

Summary
As the global Structural Genomics projects have picked up pace the number of structures annotated in the Protein Data Bank as “hypothetical protein” or “unknown function” has grown significantly. A major challenge now involves the development of computational methods to accurately and automatically assign functions to these proteins. As part of the Midwest Center for Structural Genomics (MCSG) we have developed a fully automated functional analysis server, ProFunc, which performs a battery of analyses on a submitted structure. The analyses combine a number of sequence-based and structure-based methods to identify functional clues. After the first stage of the Protein Structure Initiative (PSI) we review the success of the pipeline and the importance of structure-based function prediction. As a dataset we have chosen all structures solved by the MCSG during the 5 years of the first PSI. Our analysis suggests that two of the structure-based methods are particularly successful and provide examples of local similarity difficult to identify using current sequence methods. No one method is successful in all cases so through the use of a number of complementary sequence and structural approaches, the ProFunc server increases the chance that at least one method will find a significant hit that can help elucidate function. Manual assessment of the results is a time-consuming process and subject to individual interpretation and human error. We present a method based on the Gene Ontology schema using GO-slims that can allow the automated assessment of hits with a success rate approaching that of expert manual assessment.
Keywords: Structural Genomics, Function Prediction From Structure, Gene Ontology, GO-slims, Protein Function Prediction
Introduction
Structural genomics1 is a large-scale project aimed at experimentally determining a large number of protein 3D structures as rapidly and accurately as possible using high throughput methods. There are a number of groups funded as part of the Protein Structure Initiative (PSI) and other projects exist across the globe such as Riken (Japan), SPiNE (Europe) and the Anglo-Canadian-Swedish SGC (Structural Genomics Consortium). Each centre has individual targets and goals but major aims include:
High-throughput automation of protein production, structure determination and analysis
Increased coverage of protein fold space and hence the number of protein sequences amenable to homology modelling methods
Investigation of protein structure to elucidate function in health and disease
Reduction of the cost of structure determination
The Midwest Center for Structural Genomics (MCSG) is funded by the National Institute for General Medical Sciences (NIGMS), as part of the PSI of the National Institutes of Health. The centre aims to develop and optimise new, rapid, integrated methods for highly cost-effective determination of protein structures through X-ray crystallography. In order to achieve this goal the centre has been optimising all stages of protein structure determination: crystal growth, data collection, and structural model generation and refinement. The success of the project is indicated by the fact that as of the 30th of September 2005 (the official end of the first stage of the PSI) the MCSG had over 5000 active targets and a total of 319 structures deposited in the Protein DataBank (PDB2). However, of these deposits, over a third have no functional annotation and are described as merely “hypothetical protein” or “unknown function”. The determination of a protein’s function by experiment is expensive and time consuming and cannot be readily accommodated in a high throughput pipeline. Thus there is a need to develop automated function prediction methods to at least provide an idea of the likely function of the protein and to help guide experimental determination of its function3. The scale of the problem is clear when one considers that as of 30 September 2005 there were over 1100 proteins out of over 32,000 in the PDB labelled as “unknown function”.
In general, computational methods to infer a function for an individual protein, such as its enzymatic activity, fall into two main types: those that are sequence-based and those that are structure-based. In addition, functional information can often be inferred through comparisons of genomic organisation and gene location analysis or by methods analysing protein interaction and gene regulatory networks.
The most commonly used sequence-based approaches involve simple BLAST4 or FASTA runs which perform direct sequence-sequence comparisons of the query protein against large databases such as UniProt5 or GenBank6 in order to identify similarity with proteins of known function. More powerful and sensitive profile/pattern based methods utilise information from the sequences in whole protein families, where the family can be defined in terms of 3D structure, as in Gene3D7 and SUPERFAMILY8, or in terms of sequence similarity and function, as in Pfam9. Other useful approaches involve the investigation of phylogenetic profiles and amino acid conservation. A number of studies10,11 have indicated that significant sequence similarity (>40%) and strong profile matches are the best indicators of function although there are always exceptions to this rule12.
When the sequence-based methods fail, or provide few functional clues, the examination of the protein’s 3D structure can identify distant relationships and suggest functional roles. The structure-based methods can be classified according to the level of protein structure and specificity at which they operate, ranging from analysis of the global fold of the protein down to the identification of highly specific 3D clusters of functional residues13,14.
No single method will be successful in all cases, and there will also be proteins for which no method is useful. Accordingly, a sensible strategy may be to use as many different methods as possible, incorporating data from multiple sources, to increase the chances of obtaining some functional prediction for any given protein. To this end, the ProFunc15 server (http://www.ebi.ac.uk/thornton-srv/databases/ProFunc) has been developed at the EBI in collaboration with structural genomics consortia to explore the efficacy of combining multiple methods and data sources in a semiautomated manner. The data are presented to the depositors in order to allow them to use their expert knowledge to decide on the most likely functional clues for experimental testing. The server uses a variety of methods drawing on multiple databases:
Sequence analysis primarily involves BLAST runs against the PDB and UniProt databases to help identify functionally annotated homologues. In addition, the sequence is also scanned using InterProScan in order to identify motifs indicative of specific protein families or functional motifs.
The structure-based approaches used in ProFunc involve large-scale fold matching methods (using SSM16 and DALI), identification of smaller sub-motifs (e.g. Helix-turn-helix DNA-binding patterns17), localised pockets (surface cleft analysis and nest identification), and highly specific n-residue template methods14 (enzyme active sites, ligand binding sites, DNA-binding residues and reverse template analysis).
In addition to this, for bacterial proteins, the locus encoding the UniProt BLAST hits are located in the genome and neighbouring genes are tabulated in the hope that functional inferences can be made from the functions of the surrounding genes.
In this paper we use the MCSG structures as a test dataset to investigate the ProFunc server’s ability to determine function from structure, to identify the most successful structure-based approaches and suggest future directions and improvements.
Results
Our study into automated functional prediction using the MCSG data set is outlined as follows:
Functional coverage of the MCSG dataset.
Manual assessment of “known-function” dataset.
Identification of the best structure-based method in ProFunc.
Automated assessment of hits using GO-slims.
Analysis of specific examples.
Functional coverage of the MCSG dataset
Of the 282 non-redundant structures used in the analysis only a third have a known function (Figure 1). An additional 21% have a putative function based on sequence similarity to another protein of known function while the remainder are of unknown function. A quick way to assess how representative this dataset is of proteins in general and whether there are any biases to certain protein types is to examine its “functional space” coverage. To this end the 92 structures of known function were plotted on an “EC wheel” to estimate the functional coverage (Figure 2a). The black sector represents the 30 structures of known function that are not enzymes, 10 of which are transcriptional regulators (Table 1). Looking at the EC wheel and Table 1 together suggests there is reasonable coverage of the functional space with a slight tendency towards transcriptional regulators and hydrolases (EC 3.-.-.-). If the MCSG proteins are compared against the distribution of EC numbers across the entire PDB (Figure 2b) it is evident that the proportions for each top level EC class are similar except that there appears to be a slightly greater number of lyases and fewer oxidoreductases.
Figure 1. Breakdown of prior information for the 282 MCSG structures.
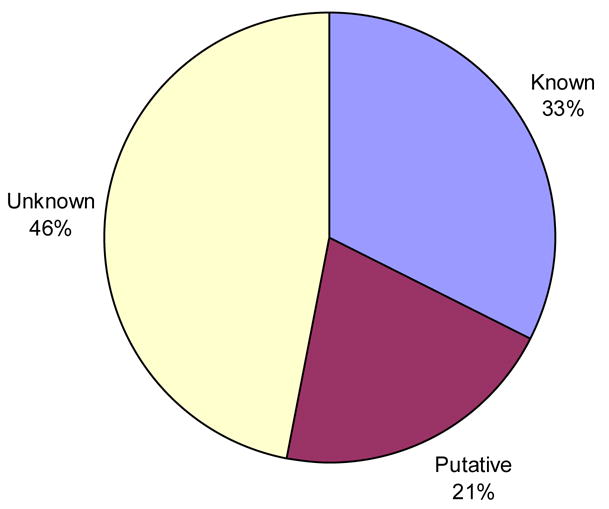
The pie chart illustrates the proportion of the 282 non-redundant structures classed as “known function”, “putative function” or “unknown function”.
Figure 2.
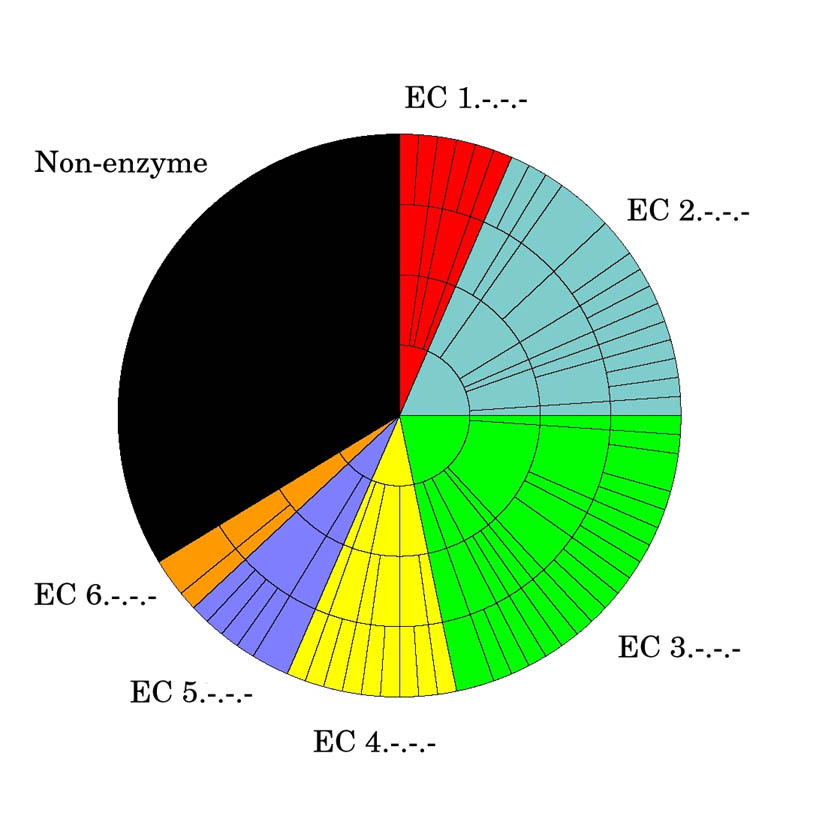
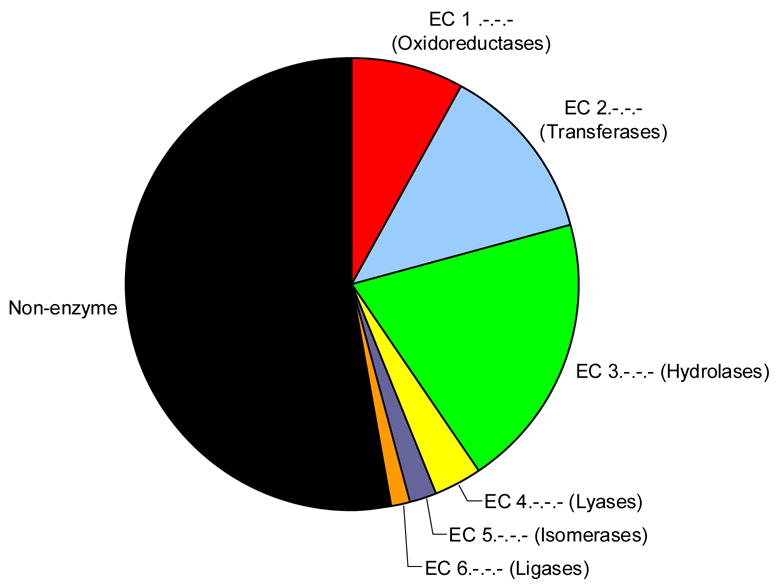
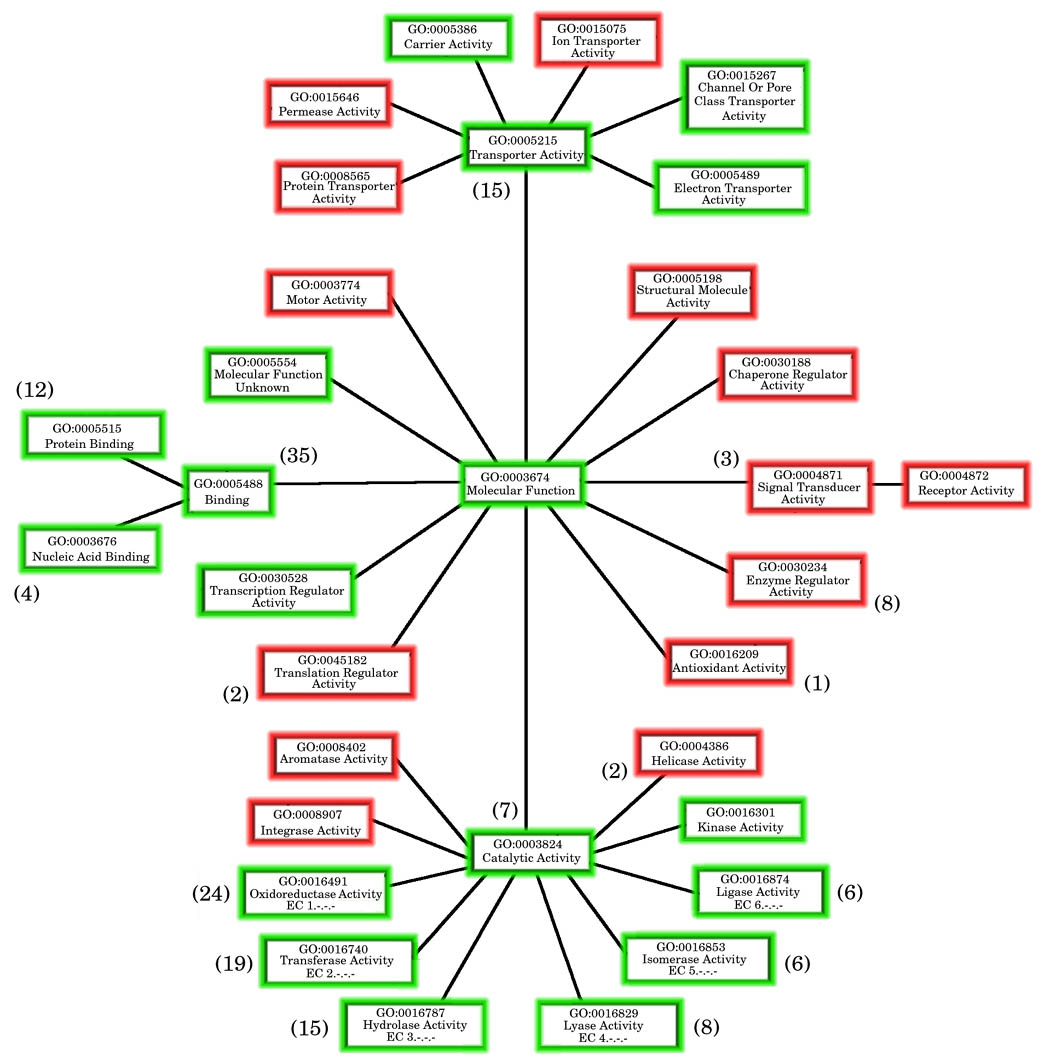
Figure 2a: EC wheel for 92 proteins of known function
- Red = E.C. 1.-.-.- (Oxidoreductases)
- Blue = E.C. 2.-.-.- (Transferases)
- Green = E.C. 3.-.-.- (Hydrolases)
- Yellow = E.C. 4.-.-.- (Lyases)
- Purple = E.C. 5.-.-.- (Isomerases)
- Orange = E.C. 6.-.-.- (Ligases)
Each shell then corresponds to the next stage down the E.C. schema through the second, third and finally the fourth level.
Figure 2b: Pie chart showing distribution of EC classes in the entire PDB
The proportions illustrated are taken from the numbers of PDB entries in the PDB with each top level E.C. number. This information is extracted from the Enzyme Structures Database at the EBI (http://www.ebi.ac.uk/thornton-srv/databases/enzymes/).
Figure 2c: Map showing the coverage of the generic GO-slim by the MCSG dataset
Any MCSG structures from the full dataset annotated with GO terms had all their GO-terms extracted and the associated GO-slim terms derived from the GOA-GOslim mapping file. All GO-slims from the “Molecular Function” branch of the gene ontology were mapped. Those GO-slim terms found in the annotations of the MCSG structures are coloured green whereas those coloured red are not covered by the MCSG dataset.
The numbers in brackets correspond to the number of terms added at that point in the hierarchy by the extended GO-slim and shows the spread of the additional information.
Table 1. Description of 30 known function proteins with no EC class.
| PDB code | Function description |
|---|---|
| 1td5 | Repressor of aceBA operon, IclR transcriptional regulator (repressor) |
| 1lj9 | Transcription regulator (MarR-like transcription factor) |
| 2a61 | Transcriptional regulator tm0710 |
| 1mkm | Transcriptional regulator, IclR family |
| 1z05 | Transcriptional regulator, ROK family |
| 1z0x | Transcriptional regulator, TetR family |
| 1zk8 | Transcriptional regulator, tetr family |
| 1sfx | HTH transcription regulator |
| 1s3j | MarR/SlyA like transcriptional factor |
| 1ylf | RRF2 family protein (Transcriptional regulator) |
| 1sr8 | Cobalamin biosynthesis protein |
| 1u7n | Fatty acid/phospholipid synthesis protein |
| 1mkz | Molybdopterin biosynthesis, protein B |
| 1xau | B and T lymphocyte attenuator |
| 1otk | Phenylacetic acid degradation protein paac |
| 1y89 | DevB protein (sol/devb family) |
| 1kr4 | Divalent cation tolerance protein |
| 1zma | Bacterocin transport accessory protein |
| 1xwm | Phosphate transport system protein phoU |
| 1zox | Clm-1 mouse myeloid receptor extracellular domain (ig-like receptor) |
| 1pqz | Murine cytomegalovirus immunomodulatory protein m144, Modulation of NK cell, immunoglobulin-like |
| 1tua | mitochondrial-type HSP70 |
| 1vzy | HSP 33 Chaperonin |
| 1r0d | I/LWEQ domain bind to actin, huntingtin interacting protein-1-related |
| 1y71 | Kinase-associated protein B |
| 1x7f | Outer surface protein |
| 1j8r | PapG Receptor-Binding, Pyelonephritic adhesin |
| 2a5l | Trp repressor binding protein wrba |
| 1mkf | Viral chemokine binding protein M3 |
| 1pzx | Signal recognition particle (DegV-like) |
Many of the MCSG structures have been annotated with GO-terms but for a more general functional description GO-slim terms can be examined. In this study the “Molecular Function” section of the gene ontology is of interest and Figure 2c shows the coverage of this area of the GO-slim hierarchy by the MCSG structures (terms shaded green are covered whereas those in red are absent), the numbers in brackets refer to the expansion of terms by extending the GO slim (discussed below).
Manual assessment using “known function” dataset
The results from the structure-based ProFunc analyses for the 92 proteins of known function in the dataset are illustrated in Figure 3 below (see Supplementary material for a spreadsheet listing all manual annotations). The results have been backdated to the release date of the query by removing hits to structures released after that date, giving a picture of what the server would have suggested had it been available at the time. The SSM results show that in approximately 55% of cases the top fold match was able to provide the correct functional assignment (almost 20% of which are strongly predicted). The standard template methods provide some success but the most accurate structure-based method is the reverse template approach (SiteSeer [SIT]), which provides the correct function in 60% of the cases (of which over 75% are strongly predicted).
Figure 3. ProFunc results for proteins of known function.
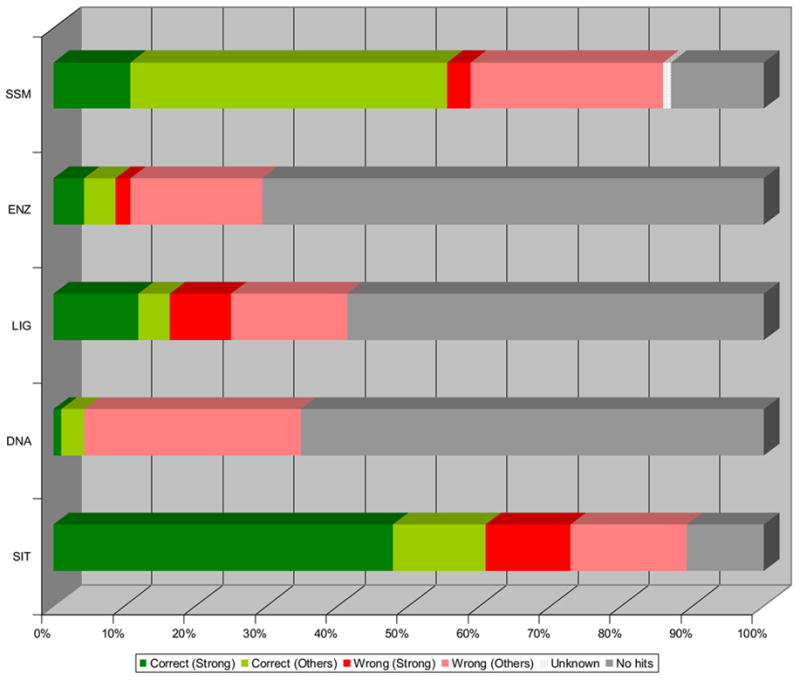
The 92 proteins classed as having “known function” in the MCSG dataset were analysed using ProFunc. The top hit (after parsing for release dates) was classified by success and strength of hit. Those hits to hypothetical proteins or members of families/domains of unknown function are classified as “unknown”. The structure-based methods used by ProFunc are as follows:
SSM – Secondary Structure Matching (MSDfold): fold comparison service.
ENZ – Enzyme template search (Catalytic Site Atlas data)
LIG – Ligand binding template search (Automatically generated templates)
DNA – DNA binding template search (Automatically generated templates)
SIT – SiteSeer (“Reverse template” method)
Identification of the best structure-based method in ProFunc
The best two structure-based methods identified by manual assessment of the ProFunc results are the reverse templates and SSM. In order further assess the methods their ROC curves were calculated (Figure 4). In order to calculate the curves a score was used as a cutoff, in the case of SSM the Z-score was of interest, whereas for the reverse templates it was the E-value.
Figure 4. ROC curves for SSM and SIT based on manual function assignment.
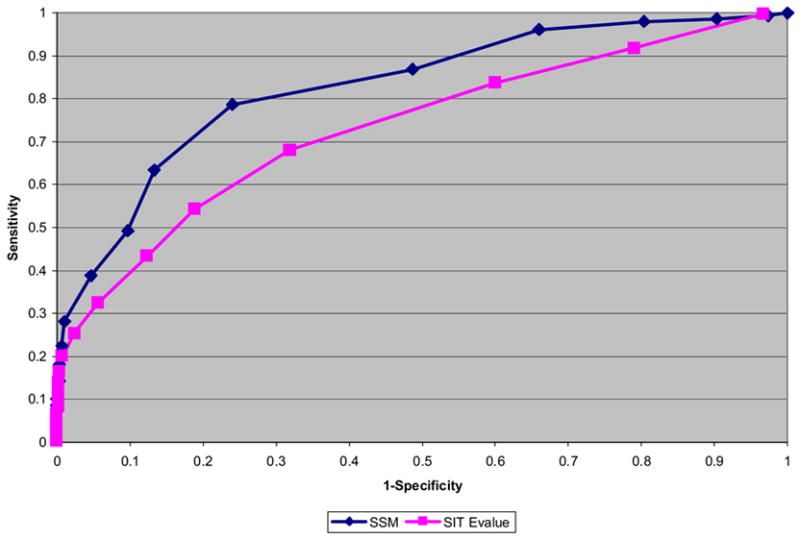
The ROC curves are plotted for SSM results and for SiteSeer (“reverse template”) results. The cut-off used by SSM is the Z-score of the hit, whereas it is the E-value that is of interest in SiteSeer (reverse templates). The ideal curve would rise vertically from the origin and then horizontally out to the right and would give an area under the curve of 1. The plot shows that the SSM Z-score appears to be a better measure for distinguishing between true and false positives than the SiteSeer (“reverse template”) measures.
Examination of the curves shows the SSM method as having the best performance, the areas under the curve being 0.83 and 0.70 respectively. An area of 1.00 corresponds to perfect prediction while 0.50 is equivalent to random prediction. One might expect the two methods to overlap to some extent – i.e. to hit the same PDB files. In fact, in only 25 of the cases did both methods return the same PDB file as their top hit. A further 25 cases matched different PDB files but still obtained identical functional predictions. Of the remaining 32 cases there were 5 where the reverse templates method found the correct match while SSM missed it, and one case where SSM gave the correct answer and the reverse templates method was wrong. This shows that, despite a significant overlap, there are a minority of cases where the one method identifies matches missed by the other. It should be noted that, even when both methods match to the same PDB entry, they provide complementary information: SSM identifies the fold similarity, while the reverse template method pinpoints local regions of high similarity and, in so doing, usually picks out the functionally important site.
Automated assessment of hits using GO-slims
One question of interest is whether GO-slim terms can be used to assess the functional predictions in an automated way rather than requiring manual assessment of true and false positives. To investigate this we used the 77 proteins with GO annotation from the 92 MCSG proteins of known function. The ProFunc results give a total of 207 structural matches. The numbers from each method are as follows:
| SSM Fold Match | 68 |
| Reverse Templates | 74 |
| Enzyme Templates | 8 |
| Ligand Templates | 47 |
| DNA Templates | 10 |
|
| |
| Total | 207 |
Comparison of the GO terms between a query and hit protein can determine whether the hit is a true or false positive. However, even for the correct matches, the terms do not usually match 100%, or one protein may have more terms than the other. So the problem of comparing GO terms is in determining how many terms need to agree before a match can be deemed a true positive. We tried a number of different cut-offs to see which gave the best agreement with the manual assignments. The cut-offs we tried were 25%, 50%, 75%, 100%, and a “constrained 50%” wherein a 100% match was required where the query protein has only 2 GO terms. We tried both the generic GO-slims (31 terms) and our hand-curated molecular function (MF) GO-slims (190 terms) which have more terms levels than the generic version. The closest agreement to the manually assessed function prediction results was obtained with a 75% cut-of on the MF-GO-slims (see Supplementary information for a detailed discussion). The generic GO-slims fared poorly due to the small number of terms. Of the 207 function predictions over 65% (136/207) involved only 2 GO-slim terms. So the overall results were significantly affected by how these cases were treated (hence the introduction of the “constrained 50%” cut-off rule). Even for the 100% cut-off rule there were identifiable errors. For example, 10 of the 16 false negatives resulted because the hit protein had fewer GO-slim terms than the query protein, making a 100% match impossible. In other cases the errors resulted from errors in annotation. Thus the match to PDB entry 1jvn (a bifunctional protein with amidotransferase and lyase activity) reported for the MCSG structure 1kxj (glutamine amidotransferase) by both SSM and the reverse templates was deemed incorrect because the GO annotation for 1jvn only covers its lyase activity. In another case, the GO annotation of an MCSG HTH transcription regulator (1sfx) is incorrectly detailed as a ligase with binding activity. The strong structural hit is to a M. jannaschii DNA-binding protein which is described in GO as a nucleic acid binder with transcription regulation activity. This hit will always be seen as a false negative match using the GO-slim method.
The MF-GO-slims performed better than the generic GO-slims, with the best agreement with the manual assessment (83% of the cases) being achieved for a cutoff of 75% (see Supplementary information). Not only do the MF-GO-slims perform better, but they also provide more specific functional annotation and hence are more useful when, say, planning any experimental verification. For example, the coverage of the E.C. hierarchy in the MF-GO-slims goes to the third level rather than only the first. Now 6% of the 207 cases have only 2 terms describing a protein, compared with the 65% for the generic GO-slims. Seven of the cases have 10 or more terms whereas the most terms per protein in the generic GO-slims is 5.
Thus the MF-GO-slims provide a greater specificity and agreement with the manual assessment than the generic GO-slims but without the problems inherent in the full gene ontology which is too complicated and unevenly distributed. In the cases where the MF-GO-slims disagree with the manual assessment the reason for the disagreement tends to be where the former overpredicts true positives.
In practice the procedure would be to first identify general similarity in function using the MF-GO-slim followed by more accurate comparisons using the full gene ontology. Clearly any GO-slim approach is of greatest use when the function of the query and hit proteins are already known and annotated with GO terms, but what of queries that are of unknown function or as yet unreleased? In this situation the method is useful for comparing all hits from all methods with one another in an attempt to find common general functions amongst the top hits.
ProFunc Typical Examples
Of course, the only sure way of verifying a functional prediction is via experiment. A major component of our collaborative effort within the MCSG is the experimental validation of functional predictions made by the ProFunc server. The three examples chosen below illustrate the various ways the server has been of use to experimentalists and how much work remains.
Example 1: Function experimentally confirmed
One example where predictions made using the server have been experimental verified has been published previously18. The example is that of the 1.5Å crystal structure of BioH protein from E. coli solved by the MCSG. Analysis of the structure using ProFunc returned a significant match (r.m.s.d. of 0.28 Å) to an enzyme active-site template for the Ser-His-Asp catalytic triad of the lipases. This prompted the experimental characterisation of this protein which was found to be a novel carboxylesterase acting on short acyl chain substrates.
Example 2: Function suggested from structure
The 1.9 Å crystal structure of hypothetical protein IsdG from staphylococcus aureus, PDB deposit 1xbw, was released on the 12th October 2004. Analysis using the ProFunc server revealed that all the BLAST hits were to other hypothetical proteins of unknown function. A separate PSI-BLAST run revealed weak similarity to antibiotic biosynthesis monooxygenases. An InterProScan run provided significant hits to two functions: the first was a PROSITE pattern match to “Peptidase, cysteine peptidase active site” and the other a Pfam domain “Antibiotic biosynthesis monooxygenase”. The genome analysis suggests a number of possible functions including oxidoreductase, methyltransferase, epimerase, transportation, possible RNA binding, and others.
When the structure-based methods were employed, we found that the strongest SSM fold matches were to hypothetical proteins and all bar one of the remaining hits were monooxygenases. There were no hits to known enzyme or ligand-binding templates and only two rather weak matches to DNA-binding templates. If the reverse templates were examined we found the majority of the top hits were to proteins of unknown function but the first significant match with an assigned function was to a monooxygenase from Streptomyces coelicolor (PDB entry 1lq9).
This is an example where the sequence-based methods provide a variety of suggested functions with similar confidence and the structure-based approaches provided additional supporting evidence that support the prediction.
Experimental analysis has characterised the protein as a haem-degrading enzyme with structural similarity to monooxygenases19.
Example 3: Function remains unknown
The 1.5Å crystal structure of a hypothetical protein (pa4017) from Pseudomonas aeruginosa, PDB deposit 2a35, was released on 9th August 2005. The structure was submitted to the ProFunc server and the results analysed. BLAST searches against the UniProt database showed similarity to other hypothetical proteins. The sequences of the majority of these hits (and that of 2a35 itself) had similarity to domains associated with NAD binding oxidoreductase activity. Structural comparisons provide additional evidence for this prediction: fold similarities to NADP-dependant reductases; ligand-binding template matches to NAD and NAP complexed structures; an enzyme template match to the short-chain dehydrogenase-reductase family; and reverse-template matches to members of the short-chain dehydrogenase-reductases and other NAD/NADP binding proteins. Further examination of the structure indicated that the 2a35 structure had its C-terminal section (about 10 residues) lying in the cleft blocking the potential NADP binding site. This means that the predictions may be invalid but it is also possible that this conformation is not the one adopted in the cell. The question then becomes if the cleft is blocked by the C-terminus, what is the new function and why?
The purified protein was used to assess the binding of a variety of small molecules (including NAD, NADH, NADP, NADPH, cAMP, ATP, ADP, nucleotide sugars, amino acids, etc), however none of the selected molecules showed significant binding. It would therefore appear that 2a35 is not capable of binding the predicted co-factors and its function may differ from those suggested by computational methods.
One interesting observation is that 2a35 shows 30% sequence similarity to Tat-interacting protein Tip30 (a human protein deposited in the PDB (2bka) that has pro-apoptotic and anti-metastatic properties). Bioinformatic analysis of this Tip30 protein shows similarity to the short-chain dehydrogenase-reductases and biochemical studies show NADPH binding specificity. The function of the Tip30 protein appears to have been adapted from a metabolic enzyme to a regulatory protein, perhaps a similar adaptation has occurred in the 2a35 protein.
Pseudomonas aeruginosa is a Gram-negative, aerobic, opportunistic pathogen affecting plants and immunocompromised humans (e.g. burns, wounds, hospital aquired infections). It is observed that hypothetical protein PA4017 showed strong structural similarities to human Tip30 protein and Arabidopsis thaliana proteins. If the plant proteins are active (as in humans) to induce apoptosis, an inactive homologue from the Pseudomonas pathogen could prevent the plant (or human) host from destroying infected cells. This hypothesis is conjecture and requires further experimental analysis, however it illustrates that even in the cases where predictions are tested but provide negative results, they can open up new avenues of research.
Discussion
The MCSG has produced a large number of structures during the first stage of the PSI (over 300 in 5 years); the structures have a wide range of functions and a number have novel folds. The MCSG structures have therefore been a useful dataset to test and develop the ProFunc server. The idea behind ProFunc is that a combined approach of sequence-based and structure-based methods, although providing the experimentalist with a lot more data, is more likely to provide the correct function or at least provide clues that can be tested.
It is widely accepted that strong sequence similarity is generally a good indicator of similarity in function. When we looked at the sequence-based methods for the dataset we found that InterProScan gave a success rate of 70% correct, BLAST vs. UniProt was 95% correct and genome analysis provided about 85% correct. It would appear from this that the sequence-based methods are all we would need, however these are likely to be an over-estimate as the results have not been backdated like the structure-based analyses. UniProt archives previous versions of sequences and each entry contains release dates and version numbers, but the backdating process is not a straightforward one. As the expectation values for BLAST hits depend on the size of the database it is not enough to just ignore the entries after the release date; a new UniProt database would be needed for each structure. This is an even greater problem for the HMM libraries as they are continually updated with limited archives. To address this problem we have initiated the collection and storage of data from ProFunc sequence and structural analyses on deposition for all MCSG structures produced during PSI2 to give an accurate reflection of the state of the databases at the time of release.
Although the sequence-based approaches are the most successful, when they fail to provide any interesting hits (such as hypothetical proteins of unknown function) or the sequences have diverged too far to detect their common ancestry, the structure can be important in narrowing down the options. Similarly, when a sequence match is weak, the information from any structural matches can increase the confidence in any tentative functional assignment that the sequence may suggest. The first stage of such functional studies is the identification of similar folds using software such as SSM. Our analysis suggests this is an effective method even in the “twilight zone” of low sequence similarity. Additional evidence for more specific functions can be provided by using local structural comparisons such as the reverse template method, that can help identify functional similarities independently of the global fold comparison. Our comparison of these methods suggests that SSM, giving a slightly better ROC curve, provides more successful function predictions overall, although the information from the reverse template method is more specific in that it usually locates the functionally important regions.
Occasionally SSM misses cases where folds have diverged but local, functional regions have been preserved over evolutionary time. These cases are picked up by the reverse template method. One such example is that of MCSG target APC5049 (PDB entry 1tjn). This structure was deposited on 6th June 2004 and is annotated as a “sirohydrochlorin cobaltochelatase” (EC 4.99.1.3). Analysis using ProFunc provided strong structural matches using the reverse templates method. The top non-self hit, with a score of 253 and an e-value of 0.005, was to PDB entry 1qgo (an anaerobic cobalt chelatase involved in cobalamin biosynthesis). This correct match was not identified using SSM and in fact its top hit, with a rather poor Z-score of 3.9, was to a “MICAREC pH 4.9, DNA-binding response regulator” (PDB entry 1nxs) and is a false positive match. Examination of the full list of SSM results for this structure reveals that the hit identified using reverse templates appears at position 65 in the SSM results at a marginally lower Z-score of 3.8. One reason that the true positive fails to achieve a higher Z-score is that the superposition of secondary structures is attempting to align a strand from the MCSG target with a helix from 1qgo. The reverse template approach is unaffected by this mismatch as it is looking at a locally conserved region distant from the mismatched secondary structures.
Another case involves a putative protein from Aquifex aeolicus (PDB entry 1t6t). The most likely function of this protein is a topoisomerase or primase with strong supporting evidence coming from sequence-based approaches. The structural analyses performed by ProFunc once again provided strong reverse template hits to primase-helicase proteins and also a reverse gyrase. The SSM results provided weak matches to a variety of proteins including sulphotransferases and PEP-dependent phosphotransferases. If the reverse template hits are examined in closer detail it becomes apparent that the putative protein is a single domain whereas the primase and topoisomerase proteins are multi-domain. As SSM is attempting to match the putative protein with the entire multi-domain structures the hits are scoring badly and are not even listed as they fall below the requisite 50% of secondary structure to be considered a match. The reverse template method once again has no such problem as it is dealing with local similarity within a 10Å radius of any putative site. One way round this issue with SSM would be to alter its search parameters but this creates additional problems with increased run-time and a far greater number of hits, the majority of which will be false positives.
The other structure-based methods are useful in different ways. When a strong match is found to one of the enzyme templates, the functional significance is greater as the templates have been created from a carefully annotated database of known enzyme reactions and catalytic residues. In the case of the ligand- and DNA-binding templates the matches can be used to identify likely substrates, cofactors or fragments of ligands that can fit in the active site. This information can be of importance to the user when trying to set up ligand binding assays or co-crystallisation experiments.
One of the biggest problems is the definition and comparison of function – how do we determine a “correct” prediction? In this analysis the assignment of whether or not a hit is correct was achieved through a laborious manual process fraught with difficulties and occasional human error. One particularly tricky case involves an ABC transporter protein that binds ATP (PDB entry 1ji0). In this example the ProFunc reverse template results provide a number of hits to other ABC transporter proteins but there are also hits to numerous other structures such as “DNA mismatch repair protein”, “gluconate kinase”, “replication factor C” and “cell division control protein”. The problem with assessing these hits is that they all have GO terms that include “ATP binding” – so are these to be marked as true positives or false positives? The question arises because the reverse template method is looking for local similarities in structure – in this case the ATP binding region. It could therefore be argued that all of these hits are “correct” as they all bind ATP, but when one looks at the function as a whole these become false positive hits. In the initial manually based analysis these cases are identified as false positives but the issue is a contentious one and illustrates the need for a clearer definition of what a “correct” hit is.
Another example is that of tartronate semialdehyde reductase (PDB entry 1tea) which was found to have 2 significant hits to “hydroxyisobutyrate dehydrogenase”. These hits were annotated as false positive based on an initial textual comparison but further examination reveals both tartronate semialdehyde reductase and hydroxyisobutyrate dehydrogenase to share the top three levels of EC classification (in this case EC 3.1.1.-). The EC class was not picked up in the procedures and illustrates some of the problems that can occur if entries are not fully annotated in the databases. In this situation, it can be argued that the manual classification should be altered to true positive as they are performing similar reactions even though substrate specificity has diverged.
A more robust method to compare the functions of two proteins is to use GO annotation from the entire gene ontology but this has its own difficulties, the greatest being that not every protein in the structure or sequence databases has GO annotation. This issue will only improve with time so this problem aside, the most pressing problems relate to the confidence of assignments: some are manually curated whereas others have been inferred from electronic annotation. The two situations do not have the same weighting or confidence and therefore this needs to be reflected in any comparisons. Additionally, the GO system is not a linear hierarchy and how exactly you compare any two terms is difficult.
Instead of using the entire ontology to compare the functions of two proteins we have shown that the use of generic GO-slim terms can bypass many of the difficulties in comparing sets of terms. In this initial study we have found that using a cutoff of 75-100% of the GO-slim terms matching between a query protein and a hit is a good indicator of a positive match. The success rate was comparable to expert manual assessment of the same data. One problem that did come to light was that the generic GO-slim is too generic - any functional comparisons made are too vague to be of use when trying to design experiments to test functional predictions. In order to bridge the gap between the two approaches we constructed a more extended molecular function GO-slim (MF-GO-slim) that allows for more detailed comparisons. This extended MF-GO-slim showed a marked improvement on the Generic GO-slim and a cut-off of 75% matching terms gives the best performance. Once a similarity in general function has been identifed by the MF-GO-slim more detailed comparisons can then be made using the full ontology. The study has shown that this very simplistic approach is useful for comparing the functions of annotated proteins but it is evident that further work will be required in order to define a quantitative measure for the similarity in GO-slim terms, perhaps using the method described by Lord et al20 for identifying semantic similarity between entries in a database. The greatest problem with the method is that it is only useful for situations where a hit has been assigned gene ontology terms – this issue will only be resolved by greater coverage by GO of the sequence and structure databases. One final question is where this approach would be used when examining results from hypothetical proteins of unknown function. The GO-slim approach can be used in this case to compare all the annotated hits from all methods with one another in order to identify commonalities in functions – the greater the similarity in function amongst the hits the more likely it is that the function is correct.
From our experiences with the ProFunc server and from the success rates described previously it is evident that, in order to improve our success rate for the second phase of the PSI, the range of analyses will need to be improved and include new predictive methods not based on homology. This is echoed by the need to look at higher level functions where we will need to take into account the cellular component, interacting partners, networks, expression, regulation, etc. The MCSG structures were a good dataset to develop and test the methods but specific benchmark datasets will be required in order to test the variety of methods and allow comparisons to be made between them rather than the current state with each method having its own “good examples”. The consideration of various functional attributes (e.g. enzyme/non-enzyme, DNA binding, metal binding, etc) and having benchmark datasets for each attribute would be a much more successful strategy than trying to build a complete dataset to test the rather vague concept of “function prediction” as a whole.
Methods
Dataset construction
The starting dataset comprised the 319 PDB deposits solved by the MCSG as of 30/09/2005. This was then culled using the PISCES server at 30% sequence identity to provide a non-redundant set of 282 structures. The resultant dataset was then split into those structures for which the functions were known, those where putative functions had been assigned by the depositors before submission to ProFunc, and those for which the function remained unknown (e.g. “hypothetical protein”).
Structural Analysis
Each structure was submitted to the ProFunc server and the results stored for analysis. The various methods within ProFunc use their own scoring scheme to rank the hits and classify them by the confidence of the match15. These scoring schemes were adopted for this analysis and used to assign confidence to the functional predictions. The parameters used to measure confidence and rank hits are described in Table 2 along with their respective ranges.
Table 2. Parameters chosen for each ProFunc method to classify hit “strength”.
| Code | “Strong” Hits | “Moderate” Hits | “Weak” Hits | |
|---|---|---|---|---|
| Structure-based Methods | ||||
| Secondary Structure Matching (SSM) | SSM | Zscore > 10 | Zscore 6–10 | Zscore <6 |
| Templates (using internal scoring scheme) | ENZ, LIG, DNA, SIT | Confidence: “Certain” (E-value <1.00 x10−6) or “Probable” (E-value 1.00 x10−6 – 0.01) | Confidence: “Possible” (E-value 0.01 – 0.10) | Confidence: “Longshot” (E-value >0.10) |
ENZ = Enzyme active site templates (CSA)
DNA = DNA based automatically generated templates
LIG = Ligand based automatically generated templates
SIT = “Reverse” template
Filtering hits
In order to compensate for any temporal bias, the structure-based results were “backdated” to the time of release of the MCSG query protein by ignoring hits to protein structures released after the MCSG structure. This allows us to see what the results would have suggested at the time of release. Note that it is not possible to backdate the sequence-based analyses in the same way hence our focus on the structure-based approaches only.
Manual functional comparison
Any free text was extracted from the PDB record along with any keywords from the corresponding PDBsum database entry for each post-filtered top hit. These were placed in a file alongside the functional annotation of the MCSG structure for comparison. The match was then assessed as a correct hit, false hit, unknown function, or no hit and noted in the file. The global sequence identity of the match was also calculated using SSEARCH21,22 in order to identify clear homologues when assessing cases of moderate structural similarity.
Comparing the best methods from manual assessment
A robust way of assessing the effectiveness of the best structure-based procedures is to calculate their Receiver Operating Characteristic (ROC) curves. The ROC curve is a graphical representation of the trade off between the false negative and false positive rates for every possible cut off value. For each structure of known function, the top hit (after the filtering process) was extracted from ProFunc. Each hit was then annotated with true positive (+), false positive (−) or unknown (?) by manual comparison of the known function with the header details and any GO annotation of the hit. Only the true and false positive results were kept (hits to unknown function cannot be grouped in either category and can be ignored) and used, alongside their scores, to create the ROC curve.
Automatic functional comparison: GO-slim method
The Gene Ontology23 is an attempt to standardise the description and definition of biological terms through three structured, controlled vocabularies. The three major sections are Cellular Component, Biological Process and Molecular Function – it is the last of these that is of interest in this study. Many recent automated function prediction methods (e.g. Phunctioner24) have utilised the Gene Ontology data in order to aid the prediction and comparison of function25–29. There are a number of ways to compare gene ontology terms but the task is made difficult by the fact that not all GO-terms are useful (e.g. “molecular function unknown”), the level of annotation differs between proteins of the same function, and any probability-based approach will be more biased towards those proteins that appear regularly in the sequence databases. In addition, the ontology is not an even hierarchy and some areas of research are over-represented, as are some species.
One way to deal with the inconsistencies in the ontology is to use the GO-slim system. A study by Dolan et. al. 30 demonstrated their use in assessing the consistency of GO annotations from different groups. GO-slims are cut down versions of the gene ontology that give a broad overview of the ontology and are useful in situations where a broad classification of a gene product function is required. The terms included in any one GO-slim can be selected by the user according to their needs, such as the aforementioned study where comparisons were made using a GO-slim consisting of only 19 terms. As standard the gene ontology consortium provides a generic, species-independent GO-slim that condenses the entire ontology into 68 key parent terms of which only 31 are in the “molecular function” class (Table 1a in supplementary material). This generic GO-slim was selected as a starting point to investigate automatic assessment of function prediction accuracy.
Procedure to compare known function with predicted function from top hit
In order to compare a query protein with any hit protein, a list of GO-slim terms was required for each. This information was obtained using various mapping files from the Gene Ontology FTP site. If a UniProt code is available for the protein, the terms were extracted from the GOA-UniProt mapping31, if a PDBcode is available then the GOA-PDB mapping file was scanned. Every GO term was then compared against the GO-slim list and, if present, added into the final list of GO-slim terms for that hit as is. If however, the term was further down the graph its GO-slim terms needed to be identified by searching the GO to GO-slim mapping file (maps all of the ontology to the GO-slim). The full list of identified “GO-slim” terms was then condensed down to a final list of unique GO-slim terms.
If a hit were correct the protein would be expected to lie in a similar “region” of the GO graph and therefore it should in theory share more GO-slim terms than would be expected of proteins with very different functions. The unique GO-slim terms from the hit were compared against the unique GO-slims from the query. If the number of terms matched was deemed to be significant it was assigned as a true hit, otherwise it was deemed false. The derivation of what constitutes a significant number of matched terms is discussed in the Results section.
Creation of Molecular Function GO-slim (“MF-GO-slim”)
One problem with using the generic GO-slim is its generality (7844 molecular function GO terms slimmed down to 31 key parent terms) which is exemplified by the enzymes. The generic GO-slim condenses the gene ontology at a level that is equivalent to the top level of the EC schema (e.g. E.C. 1.-.-.- : Oxidoreductases). In order to derive an extended GO-slim that is more specific for molecular function prediction the ontology needed to be edited. The gene ontology consortium offers the DAG-edit tool to view the entire ontology and allow users to select terms of interest to put into a new GO-slim. A perl script supplied by the GO team was then used to map the entire ontology to the newly created extended GO-slim (MF-GO-slim) so that it could be used in place of the generic GO-slim. The 190 “molecular function” GO-terms selected for inclusion as part of the MF-GO-slim are listed in the supplementary material (Table 1b in supplementary material).
Supplementary Material
Acknowledgments
This work was performed with funding from the National Institutes of Heath, grant number GM62414, the US DoE under contract W-31-109-Eng-38. The authors wish to thank the Gene Ontology group at the EBI for their helpful discussions and assistance with the creation of the modified GOslims.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Blundell TL, Mizuguchi K. Structural genomics: an overview. Prog Biophys Mol Biol. 2000;73:289–295. doi: 10.1016/s0079-6107(00)00008-0. [DOI] [PubMed] [Google Scholar]
- 2.Chen L, Oughtred R, Berman HM, Westbrook J. TargetDB: a target registration database for structural genomics projects. Bioinformatics. 2004;20:2860–2862. doi: 10.1093/bioinformatics/bth300. [DOI] [PubMed] [Google Scholar]
- 3.Watson JD, et al. Target selection and determination of function in structural genomics. IUBMB Life. 2003;55:249–255. doi: 10.1080/1521654031000123385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bairoch A, et al. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2005;33:D34–D38. doi: 10.1093/nar/gki063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeats C, et al. Gene3D: modelling protein structure, function and evolution. Nucleic Acids Res. 2006;34:D281–D284. doi: 10.1093/nar/gkj057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gough J, Chothia C. SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments. Nucleic Acids Res. 2002;30:268–272. doi: 10.1093/nar/30.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonnhammer EL, Eddy SR, Birney E, Bateman A, Durbin R. Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 1998;26:320–322. doi: 10.1093/nar/26.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Todd AE, Orengo CA, Thornton JM. Plasticity of enzyme active sites. Trends Biochem Sci. 2002;27:419–426. doi: 10.1016/s0968-0004(02)02158-8. [DOI] [PubMed] [Google Scholar]
- 11.Watson JD, Laskowski RA, Thornton JM. Predicting protein function from sequence and structural data. Curr Opin Struct Biol. 2005;15:275–284. doi: 10.1016/j.sbi.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Whisstock JC, Lesk AM. Prediction of protein function from protein sequence and structure. Q Rev Biophys. 2003;36:307–340. doi: 10.1017/s0033583503003901. [DOI] [PubMed] [Google Scholar]
- 13.Stark A, Russell RB. Annotation in three dimensions. PINTS: Patterns in Non-homologous Tertiary Structures. Nucleic Acids Res. 2003;31:3341–3344. doi: 10.1093/nar/gkg506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laskowski RA, Watson JD, Thornton JM. Protein function prediction using local 3D templates. J Mol Biol. 2005;351:614–626. doi: 10.1016/j.jmb.2005.05.067. [DOI] [PubMed] [Google Scholar]
- 15.Laskowski RA, Watson JD, Thornton JM. ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res. 2005;33:W89–W93. doi: 10.1093/nar/gki414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 17.Jones S, Barker JA, Nobeli I, Thornton JM. Using structural motif templates to identify proteins with DNA binding function. Nucleic Acids Res. 2003;31:2811–2823. doi: 10.1093/nar/gkg386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanishvili R, et al. Integrating structure, bioinformatics, and enzymology to discover function: BioH, a new carboxylesterase from Escherichia coli. J Biol Chem. 2003;278:26039–26045. doi: 10.1074/jbc.M303867200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu R, et al. Staphylococcus aureus IsdG and IsdI, heme-degrading enzymes with structural similarity to monooxygenases. J Biol Chem. 2005;280:2840–2846. doi: 10.1074/jbc.M409526200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lord PW, Stevens RD, Brass A, Goble CA. Investigating semantic similarity measures across the Gene Ontology: the relationship between sequence and annotation. Bioinformatics. 2003;19:1275–1283. doi: 10.1093/bioinformatics/btg153. [DOI] [PubMed] [Google Scholar]
- 21.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 22.Pearson WR. Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms. Genomics. 1991;11:635–650. doi: 10.1016/0888-7543(91)90071-l. [DOI] [PubMed] [Google Scholar]
- 23.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pazos F, Sternberg MJ. Automated prediction of protein function and detection of functional sites from structure. Proc Natl Acad Sci USA. 2004;101:14754–14759. doi: 10.1073/pnas.0404569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo X, Shriver CD, Hu H, Liebman MN. Analysis of metabolic and regulatory pathways through Gene Ontology-derived semantic similarity measures. AMIA Annu Symp Proc. 2005;972 [PMC free article] [PubMed] [Google Scholar]
- 26.Vinayagam A, et al. Applying Support Vector Machines for Gene Ontology based gene function prediction. BMC Bioinformatics. 2004;5:116. doi: 10.1186/1471-2105-5-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smid M, Dorssers LC. GO-Mapper: functional analysis of gene expression data using the expression level as a score to evaluate Gene Ontology terms. Bioinformatics. 2004;20:2618–2625. doi: 10.1093/bioinformatics/bth293. [DOI] [PubMed] [Google Scholar]
- 28.Lee V, Camon E, Dimmer E, Barrell D, Apweiler R. Who tangos with GOA?-Use of Gene Ontology Annotation (GOA) for biological interpretation of '-omics' data and for validation of automatic annotation tools. In Silico Biol. 2005;5:5–8. [PubMed] [Google Scholar]
- 29.Carroll S, Pavlovic V. Protein classification using probabilistic chain graphs and the Gene Ontology structure. Bioinformatics. 2006 doi: 10.1093/bioinformatics/btl187. [DOI] [PubMed] [Google Scholar]
- 30.Dolan ME, Ni L, Camon E, Blake JA. A procedure for assessing GO annotation consistency. Bioinformatics. 2005;21 (Suppl 1):i136–i143. doi: 10.1093/bioinformatics/bti1019. [DOI] [PubMed] [Google Scholar]
- 31.Camon E, et al. The Gene Ontology Annotation (GOA) Database: sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res. 2004;32:D262–D266. doi: 10.1093/nar/gkh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.