

Abstract
In this video, we demonstrate how to use the neuron microfluidic device without plasma bonding. In some cases it may be desirable to reversibly bond devices to the Corning No. 1 cover glass. This could be due, perhaps, to a plasma cleaner not being available. In other instances, it may be desirable to remove the device from the glass after the culturing of neurons for certain types of microscopy or for immunostaining, though it is not necessary to remove the device for immunostaining since the neurons can be stained in the device. Some researchers, however, still prefer to remove the device. In this case, reversible bonding of the device to the cover glass makes that possible. There are some disadvantages to non-plasma bonding of the devices in that not as tight of a seal is formed. In some cases axons may grow under the grooves rather than through them. Also, because the glass and PDMS are hydrophobic, liquids do not readily enter the device making it necessary at times to force media and other reagents into the device. Liquids will enter the device via capillary action, but it takes significantly longer as compared to devices that have been plasma bonded. The plasma cleaner creates temporary hydrophilic charges on the glass and device that facilitate the flow of liquids through the device after bonding within seconds. For non-plasma bound devices, liquid flow through the devices takes several minutes. It is also important to note that the devices to be used with non-plasma bonding need to be sterilized first, whereas plasma treated devices do not need to be sterilized prior to use because the plasma cleaner will sterilize them.
Protocol
Preparing Microfluidic Devices For Neuron Cell Culture
1) Clean Corning No.1 24 mm X 40 mm glass slides
Sonicate the glass slides in a water bath sonicator for 30 minutes
Rinse the glass slides with 70% EtOH
Wash slides three times with dH2O
Dry slides in a tissue culture hood for several hours, preferably overnight
Note: We have special metal trays that we load with the slides. The slides are separated individually and placed in slots in this metal rack. We can then place these metal loading trays in a glass dish that we fill with water, place in the water bath sonicator, and sonicate for 30 minutes. Subsequent washes are carried out in this dish.
2) Preparing the PDMS devices
Cut out the PDMS mold from the silicon wafer, punch holes in the PDSM cast and quarter it
Clean the PDMS devices by first blowing inert gas (Argon or Nitrogen) across them. Then use 3M Scotch Brand 471 tape to lift off any remaining debris
Note: It is important to keep the devices face up. Initially, when we cut the PDMS away from the silicon wafer, the side in contact with the wafer is the clean side: it contains the microfluidic channels. We try to be as careful as we can as to not damage the device, and keep the clean side face up until we are ready for plasma bonding.
Autoclave devices in plastic containers
After autoclaving, clean No. 1 Corning glass slides and plasma-treat devices using a Harrick brand plasma cleaner, www.harrickplasma.com
Place the devices clean side down on the glass slide.
The device is now plasma bonded to the glass slide.
3) Coating The Devices With Poly L-Lysine (PLL)
PLL is added to a well on each side of the device and allowed to flow through into the next well
=> 150 µl of PLL for the large wells and 30 µ l PLL for the smaller wells. It does not have to be exact as long as the wells are full.
Note: If you look at a device you will see 4 wells: each two are connected. You want to put the PLL in one well so that it will flow through the device into the next well.
Allow the PLL to flow through the device for about 10 minutes and then add more PLL to the wells to fill them up
Place the devices containing PLL in an incubator at 37°C for a minimum of 4 hours (overnight is preferable)
Vacuum out the excess PLL after treatment, but be careful not to suck all the liquid out of the device
Note: You do not want to introduce air bubbles to the channels or chamber. Just vacuum out the excess PLL in the wells.
Add autoclaved dH2O to each well on either side of the device, and allow to flow to the other well by capillary action
Figure 1 is a diagram of a microfluidic device. Notice how the Blue (Soma side) is connected and the Red (Axonal side) are connected. When we say put the PLL, or water or media, on one well on each side of the device, what we mean, for example, is: Place 150 ul of autoclaved dH2O in one Blue Well then place 150 µl of autoclaved dH20 into one Red Well. The water (or PLL, or Media, or Cells) will flow through the device to the other corresponding well.
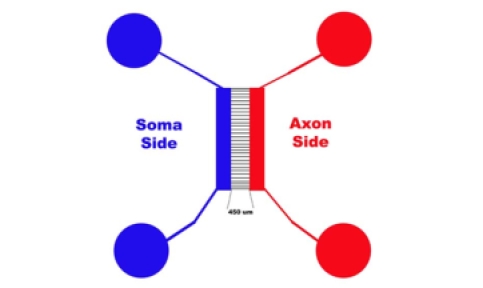
Figure 1
Note: Please note that, from now on, when I say "place media, cells, water or PLL in the TOP wells", I am referring to the diagram above where the Soma would be on the left and the Axons will grow to the right.
Aspirate off the water (again being careful not to fully remove all the liquid from the device), then add another 150 µl of autoclaved dH2O to one of each connected well and allow it to flow through to the corresponding well.
Place the device containing dH2O in an incubator at 37°C for 1 hour, then repeat the wash step again. Wash the devices three times with dH2O for one hour each, which is the total.
Note: Due to the possibility that borate from the PLL can absorb into the PDMS, some in the Jeon Lab recommend incubating the devices overnight with autoclaved dH2O after a couple of initial quick rinses. This will ensure that all free borate will leach out of the PDMS prior to the loading of cells. If this is not done, there may be a release of borate into the media over time, which could lead to cytotoxicity.
Add Neural Basal Media (NBM) with the necessary factors (glutamax, B27, PenStrep) to the top wells
Note: The devices can be incubated overnight and plated with cells the next day, or incubated a minimum of 3 hours with NMB + factors before plating the cells.
Preparing The Neurons For Loading Into The Devices
We use 18 day old fetal rat cortex that we either buy from a company called Brain Bits or that is prepared here on campus for us. We prefer to get the tissue fresh.
Obtain 1 fetal brain cortex (2 pieces of tissue) stored in Hibernate E
Take 3 long glass pipettes and fire them each into successively smaller sizes using an alcohol lamp
remove tissues from Hibernate E with a glass pipette and place in 2 ml of NMB + Factors in a sterile 15 ml tube.
-
Trituration: Using a bulb and glass pipette the cortex is passed up and down about 5 to 10 times. Then, the next smaller pipette is used to pipette up and down the tissue. Finally, the smallest pipette is used to pipette up and down the tissue. We like to make sure that there are no visible chunks.
Note: Recently, the Jeon Lab began comparing cells from tissue stored in Hibernate E to cells that were prepared from Tissue initially treated with 0.125% Trypsin in 50% Ca++ free and Mg++ free dissection buffer. The consensus is that tissue prepared using trypsin yield more viable cells than tissue placed initially in Hibernate E. If you are NOT going to use the tissue right away, then you need to store the tissue in Hibernate E.
If you are going to use the tissue right away and would like increased viability, then treat the tissue with Trypsin prior to mechanical trituration as following: 1) dilute 1 ml 0.25% Trypsin (Gibco, Invitrogen) with 1 ml of Ca++ free Mg++ free dissection buffer (final Trypsin = 0.125%) into a 15 ml tube (keep ice cold); 2) place tissue in buffer and incubate immediately at 37°C for 8 minutes; 3) add 10 ml DMEM/10% FBS to stop the Trypsin reaction and continue protocol below.
Centrifuge cells at 1100 rpm for 1 minute
Aspirated media carefully off the cell pellet
Add 1 ml of NMB + factors to the pellet and re-suspended it by pipetting up and down
Pass the re-suspended cells by gravity flow through a filter to remove any clumps (BD Falcon cell strainer 40 um nylon)
- Count and plate cells:
- Into a microfuge tube, add 60 µl NMB + factors, 20 µl of cell suspension, and 20 µl of trypan blue then mix
- Add about 10 µl of this solution to a hemocytometer and count the live cells (not blue)
-
Adjust concentration to 2.5 - 4.5 x106 cells/mlNote: We generally obtain a concentration of about 2.5 - 4.5 x 106 cells/ml. depending on the volume the cells are resuspended in (normally 1 ml NBM + B27/Glutamax/PenStrep percortex). If the tissue was prepared with trypsin, the yield will likely be increased.
Plating The Devices
Load cells (primary neurons) into one well of the device
-
Plate 5 µl per small device and 20 µl per big device
Note: You can load 10 µl of cells on the top of the device and 10 µl on the bottom of the device (half total volume), or directly all 20 µl of cells in the top somal well (5 µl in the top somal well for the small devices).
Incubate for 10 minutes in a 37°C incubator so cells can start to attach
-
Add approximately 150/30 µl of NMB + factors to each well depending on device size
Note: Volumes may vary depending on the thickness of the PDMS. All that matters is that the wells are full.
Supplemental On Using The Devices Without Plasma Bonding
Without plasma treating, Christina Tu, a technician, who does it successfully every week without plasma, says to just load the cells right away.
Remember to remove the media from both wells on the soma side (axon side won't matter if you leave the media in). It's the fluid flow that allows the cells to enter the main channel. If you have media in the wells, you won't get fluid flow.
Discussion
Here we demonstrated how to use the neuron microfluidic device without needing a plasma cleaner. We refer to this as non-plasma bonding.
References
- Park JW, Vahidi B, Taylor AM, Rhee SW, Jeon NL. Microfluidic culture platform for neuroscience research. Nat Protoc. 2006;1(4):2128–2136. doi: 10.1038/nprot.2006.316. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2(8):599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]