

Abstract
Mutations in COCH (coagulation factor C homology) are etiologic for the late-onset, progressive, sensorineural hearing loss and vestibular dysfunction known as DFNA9. We introduced the G88E mutation by gene targeting into the mouse and have created a CochG88E/G88E mouse model for the study of DFNA9 pathogenesis and cochlin function. Vestibular-evoked potential (VsEP) thresholds of CochG88E/G88E mice were elevated at all ages tested compared with wild-type littermates. At the oldest ages, two out of eight CochG88E/G88E mice had no measurable VsEP. Auditory brainstem response (ABR) thresholds of CochG88E/G88E mice were substantially elevated at 21 months but not at younger ages tested. At 21 months, four of eight CochG88E/G88E mice had absent ABRs at all frequencies tested and two of three CochG88E/+ mice had absent ABRs at three of four frequencies tested. Distortion product otoacoustic emission amplitudes of CochG88E/G88E mice were substantially lower than Coch+/+ mice and absent in the same CochG88E/G88E mice with absent ABRs. These results suggest that vestibular function is affected beginning as early as 11 months when cochlear function appears to be normal, and dysfunction increases with age. Hearing loss declines substantially at 21 months of age and progresses to profound hearing loss at some to all frequencies tested. This is the only mouse model developed to date where hearing loss begins at such an advanced age, providing an opportunity to study both progressive age-related hearing loss and possible interventional therapies.
INTRODUCTION
COCH (coagulation factor C homology) and its encoded protein, cochlin, are expressed at high levels in the inner ear as initially shown by northern blot, tissue in situ hybridization, and immunohistochemistry (1,2). Cochlin is detected by proteomic analysis as the most abundant protein in the bovine (3), as well as mouse and human inner ears (4). A growing number of mutations in COCH, causing late-onset, progressive hearing loss and vestibular dysfunction characteristic of DFNA9 are being diagnosed (Fig. 1 and Table 1). In addition to the original mutations clustered in the FCH(factor C homology)/LCCL(limulus factor C, cochlin, lung gestational protein) domain (5–8), there are now three mutations reported in the vWFA2 domain (von Willebrand factor A-like domain) (9,10), as well as another recently reported mutation (in the FCH/LCCL domain) in a Dutch kindred (11).
Figure 1.

Schematic representation of the deduced amino acid structure of human COCH, encoding the protein cochlin, shows a predicted signal peptide (SP), followed by a domain initially designated as FCH, also known as the LCCL domain, followed by an intervening domain, and two domains (vWFA1 and vWFA2) separated by an intervening domain. Twelve mutations (all missense except for one in-frame deletion), causing DFNA9 deafness and vestibular disorder, are indicated. The positions of all cysteine residues are shown as ‘C’.
Table 1.
COCH mutations in DFNA9
| Origin | Exon with mutation | Nucleotide change | Amino acid change | Protein domain | Reference |
|---|---|---|---|---|---|
| Belgium and The Netherlands | 4 | C207T | P51S | FCH/LCCL | (6,7) |
| United States | 4 | T253G | V66G | FCH/LCCL | (5) |
| The Netherlands | 5 | G315T | G87W | FCH/LCCL | (28) |
| United States and The Netherlands | 5 | G319A | G88E | FCH/LCCL | (5,15) |
| Hungary | 5 | 366_368delGTA | V104del | FCH/LCCL | (29) |
| The Netherlands | 5 | T382C | I109T | FCH/LCCL | (11) |
| Australia | 5 | T382A | I109 N | FCH/LCCL | (8) |
| United States | 5 | T405C | W117R | FCH/LCCL | (5) |
| Japan | 5 | G411A | A119T | FCH/LCCL | (30) |
| China | 12 | T1591C | M512T | vWFA2 | (10) |
| United States | 12 | G1681T | C542F | vWFA2 | (9) |
| China | 12 | G1681A | C542Y | vWFA2 | (10) |
Prevalence of COCH mutations is not known, as there is no systematic genetic screening worldwide in individuals with late-onset hearing loss. However, the finding of COCH mutations on four continents (Table 1), and the observation of four distinct mutations in the Netherlands alone, suggests that the true prevalence of COCH mutations may be substantially higher than currently recognized. In addition, the possibility of COCH playing important roles in presbycusis and disorders of imbalance has been considered (6,7), and cochlin has also been shown as a major target antigen for autoimmune sensorineural hearing loss by both antibody and T-cell-mediated mechanisms (12–14).
Temporal bones from individuals with DFNA9 exhibit an intriguing and characteristic histopathology. The number of fibrocytes in the spiral ligament and in the spiral limbus is reduced and a homogeneous eosinophilic material is present in these structures that stain with anti-cochlin antibodies (4). The relationship between the finding of cochlin-staining eosinophilic material and lack of cellularity remains to be elucidated. In an effort to understand the function of COCH in the inner ear and its role in pathogenesis of DFNA9 hearing and balance disorder, we created a mouse knock-in model designated CochG88E/G88E. Because all mutations known to date in COCH are either missense or an in-frame deletion, and none lead to premature termination and truncation of the protein, we chose to create the targeted missense mutation in the mouse rather than a null mutant.
The G88E mutation was initially found in a US family (5) and subsequently in the Netherlands with likely different founders (15). In vitro studies of the FCH/LCCL domain of bacterially expressed cochlin with the G88E mutation (as well as several others of the mutations) have revealed misfolding of this domain by nuclear magnetic resonance (NMR) structural analyses (16). Herein, we report the generation of a mouse model bearing the G88E mutation, and describe the results of functional hearing and vestibular testing as well as histological findings. With age-related hearing loss being the most common sensory problem in humans, our mouse model provides potentially an excellent system for its study; this is the only model to date with onset of hearing loss at this advanced age.
RESULTS AND DISCUSSION
Verification of the CochG88E/G88E mouse model
RT–PCR analysis
To confirm successful transcription of mutant Coch, we performed reverse transcription polymerase chain reaction (RT–PCR) of ribonucleic acid (RNA) isolated from a CochG88E/+ mouse. An expected product of 1.7 kb representing the full-length coding region of Coch was obtained. Sequencing of the PCR product was performed, showing presence of the introduced Coch mutation as well as correct splicing of introns 4–6, confirming that the remaining loxP site in intron 5 after deletion of neo (neomycin) does not disrupt accurate splicing.
Immunohistochemistry
To confirm translation of the mutant CochG88E/G88E transcript, we performed immunohistochemistry on cochlear and vestibular sections from a 3-month-old CochG88E/G88E mouse, using an anti-cochlin antibody, which showed intense and specific cochlin staining in the structures where cochlin expression is normally seen in the wild-type inner ear (4) (Fig. 2). Specifically, cochlin immunostaining was prominent throughout the spiral ligament and spiral limbus in the cochlea and in the area of stromal fibrocytes in the crista ampullaris of a semi-circular canal. All immunostaining represents mutant cochlin, confirming successful translation of the mutant cochlin protein. Of note, inner ear sections from Coch−/− immunostained with the same antibody show complete lack of staining (4).
Figure 2.

Immunohistochemistry on postnatal (3-month-old) CochG88E/G88E (C, G) and Coch+/+ (A, E) mouse cochlea (A, C ×150) and crista of the posterior ampulla in the vestibular system (E, G ×200) with anti-cochlin. Immunostaining (in the left panels) appears as a reddish brown DAB reaction product; no counterstain was applied on these sections. Serial sections stained with H&E are shown in parallel in the right panels. The CochG88E/G88E mice contain only mutant Coch, therefore all immunostaining represents mutant cochlin, confirming successful translation of the mutant cochlin protein. The pattern of immunostaining in the CochG88E/G88E inner ear is similar to that of the Coch+/+ mice, as previously described (4). Prominent cochlin immunostaining is present in the fibrocytes and in the extracellular matrix throughout the spiral ligament [filled arrows in (A)] and spiral limbus [open arrow in (A)] and in the fibrocytes and stroma underlying the sensory epithelium in the crista [open arrow in (E)], as well as in the ampullary wall [filled arrow in (E)].
Functional studies: VsEP, ABR and DPOAE measurements
Vestibular-evoked potential (VsEP) measurements are a reflection of vestibular function, more specifically of gravity receptor or otolithic organs, the saccule and utricle. VsEP thresholds for CochG88E/G88E, CochG88E/+ and Coch+/+ mice were obtained at five ages spanning 11–21 months (Fig. 3). VsEP thresholds of CochG88E/G88E mice were elevated at all ages tested compared with the Coch+/+ littermates. At 19 and 21 months, two of the CochG88E/G88E mice had no measurable VsEP. VsEP thresholds for CochG88E/G88E and Coch+/+ mice at 11, 17, 19 and 21 months were analyzed using a two-factor analysis of variance (ANOVA, Age X Genotype). The two factor ANOVA revealed no significant interaction; however significant main effects were seen for age [F(3,35) = 4.349, P = 0.01] and genotype [F(1,35) = 15.749, P < 0.001]. VsEP thresholds for CochG88E/G88E mice were significantly higher than for Coch+/+ mice and the oldest age groups (17, 19 and 21 months) each had significantly higher VsEP thresholds than 11-month-old mice (post-hoc least significant difference, P = 0.015, 0.006 and 0.0001, respectively). These results suggest that gravity receptor function declines with advancing age for both genotypes, but CochG88E/G88E mice have a greater deficit at all ages tested. Interestingly, VsEP thresholds for the CochG88E/+ mice were similar to the Coch+/+ mice at the youngest and oldest ages tested.
Figure 3.
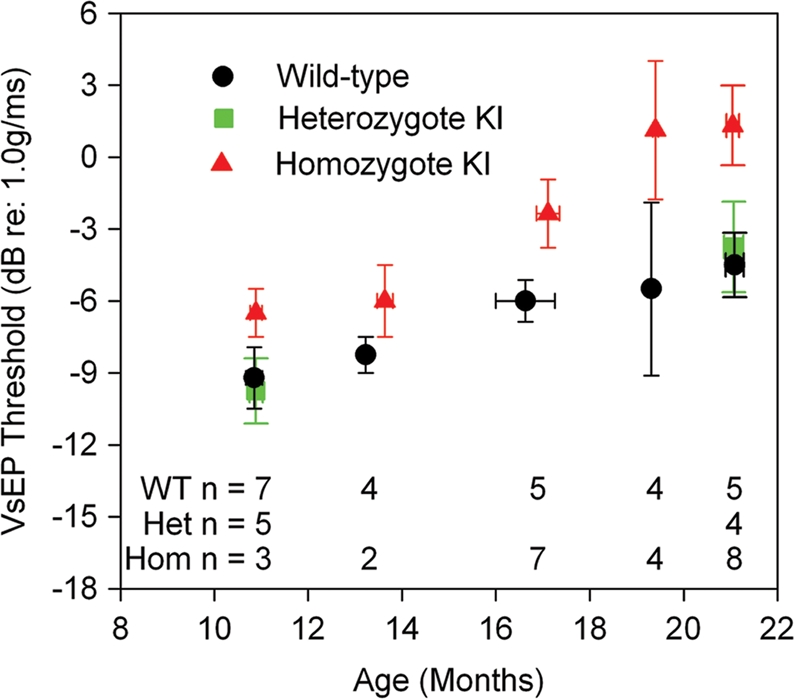
Means and standard errors for VsEP thresholds as a function of age. Coch+/+ and CochG88E/G88E mice are represented at all ages tested. CochG88E/+ mice were tested at the youngest and oldest ages. Although age-related decline in gravity receptor sensitivity can be seen for all genotypes, VsEP thresholds for CochG88E/G88E mice were significantly higher than for Coch+/+ mice at all ages tested, indicating poorer vestibular function for CochG88E/G88E mice. In fact, at 19 and 21 months of age, two of the CochG88E/G88E mice had no measurable VsEP.
Although gravity receptor (otolithic organs) function for the CochG88E/G88E mice appeared to be affected as early as 11 months, auditory function at 11 months was identical for all genotypes (Figs 4 and 5). Distortion product otoacoustic emission (DPOAE) amplitudes of CochG88E/G88E mice at 19 months were slightly reduced for the mid-frequencies (5–10 dB smaller) (Fig. 5). One 19-month-old CochG88E/G88E mouse had much higher ABR thresholds than the other CochG88E/G88E mice (data not shown) and had essentially absent DPOAEs indicating that it was more substantially affected with hearing loss. This mouse also had absent VsEPs, indicating concomitantly more severe phenotypes in both the hearing and gravity receptor systems. By 21 months of age, DPOAE amplitudes for the CochG88E/G88E mice were substantially smaller than for Coch+/+ mice (up to 30 dB smaller) and ABR thresholds were substantially higher than for wild-types. Indeed, six of the eight CochG88E/G88E mice tested at 21 months had absent DPOAEs and absent ABRs at the maximum stimulus levels of 32 and 41 kHz frequencies, and four of eight CochG88E/G88E mice tested had absent ABRs at all frequencies; therefore, the ABR mean thresholds shown in Figure 4 likely underestimate actual auditory thresholds. CochG88E/+ mice at 21 months showed elevated ABR thresholds at the same level as the CochG88E/G88E mice. The CochG88E/+ mice also displayed reduced DPOAE amplitudes.
Figure 4.

Means and standard errors for ABR thresholds at 8, 16, 32 and 41.2 kHz. Data for all genotypes at two ages are shown. At 21 months, four of eight CochG88E/G88E mice had absent ABRs at all frequencies tested, and two of three CochG88E/+ mice had absent ABRs in three of four frequencies tested; therefore, symbols for these groups represent a conservative estimate of ‘average’ thresholds. Arrows denote that true thresholds may be higher than estimated. Overall, the data demonstrate an age-related decline in hearing among all genotypes, but the most substantial declines were seen in the CochG88E/G88E mice where half of the animals tested had profound hearing loss at all frequencies. CochG88E/+ mice also show decline in auditory function at a level similar to that of the CochG88E/G88E mice.
Figure 5.

Average DPOAE amplitudes are shown at 19 and 21 months. Error bars represent standard errors. Primaries (f2/f1 = 1.25) from 5 to 55 kHz were presented at 65 dB SPL. At 19 months, DPOAEs were present for CochG88E/G88E and Coch+/+ mice although emissions for CochG88E/G88E mice were 10–15 dB smaller than those of the Coch+/+ mice. At 21 months, smaller DPOAE amplitudes were recorded for all genotypes suggesting age-related changes; however, emissions for the CochG88E/G88E mice decreased by up to 20 dB while Coch+/+ mice emissions dropped only by ∼10 dB. Emission amplitudes for CochG88E/G88E mice were substantially smaller than those of Coch+/+ mice at 21 months. Consistent with the ABR results, half of the CochG88E/G88E mice (i.e. those with absent ABRs) also had absent DPOAEs (i.e. DPOAE amplitudes indistinguishable from the noise floor).
Overall, the functional results suggest that vestibular and cochlear dysfunction occur in the DFNA9 mouse model, but not simultaneously. Vestibular function, particularly gravity receptor function, is affected beginning as young as 11 months when cochlear function appears to be normal. Vestibular deficit increases with advancing age, including profound deficits identified in two elderly CochG88E/G88E mice. Cochlear function begins to deteriorate at advanced ages. Since few studies have systematically evaluated vestibular function in patients with DFNA9, the extent of vestibular dysfunction and age of onset are not well characterized. However, some clinical reports of DFNA9 patients do indicate onset of vestibular malfunction to precede hearing loss, similar to the situation observed in this mouse model. Studies on a Dutch family bearing this mutation revealed start of hearing deterioration at age 46–49 and onward, and onset of vestibular dysfunction at ∼46 years of age (15). Similarly, a large study of 74 individuals in the Netherlands bearing the P51S mutation, in the same domain of cochlin as the G88E mutation, also showed the onset of vestibular malfunction (34 years of age) to precede onset of hearing deterioration (43 years of age, onwards) (17). Another study of DFNA9 patients, with the C542F mutation (9), demonstrates that VEMPs (vestibular-evoked myogenic potentials) were absent in two of three DFNA9 patients tested, indicating saccular otolithic deficits. These results are in agreement with our mouse VsEP results, which also demonstrate otolithic deficits. In this study (9), canal dysfunctions in some subjects [bilateral weakness for calorics and reduced VOR (vestibular ocular reflex) gains in rotary chair testing] were implicated. We cannot rule out canal dysfunction in the mouse model at this time, as VOR testing has not been performed.
Onset of hearing loss in patients with DFNA9 is quite late, up to the fifth decade. Late-onset hearing loss in this mouse model is consistent with findings reported in human patients. Earlier onset of vestibular malfunction in this mouse model, as well as the human VOR data (15,17), strongly indicate that vestibular testing, including measures that evaluate ampullar and macular end organs, should be considered in the clinical evaluation of patients with DFNA9.
Histopathology
Initial examination of CochG88E/G88E mice at 1 year of age revealed some deterioration of type IV fibrocytes of the spiral ligament seen as loss of nuclei (data not shown). However, at more advanced ages, the loss of type IV fibrocytes observed in the CochG88E/G88E were similar to changes seen in the Coch+/+ mice. In CochG88E/G88E mice of 21 (n = 8) and 26 (n = 2) months of age, histology did not reveal obvious deterioration of cochlear and vestibular sensory epithelia (Fig. 6). Interestingly, however, CochG88E/G88E mice at this age have pronounced hearing and vestibular symptoms.
Figure 6.

Photomicrograph of (A) the cochlear duct of a 26-month-old CochG88E/G88E mouse, and (B) the utricular macula and (C) saccular macula of a 21-month-old CochG88E/G88E mouse. Some loss of type IV fibrocytes in the cochlea in these mice is observed, but similar changes are present in the Coch+/+ mice (data not shown). There is no obvious deterioration of cochlear and vestibular sensory epithelia.
In addition, our mouse model did not show the eosinophilic deposits characteristic of DFNA9 suggesting that there is no detectable cochlin aggregation in the mouse at the ages tested. It is possible that these microfibrillar deposits have not yet formed to the degree of visibility and are at a lower level than detectable at the time of analysis in the mouse. It is also possible that the histopathology in the moue exhibits different expressivity from that in the human where eosinophilic deposits are observed in end-stage DFNA9.
MATERIALS AND METHODS
Construction of the targeting vector
To construct a mutant Coch targeting vector (Fig. 7A), we screened a mouse 129 genomic library in EMBL3 phage (gift of Dr R. Maas, Boston, MA, USA) (18), using radiolabeled Coch cDNA probes by standard methods. Coch genomic clones were isolated and characterized by restriction mapping. A 5.58 kb fragment spanning introns 3 to 8 of Coch was subcloned into pBluescript II SK vector (Stratagene, La Jolla, CA, USA). A nucleotide change in exon 5 (A to G), translating into the p.G88E mutation was introduced using QuikChange site-directed mutagenesis following the manufacturer’s protocol (Stratagene). A unique AvrII site was introduced into intron 5 by the same method, to allow subsequent cloning of a neomycin-resistant gene, pgk-neo, flanked by loxP sites, for positive selection of embryonic stem (ES) cells transfected with the construct. The 5.58 kb Coch genomic fragment containing the mutation was cloned into a vector containing the thymidine kinase (tk) gene for negative selection (gift of Dr R. DePinho, Boston, MA, USA), with the tk gene positioned upstream of the 5′ end of the Coch fragment. A 2 kb loxP–neo–loxP cassette was then cloned into intron 5. The resulting targeting vector (Fig. 7A) has a shorter homologous recombinant arm of 1.68 kb derived from the 5′ end of the Coch fragment, upstream of the neo cassette, and a longer homologous recombinant arm of 3.9 kb from the 3′ side of the Coch fragment, downstream of neo.
Figure 7.

(A) Representation of the Coch targeting construct, homologous recombination with the wild-type allele (targeted locus), screening strategy for recombinants by PCR analysis, and removal of the neo cassette in the targeted allele, followed by PCR analysis to identify recombinants with neo excised. (B) Screening strategy for homologous recombinants by Southern blot analysis.
Generation of CochG88E/G88E mutant mice
The targeting construct was linearized with a unique AscI site, 3′ of the Coch construct and electroporated into J1 (129-derived) ES cells. Homologous recombinants were identified by both PCR and Southern blot analysis. For PCR analysis (Fig. 7A), a forward primer upstream of the 5′ Coch homology arm in the targeting construct (5′-GTG CTG CCT TCC ATA TTC CTT TGG-3′) and a reverse primer in neo (5′-CGC ATT GTT AGA TTT CAT ACA CGG-3′) were used, yielding a 2.26 kb fragment. For the long homologous arm, a forward primer in neo (5-′TAT ACG AAG TTA TTA GGT CCA GCG-3′) and a reverse primer in the 3′ region external to the homologous arm of the targeting construct (5′-CAT GTA AAG AGA GCT AGT GTG ATG-3′) were used, yielding a 4.2 kb fragment. The amplified products were sequenced to confirm homologous recombination of the construct into the genomic Coch targeted locus, as well as presence of the targeted mutation and accuracy of all exons, exon–intron junctions and neo sequence.
Southern blot analysis (Fig. 7B) using probes external to the genomic DNA in the targeting vector was performed for both the 5′ and 3′ sides of the homologous regions. Southern blots of BamHI and HindIII digests of the ES cell genomic DNA probed with a 5′ probe yielded 4.57 and 7.8 kb band, respectively, in the wild-type, and a 3.77 and 6.45 kb band, respectively, in the targeted allele. BglII and PstI genomic digests probed with a 3′ probe yielded 9.16 and 7.5 kb bands, respectively, in the wild-type, and 11.2 and 5.0 kb bands, respectively, in the targeted allele.
Two rounds of transfections of the mutant Coch targeting construct into J1 ES cells were performed, yielding ∼240 ES cell clones, which were screened for homologous recombination. A total of four ES cell clones were found to be homologous recombinants. Integrity of all exons, the introduced mutation, loxP sites, and accurate recombination of homologous arms were screened by PCR, Southern blots and sequencing. One homologous recombinant clone had a small insertion from the vector at the end of one homologous arm, and another had not incorporated the mutant base pair; therefore these clones were not used for blastocyst injections. The third clone, which appeared not to have any aberrations in homologous recombination, yielded very few chimera, none of which showed germline transmission of the mutant allele. Subsequently this ES cell clone was karyotyped and shown to be mosaic for trisomy 8, which may have been the cause of unsuccessful germline transmission. The fourth ES cell clone showed homologous recombination and no other defects as verified further by sequencing of the mutation, exons and borders of the homologous arms. This ES cell clone was used for microinjection into C57/6 blastocysts, which were implanted into pseudopregnant mice and resulted in several chimera which showed germline transmission of the Coch G88E allele.
Male chimeric mice with high levels of ES cell contribution were backcrossed to C57BL/6 females. F1 heterozygous offspring with agouti coat color were further tested by genotyping using tail DNA for PCR and Southern blot analysis to confirm germline transmission of the mutant Coch construct. For PCR analysis of F1 genotyping, we employed a forward primer in exon 5 (5′-TTC CTG GTC GAG AGA ACT AC-3′) and two reverse primers, one in neo (5′-CGC ATT GTT AGA TTT CAT ACA CGG-3′) and the other in exon 6 (5′-CTT CCT GGG TAC TGC TTT TGC-3′), yielding a 400 and a 550 bp amplified product in the targeted and wild-type alleles, respectively.
The neo cassette flanked by loxP sites was then excised from the targeted locus by breeding F1 heterozygotes with a transgenic mouse carrying EIIa-cre (19), leaving a loxP site in intron 5 of Coch (Fig. 7A). Deletion of the neo gene in the F1 offspring was assayed by PCR of tail DNA using forward and reverse primers (5′-TCAACCATGGGACAGAGTTACC-3′ and 5′-TTGATGTATTTGAAAAGCAC-3′) flanking the remaining loxP site, yielding a 276 and a 176 bp amplified product in the targeted and the wild-type alleles, respectively (Fig. 7A). Heterozygotes with excised neo were then crossed with the CBA/CaJ strain, and those with germline transmission of the excised neo and loss of the cre transgene were identified. Breeding to the CBA/CaJ strain for at least 10 generations was continued, because this strain does not exhibit significant hearing loss at advanced ages, and therefore does not confound hearing analysis of the mutant mouse at advanced ages. Intercrosses from the first and third generations of CBA/CaJ crosses were performed to generate cohorts of mice with all three genotypes for functional and histological analyses.
Tissues
Mouse tissues were obtained according to guidelines and protocols approved by the Harvard Medical School Standing Committee on Animals (Boston, MA, USA). For RNA isolation, spleen tissue was dissected from a CochG88E/+ mouse and processed as described in the following section. For histology and immunohistochemistry, mice were perfused intracardially and tissues fixed in 4% paraformaldehyde. After removal of the stapes from the oval window, and piercing of the round window, 4% paraformaldehyde fixative was perfused gently through the cochlea. Inner ears were immersed in fixative for 24 h, followed by decalcification in 120 mm ethylenediaminetetraacetic acid (EDTA) for 1 week at room temperature, and embedded in paraffin by standard histologic procedures. Serial sections were obtained at 5–8 microns thickness and used for staining with hematoxylin and eosin (H&E) and for immunohistochemistry.
Also for histologic evaluation, osmium tetroxide staining and embedding in Araldite resins (Polysciences, Warrenton, PA, USA) was performed, as this technique yields better morphology of tissues. Mice were perfused intracardially with 2.5% glutaraldehyde and 1.5% paraformaldehyde in a 65 mm phosphate buffer. Both petrous temporal bones were extracted and the round and oval windows were opened to allow intralabyrinthine perfusion of fixative. After overnight postfixation in the same fixative at room temperature, cochleas were osmicated (1% OsO4 in dH2O) for 1 h and decalcified (100 mm EDTA with 0.4% glutaraldehyde) for 3 days. After decalcification, cochleas were dehydrated in ethanols and propylene oxide and then embedded in Araldite resins and sectioned at 40 µm with a carbide steel knife. A total of 10 CochG88E/G88E mice (eight at 21 months old, and two at 26 months old), and seven Coch+/+ mice (five at 21 months old, and two at 26 months old) were analyzed for histology.
RNA isolation and RT–PCR
Total cellular RNAs were isolated (20) from a CochG88E/+ mouse spleen and reverse transcribed using SuperScript III (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. PCR of the cDNA was performed for 30 cycles under standard conditions, with a 53°C annealing, using forward (5′-TCC ACG GAG ATC CTC GCC ATG-3′) and reverse (5′-TGC ACG TAT TCC TTG AGT TGT CAG-3′) primers in exons 2 and 12, respectively. The amplified product was gel isolated and sequenced.
Immunohistochemistry
Immunostaining was performed using an anti-cochlin antibody generated against a peptide in the vWFA1 domain of cochlin (Fig. 1), corresponding to amino acid residues 163–181 of human cochlin, identical to the residues in murine and bovine cochlin (21). Anti-serum was purified through a protein A sepharose column, followed by peptide-affinity chromatography. This antibody (anti-cochlin/vWFA1 domain) recognizes all three different-sized isoforms of cochlin.
Immunohistochemistry was performed as previously described (4). Briefly, paraffin-embedded sections from postnatal CochG88E/G88E and Coch+/+ mice, were incubated with anti-cochlin/vWFA1 domain antibody overnight at room temperature, washed, and incubated with a secondary biotinylated anti-rabbit IgG (Vector Labs, Burlingame, CA, USA). Immunostaining was visualized by incubation with the Vectastain ABC reagent (Vector Labs) followed by 3,3′-diaminobenzidine (DAB). Sections were not counterstained.
VsEP, ABR and DPOAE measurements
Animals and animal preparation
The use of animals for these studies was approved by the Institutional Animal Care and Use Committee at East Carolina University. Homozygous, CochG88E/G88E (n = 17), heterozygous, CochG88E/+ (n = 14) and Coch+/+ wild-type littermates (n = 15) were studied at five ages: 11, 13, 17, 19 and 21 months. Mice were anesthetized with a ketamine (18 mg/ml) and xylazine (2 mg/ml) solution (5–7 µl per gram body weight injected intraperitoneally). Core body temperature was maintained at 37.0 ± 0.1°C using a homeothermic heating blanket system (FHC, Inc., Bowdoin, ME, USA). VsEPs were measured at all ages, auditory brainstem responses (ABRs) are reported at 11 and 21 months of age, and DPOAEs were measured at 19 and 21 months only. Six animals were measured at two or more ages.
Vestibular stimulus and stimulus coupling
VsEP recordings were based on methods for mice published by Jones and Jones (22) and Jones et al. (23–25), and are briefly described later. The published methods were modified to utilize a non-invasive coupling system for securing the head to the mechanical shaker. Linear acceleration pulses, 2 ms duration, were presented to the cranium in the naso-occipital axis using two stimulus polarities, normal (+Gx axis) and inverted (−Gx axis). Stimuli were presented at a rate of 17 pulses/s. Stimulus amplitude ranged from +6 to –18 dB re: 1.0 g/ms (where 1 g = 9.8 m/s2) adjusted in 3 dB steps.
Stimuli were delivered to the head using a voltage-controlled mechanical shaker. The head was coupled to a custom platform with a custom head clip, a lightweight plastic spring clip with tines modified to encircle the head anterior to the pinnae. The spring clip was screwed to the custom platform mounted to the mechanical shaker. Although previously published reports using VsEPs utilized an invasive surgical approach to couple the cranium to the shaker, the coupling used here was non-invasive.
ABR stimulus and stimulus coupling
For ABR testing, tone burst stimuli were generated by a signal generator (Agilent HP 23201A or Stanford Research Systems SRS785 signal analyzer) and controlled using custom software and Tucker Davis Technologies (TDT, Gainesville, FL, USA) modules (TG6, SW2, PA4). Tone bursts at 8, 16, 32 and 41.2 kHz had 1.0 ms rise–fall times with 1.0 ms plateau (3 ms total duration). Stimuli for ABR testing were calibrated using a Bruel & Kjaar 1/4 in. microphone and Nexus amplifier. Stimuli were calibrated in dB peSPL and were presented via high frequency transducers (TDT ED1 driver, EC1 speakers) coupled at the left ear via PE tubing. Auditory stimuli were presented at a rate of 17 stimuli/s.
VsEP and ABR recording parameters
Stainless steel wire was placed subcutaneously at the nuchal crest to serve as the non-inverting electrode. Needle electrodes were placed posterior to the left pinnae and at the ventral neck for inverting and ground electrodes, respectively. Traditional signal averaging was used to resolve responses in electrophysiological recordings. Ongoing electroencephalographic activity was amplified (×200 000), filtered (300 to 3000 Hz, −6 dB amplitude points) and digitized (1024 points, 10 µs/pt). Two hundred and fifty six primary responses were averaged for each VsEP or ABR response waveform. All responses were replicated. VsEP recordings began at the maximum stimulus intensity (i.e. +6 dB re: 1.0 g/ms) with and without acoustic masking, then intensity was dropped to −18 dB and raised in 3 dB steps to complete an intensity profile. A broad band forward masker (50–50 000 Hz, 97 dB SPL) was presented during VsEP measurements to verify absence of cochlear responses (22). ABR intensity series was collected with a descending series of stimulus intensities (12 dB steps) beginning at ∼100 dB peSPL.
DPOAE stimulus and recording
Methods for recording DPOAEs were similar to those previously described (26,27). Stimuli for DPOAEs were generated and controlled with modules from TDT. Pure tone frequencies (f1, f2, f2/f1 ratio = 1.25), at equal levels (L1 = L2 = 60 dB SPL), 150 ms duration, were generated with independent sources (Agilent HP 23201A signal generators) and routed through separate drivers to mix acoustically in the ear canal (via plastic tubing placed securely at the external acoustic meatus). Stimuli were calibrated in a 0.1 ml coupler, which simulated the mouse ear canal volume. Stimulus frequencies for the primaries were such that geometric mean [GM = (f1 × f2)0.5] frequencies ranged from 6.0 to 48.5 kHz (at least eight frequencies per octave). Ear canal sound pressure levels were recorded with a low noise probe microphone (Etymotic ER 10B+). The microphone output was amplified and routed to a dynamic signal analyzer (Stanford Research Systems SRS785) for sampling (at 200 kHz) and fast Fourier transform (FFT). The amplitude of f1, f2, and the 2f1–f2 distortion product were measured from the FFT waveform. The noise floor was measured from the amplitudes in the five frequency bins above and below the 2f1–f2 component. The recording system was also tested periodically in the 0.1 ml coupler to assess the presence of artifact distortion products. Frequencies where distortion products were present in the test cavity were excluded from further analyses; however no artifactual distortion was identified.
Data analysis
The first three positive and negative response peaks were scored for VsEPs. Response peak latencies (measured in milliseconds, ms), peak to peak amplitudes (measured in microvolts, μV) and thresholds (measured in dB re: 1.0 g/ms) were quantified for VsEPs. Threshold (in dB peSPL) were obtained for ABR waveforms and amplitudes were quantified for DPOAEs. Descriptive statistics were calculated for each measure of each genotype. Animals with absent-evoked potentials contributed to an estimated mean of the group by replacing the ‘no response’ data points with the maximum possible stimulus level (+9 dB for VsEP and 100 dB peSPL for ABR). All DPOAE amplitudes contributed to the DPOAE mean data.
FUNDING
This work was supported by National Institutes of Health grants DC03402 (to C.C.M.), DC0118 and DC006443 (to S.M.J.), and P30-05209 (to M.C.L.).
ACKNOWLEDGEMENTS
We would like to thank Dr Roderick Bronson and Li Zhang at the Dana Farber/Harvard Cancer Center Rodent Histopathology Core and B. Mock for assistance with functional data collection.
Conflict of Interest statement. None to declare.
REFERENCES
- 1.Robertson N.G., Khetarpal U., Gutiérrez-Espeleta G.A., Bieber F.R., Morton C.C. Isolation of novel and known genes from a human fetal cochlear cDNA library using subtractive hybridization and differential screening. Genomics. 1994;23:42–50. doi: 10.1006/geno.1994.1457. [DOI] [PubMed] [Google Scholar]
- 2.Robertson N.G., Resendes B.L., Lin J.S., Lee C., Aster J.C., Adams J.C., Morton C.C. Inner ear localization of mRNA and protein products of COCH, mutated in the sensorineural deafness and vestibular disorder, DFNA9. Hum. Mol. Genet. 2001;10:2493–2500. doi: 10.1093/hmg/10.22.2493. [DOI] [PubMed] [Google Scholar]
- 3.Ikezono T., Omori A., Ichinose S., Pawankar R., Watanabe A., Yagi T. Identification of the protein product of the Coch gene (hereditary deafness gene) as the major component of bovine inner ear protein. Biochim. Biophys. Acta. 2001;1535:258–265. doi: 10.1016/s0925-4439(00)00101-0. [DOI] [PubMed] [Google Scholar]
- 4.Robertson N.G., Cremers C.W., Huygen P.L., Ikezono T., Krastins B., Kremer H., Kuo S.F., Liberman M.C., Merchant S.N., Miller C.E., et al. Cochlin immunostaining of inner ear pathologic deposits and proteomic analysis in DFNA9 deafness and vestibular dysfunction. Hum. Mol. Genet. 2006;15:1071–1085. doi: 10.1093/hmg/ddl022. [DOI] [PubMed] [Google Scholar]
- 5.Robertson N.G., Lu L., Heller S., Merchant S.N., Eavey R.D., McKenna M., Nadol J.B., Jr, Miyamoto R.T., Linthicum F.H., Jr, Lubianca Neto J.F., et al. Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nature Genet. 1998;20:299–303. doi: 10.1038/3118. [DOI] [PubMed] [Google Scholar]
- 6.de Kok Y.J.M., Bom S.J.H., Brunt T.M., Kemperman M.H., van Beusekom E., van der Velde-Visser S.D., Robertson N.G., Morton C.C., Huygen P.L.M., Verhagen W.I.M., et al. A Pro51Ser mutation in the COCH gene is associated with late onset autosomal dominant progressive sensorineural hearing loss with vestibular defects. Hum. Mol. Genet. 1999;8:361–366. doi: 10.1093/hmg/8.2.361. [DOI] [PubMed] [Google Scholar]
- 7.Fransen E., Verstreken M., Verhagen W.I.M., Wuyts F.L., Huygen P.L.M., D′Haese P., Robertson N.G., Morton C.C., McGuirt W.T., Smith R.J.H., et al. High prevalence of symptoms of Meniere’s disease in three families with a mutation in the COCH gene. Hum. Mol. Genet. 1999;8:1425–1429. doi: 10.1093/hmg/8.8.1425. [DOI] [PubMed] [Google Scholar]
- 8.Kamarinos M., McGill J., Lynch M., Dahl H. Identification of a novel COCH mutation, I109N, highlights the similar clinical features observed in DFNA9 families. Hum. Mutat. 2001;17:351. doi: 10.1002/humu.37. [DOI] [PubMed] [Google Scholar]
- 9.Street V.A., Kallman J.C., Robertson N.G., Kuo S.F., Morton C.C., Phillips J.O. A novel DFNA9 mutation in the vWFA2 domain of COCH alters a conserved cysteine residue and intrachain disulfide bond formation resulting in progressive hearing loss and site-specific vestibular and central oculomotor dysfunction. Am. J. Med. Genet. A. 2005;139:86–95. doi: 10.1002/ajmg.a.30980. [DOI] [PubMed] [Google Scholar]
- 10.Yuan H.J., Han D.Y., Sun Q., Yan D., Sun H.J., Tao R., Cheng J., Qin W., Angeli S., Ouyang X.M., et al. Novel mutations in the vWFA2 domain of COCH in two Chinese DFNA9 families. Clin. Genet. 2008;73:391–394. doi: 10.1111/j.1399-0004.2008.00972.x. [DOI] [PubMed] [Google Scholar]
- 11.Pauw R.J., Huygen P.L., Collin R.W., Cruysberg J.R., Hoefsloot L.H., Kremer H., Cremers C.W. Phenotype description of a novel DFNA9/COCH mutation, I109T. Ann. Otol. Rhinol. Laryngol. 2007;116:349–357. doi: 10.1177/000348940711600506. [DOI] [PubMed] [Google Scholar]
- 12.Boulassel M.R., Tomasi J.P., Deggouj N., Gersdorff M. COCH5B2 is a target antigen of anti-inner ear antibodies in autoimmune inner ear diseases. Otol. Neurotol. 2001;22:614–618. doi: 10.1097/00129492-200109000-00009. [DOI] [PubMed] [Google Scholar]
- 13.Baek M.J., Park H.M., Johnson J.M., Altuntas C.Z., Jane-Wit D., Jaini R., Solares C.A., Thomas D.M., Ball E.J., Robertson N.G., et al. Increased frequencies of cochlin-specific T cells in patients with autoimmune sensorineural hearing loss. J. Immunol. 2006;177:4203–4210. doi: 10.4049/jimmunol.177.6.4203. [DOI] [PubMed] [Google Scholar]
- 14.Tebo A.E., Szankasi P., Hillman T.A., Litwin C.M., Hill H.R. Antibody reactivity to heat shock protein 70 and inner ear-specific proteins in patients with idiopathic sensorineural hearing loss. Clin. Exp. Immunol. 2006;146:427–432. doi: 10.1111/j.1365-2249.2006.03227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kemperman M.H., De Leenheer E.M.R., Huygen P.L.M., van Duijnhoven G., Morton C.C., Robertson N.G., Cremers F.P.M., Kremer H., Cremers C.W.R.J. Audiometric, vestibular, and genetic aspects of a DFNA9 family with a G88E COCH mutation. Otol. Neurotol. 2005;26:926–933. doi: 10.1097/01.mao.0000185062.12458.87. [DOI] [PubMed] [Google Scholar]
- 16.Liepinsh E., Trexler M., Kaikkonen A., Weigelt J., Banyai L., Patthy L., Otting G. NMR structure of the LCCL domain and implications for DFNA9 deafness disorder. EMBO J. 2001;20:5347–5353. doi: 10.1093/emboj/20.19.5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bischoff A.M., Huygen P.L., Kemperman M.H., Pennings R.J., Bom S.J., Verhagen W.I., Admiraal R.J., Kremer H., Cremers C.W. Vestibular deterioration precedes hearing deterioration in the P51S COCH mutation (DFNA9): an analysis in 74 mutation carriers. Otol. Neurotol. 2005;26:918–925. doi: 10.1097/01.mao.0000185048.84641.e3. [DOI] [PubMed] [Google Scholar]
- 18.Satokata I., Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nature Genet. 1994;6:348–356. doi: 10.1038/ng0494-348. [DOI] [PubMed] [Google Scholar]
- 19.Lakso M., Pichel J.G., Gorman J.R., Sauer B., Okamoto Y., Lee E., Alt F.W., Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl Acad. Sci. USA. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chirgwin J.R., Przybyla A.E., MacDonald R.J., Rutter W.J. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 21.Ikezono T., Shindo S., Li L., Omori A., Ichinose S., Watanabe A., Kobayashi T., Pawankar R., Yagi T. Identification of a novel Cochlin isoform in the perilymph: insights to Cochlin function and the pathogenesis of DFNA9. Biochem. Biophys. Res. Commun. 2004;314:440–446. doi: 10.1016/j.bbrc.2003.12.106. [DOI] [PubMed] [Google Scholar]
- 22.Jones T.A., Jones S.M. Short latency compound action potentials from mammalian gravity receptor organs. Hear. Res. 1999;136:75–85. doi: 10.1016/s0378-5955(99)00110-0. [DOI] [PubMed] [Google Scholar]
- 23.Jones S.M., Erway L.C., Bergstrom R.A., Schimenti J.C., Jones T.A. Vestibular responses to linear acceleration are absent in otoconia-deficient C57BL/6JEi-het mice. Hear. Res. 1999;135:56–60. doi: 10.1016/s0378-5955(99)00090-8. [DOI] [PubMed] [Google Scholar]
- 24.Jones S.M., Subramanian G., Avniel W., Guo Y., Burkard R.F., Jones T.A. Stimulus and recording variables and their effects on mammalian vestibular evoked potentials. J. Neurosci. Methods. 2002;118:23–31. doi: 10.1016/s0165-0270(02)00125-5. [DOI] [PubMed] [Google Scholar]
- 25.Jones S.M., Erway L.C., Johnson K.R., Yu H., Jones T.A. Gravity receptor function in mice with graded otoconial deficiencies. Hear. Res. 2004;191:34–40. doi: 10.1016/j.heares.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Jimenez A.M., Stagner B.B., Martin G.K., Lonsbury-Martin B.L. Age-related loss of distortion product otoacoustic emissions in four mouse strains. Hear. Res. 1999;138:91–105. doi: 10.1016/s0378-5955(99)00154-9. [DOI] [PubMed] [Google Scholar]
- 27.Guimaraes P., Zhu X., Cannon T., Kim S., Frisina R.D. Sex differences in distortion product otoacoustic emissions as a function of age in CBA mice. Hear. Res. 2004;192:83–89. doi: 10.1016/j.heares.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 28.Collin R.W., Pauw R.J., Schoots J., Huygen P.L., Hoefsloot L.H., Cremers C.W., Kremer H. Identification of a novel COCH mutation, G87W, causing autosomal dominant hearing impairment (DFNA9) Am J Med Genet A. 2006;140:1791–1794. doi: 10.1002/ajmg.a.31354. [DOI] [PubMed] [Google Scholar]
- 29.Nagy I., Horvath M., Trexler M., Repassy G., Patthy L. A novel COCH mutation, V104del, impairs folding of the LCCL domain of cochlin and causes progressive hearing loss. J Med Genet. 2004;41:e9. doi: 10.1136/jmg.2003.012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usami S., Takahashi K., Yuge I., Ohtsuka A., Namba A., Abe S., Fransen E., Patthy L., Otting G., Van Camp G. Mutations in the COCH gene are a frequent cause of autosomal dominant progressive cochleo-vestibular dysfunction, but not of Meniere's disease. Eur J Hum Genet. 2003;11:744–748. doi: 10.1038/sj.ejhg.5201043. [DOI] [PubMed] [Google Scholar]