, Amano R, Saneyoshi M, Maruyama T, Yamaguchi T. Inhibition of vertebrate telomerases by the triphosphate derivatives of carbocyclic oxetanocin analogs. Nucleic. Acids Res. Suppl. 2003;3:285–286. doi: 10.1093/nass/3.1.285. [DOI] [PubMed] [Google Scholar]
- 43.Hayashi S, Norbeck DW, Rosenbrook W, Fine RL, Matsukura M, Plattner JL, Broder S, Mitsuya H. Cyclobut-A and cyclobut-G, carbocyclic oxetanocin analogs that inhibit the replication of human immunodeficiency virus in T cells and monocytes and macrophages in vitro. Antimicrob Agents Chemother. 1990;34:287–294. doi: 10.1128/aac.34.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chandler LE, Bartsevich VV, Pakrasi HB. Regulation of Manganese Uptake in Synechocystis 6803 by RfrA, A Member of a Novel Family of Proteins Containing a Repeated Five-Residue Domain. Biochemistry. 2003;42:5508–5514. doi: 10.1021/bi027113a. [DOI] [PubMed] [Google Scholar]
- 45.Kamei A, Yoshihara S, Yuasa T, Geng X, Ikeuchi M. Biochemical and Functional Characterization of a Eukaryotic-type Protein Kinase, SpkB, in the Cyanobacterium, Synechocystis sp. PCC 6803. Current Microbiol. 2003;46:296–301. doi: 10.1007/s00284-002-3887-2. [DOI] [PubMed] [Google Scholar]
- 46.Daigle DM, McKay GA, Thompson PR, Wright GD. Aminoglysocide antibiotic phosphotransferases are also Serine Protein Kinases. Chemistry & Biology. 1999;6:11–18. doi: 10.1016/S1074-5521(99)80016-7. [DOI] [PubMed] [Google Scholar]
- 47.Vetting MW, Magnet S, Nieves E, Roderick SL, Blanchard JS. A Bacterial Acetyltransferase Capable of Regioselective N-Acetylation of Antibiotics and Histones. Chemistry & Biology. 2004;11:565–573. doi: 10.1016/j.chembiol.2004.03.017. [DOI] [PubMed] [Google Scholar]