

Abstract
The proteasome constitutes the central proteolytic component of the highly conserved ubiquitin–proteasome system, which is required for the maintenance and regulation of basic cellular processes, including differentiation, proliferation, cell cycling, gene transcription and apoptosis. Here we show that inhibition of proteasomal proteolytic activity by the proteasome inhibitors bortezomib and lactacystin suppresses essential immune functions of human CD4+ T cells activated by allogeneic dendritic cells (DCs). In activated CD4+ T cells, proteasome inhibition induces apoptosis accompanied by rapid accumulation and stabilization of the tumour suppressor protein p53. Activated CD4+ T cells surviving proteasome inhibition undergo inhibition of proliferation by induction of G1 phase cell-cycle arrest. Induction of G1 arrest is accompanied by the accumulation of cyclin-dependent kinase inhibitors p21WAF1/CIP1 and p27KIP1 and the disappearance of cyclin A, cyclin D2 and proliferating cell nuclear antigen, proteins known to regulate G1 to S phase cell-cycle transitions. Expression of the activation-associated cell surface receptors CD25, CD28, CD120b and CD134 as well as production of interferon-γ (IFN-γ), tumour necrosis factor-α (TNF-α), interleukin-4 (IL-4) and IL-5 is suppressed in response to proteasome inhibition in CD4+ T cells activated by DCs. Expression of CD25, IFN-γ, TNF-α, IL-4 and IL-5 is known to be mediated by the transcriptional activity of nuclear factor of activated T cells (NFAT), and we show here that proteasome inhibition suppresses activation and nuclear translocation of NFATc2 in activated CD4+ T cells. Thus, the proteasome is required for essential immune functions of activated CD4+ T cells and can be defined as a molecular target for the suppression of deregulated and unwanted T-cell-mediated immune responses.
Keywords: immunosuppression, proteasome, proteasome inhibitors, T cells
Introduction
The ubiquitin–proteasome system (UPS) constitutes an evolutionarily conserved and highly organized cellular machinery for cytosolic and nuclear protein degradation in eukaryotic cells.1,2 A plethora of cell proteins essential for the regulation of development, differentiation, proliferation, cell cycling, apoptosis, gene transcription, signal transduction, senescence, inflammation and stress response have been shown to undergo limited and controlled proteolytic degradation by the UPS.1,3,4 The UPS therefore plays a key role in the regulation and maintenance of essential cellular processes.
The central proteolytic unit of the UPS is the 26S proteasome, a large multicatalytic multisubunit protease composed of a 20S barrel-shaped catalytic core complex and two flanking 19S regulatory complexes.5,6 The proteolytic activities of the 26S proteasome occur in the 20S complex, built up of four axially stacked rings. Each outer ring contains seven different α-subunits, and each of the two inner rings is formed by seven different but related β-subunits. Only β1, β2 and β5 subunits and their corresponding immunosubunits β1i, β2i and β5i are proteolytically active and harbour proteolytic sites formed by N-terminal threonine residues that face the central cavity of the 20S complex.5–7 Based on their specificity towards oligopeptidyl substrates, β1, β2 and β5 subunits have been described as possessing caspase-like, trypsin-like and chymotrypsin-like peptidase activity, respectively.8,9
The identification and use of synthetic and biological inhibitors with different inhibitory profiles towards the peptidase activities of β1, β2 and β5 proteolytic subunits have principally contributed to the identification of essential functions of the 26S proteasome in various processes and pathways of eukaryotic cells.3,10–13 In particular, inhibition of proteasome activities by proteasome inhibitors induces apoptosis preferentially in rapidly proliferating and neoplastic cells.3,14–16 These findings have recently paved the way for the use of proteasome inhibitors in cancer therapy.17–19 Moreover, proteasome inhibition has recently been shown to interfere with essential immune functions of human dendritic cells (DCs),20,21 and several studies in rodents suggest that proteasome inhibitors can be used as immunosuppressive agents for the treatment of deregulated and unwanted T-cell-mediated immune responses, including those that contribute to the pathogenesis of polyarthritis, psoriasis, allograft rejection and graft-versus-host disease.22–27
It was recently demonstrated that proteasome inhibitors induce apoptosis in activated and proliferating, but not in resting, human T cells,28,29 providing one possible mechanism for the suppression of T-cell-mediated immune responses by proteasome inhibitors. We have now investigated the effects and mechanisms induced by proteasome inhibition in human CD4+ T cells activated in a physiological manner by allogeneic DCs. Using inhibitors that target the proteolytic activity of β1, β1i, β2, β2i, β5 and β5i proteasome subunits, we show here that proteasome inhibition suppresses the activation, proliferation, survival and important immune functions of human CD4+ T cells, revealing an essential role of the proteasome in the regulation of T-cell-mediated immune responses.
Materials and methods
Antibodies and reagents
Recombinant human (rh) granulocyte–macrophage colony-stimulating factor (GM-CSF), interleukin-4 (rhIL-4), and interferon-γ (rhIFN-γ) were all purchased from AL-Immunotools (Friesoythe, Germany). Lipopolysaccharide (LPS) from Escherichia coli 055:B5 was obtained from Sigma (Heidelberg, Germany). The biotinylated proteasome-specific affinity probe AdaK(Bio)AhX3L3VS (BioALVS) was purchased from Calbiochem (Darmstadt, Germany).
The following monoclonal antibodies (mAbs) were used: fluorescein isothiocyanate (FITC)-conjugated mouse immunoglobulin G1 (IgG1) isotype control (clone MOPC-21), anti-CD3 (SK1) and anti-CD209 (DCN46) from BD Pharmingen (Heidelberg, Germany), anti-CD28 (KOLT-1) and anti-CD95 (FAS 19) from AL-ImmunoTools, phycoerythrin (PE)-conjugated IgG1 isotype control (MOPC-21), anti-CD4 (SK3), anti-CD8 (SK7), anti-CD25 (2A3) and anti-CD134 (OX-40; ACT35) from BD Pharmingen and anti-CD120b (MR2-1) from Serotec (Düsseldorf, Germany). An antibody against mono- and poly-ubiquitinylated proteins (clone FK2, mouse mAb), anti-proteasome β1 subunit (MCP421, mouse mAb), anti-proteasome β1i subunit (LMP2-13, mouse mAb), anti-proteasome β2 subunit (MCP165, mouse mAb), anti-proteasome β2i subunit [rabbit polyclonal antibody (pAb)], anti-proteasome β5 subunit (rabbit pAb) and anti-proteasome β5i subunit (LMP7-1, mouse mAb) were all purchased from Biomol (Hamburg, Germany), anti-p21WAF/CIP1 (C-19, rabbit pAb), anti-p27KIP1 (F-8, mouse mAb), anti-p53 (DO-1, mouse mAb), anti-proliferating cell nuclear antigen (PCNA; FL-261, rabbit pAB), anti-cyclin A (H-432, rabbit pAb), anti-cyclin D2, anti-nuclear factor of activated T cells (NFAT) c2 (4G6-G5, mouse mAb), anti-lamin A/C (636, mouse mAb) were obtained from Santa Cruz (Heidelberg, Germany), secondary horseradish peroxidase (HRP) -conjugated anti-rabbit, anti-mouse antibodies were provided by Pierce (Bonn, Germany), and β-actin (AC-15, mouse mAb) was purchased from Sigma. The proteasome inhibitors bortezomib (PS-341, Velcade®) from Millennium Pharmaceuticals (Cambridge, MA) and lactacystin from Biomol (Hamburg, Germany), were dissolved in dimethylsulphoxide (DMSO) and stored at −20°.
Generation of DCs
The DCs were generated from human healthy donor CD14+ monocytes, isolated from heparinized peripheral blood by Ficoll (Innotrain, Kronberg, Germany) density gradient centrifugation and subsequent negative isolation with immunomagnetic beads (Dynal/Invitrogen, Karlsruhe, Germany) as described before.21 Briefly, CD14+ monocytes were cultured for 48 hr (day 1 to day 3) in serum-free culture medium (CM) consisting of RPMI-1640 (Gibco/Invitrogen, Karlsruhe, Germany), 2 mm l-glutamine (Sigma), 100 IU penicillin and 100 μg/ml streptomycin (Gibco/Invitrogen) in the presence of 50 ng GM-CSF and 100 ng rhIL-4 to generate immature DCs (iDCs). To induce final maturation of iDCs, cells were treated on day 3 with 100 ng/ml LPS and IFN-γ (1000 U/ml). Purity of the cells generated in this manner ranged between 90% and 95%. Mature DCs (mDCs) were defined by their profile of cell surface molecule expression, examined and quantified by flow cytometry.
T-cell purification and activation
CD4+ T cells were isolated from the peripheral blood of human healthy donors by Ficoll density gradient centrifugation and subsequent negative isolation with immunomagnetic beads (Invitrogen). Purity of cells was defined by flow cytometric analysis of CD3/CD4/CD8 surface molecule expression and ranged between 90% and 98%.
For activation of T cells, 2·25 × 106 cells/ml freshly isolated CD4+ T cells were cocultured for 5 days with allogeneic mDCs in CM supplemented with 10% fetal calf serum (FCS, Gibco-Invitrogen) in a DC : T-cell ratio of 1 : 10. Proteasome inhibitors or DMSO control were added at day 4 to the activation culture for the last 24 hr or for the times indicated. In all coculture experiments of DC-mediated CD4+ T-cell stimulation, the purity of CD4+ T cells was higher than 93% and the ratio of DCs was lower than 1% after coculture for 5 days. Non-activated, resting T cells were cultured in CM with 10% FCS without further additions.
Restimulation of T cells
For restimulation assays, activated T cells generated by 4-day culture with allogeneic mDCs were washed, counted and resuspended in fresh CM supplemented with 10% FCS at a concentration of 1 × 106 cells/ml. Cells were exposed to the indicated concentrations of bortezomib or DMSO control. After an incubation period of 24 hr, cells were washed extensively with warm phosphate-buffered saline (PBS), and only viable and non-apoptotic T cells were counted using trypan blue exclusion staining. Cells were resuspended in fresh CM supplemented with 10% FCS at a concentration of 0·5 × 106 cells/ml and restimulated for 24 hr with either 20 μg/ml lectin from Phytolacca americana (Sigma), the same allogeneic mDCs used for initial stimulation or third-party allogeneic mDC in a DC : T-cell ratio of 1 : 20.
Probing proteasome β-subunit activity using activity probe
Affinity labelling of active proteasome subunits using the proteasome-specific affinity probe BioALVS was performed as described previously.30,31 Briefly, activated T cells were incubated with the indicated amounts of each inhibitor for 1 hr. The cells were washed twice and pelleted with ice-cold PBS. A volume of glass beads (≥ 106 μm, acid-washed, Sigma) equivalent to the volume of the pellet was added, followed by a similar volume of homogenization buffer (50 mm Tris–HCl pH 7·4, 1 mm dithiothreitol, 5 mm MgCl2, 2 mm ATP, 250 mm sucrose). Cells were vortexed at high speed for 1 min, and the beads and cell debris were removed by centrifugation at 10 000 g for 5 min at 4°. The resulting supernatant was centrifuged at 10 000 g for 20 min at 4° to remove intact cells and nuclei. Protein concentration was determined using a DC-Protein assay (Bio-Rad, Munich, Germany). Equal amounts of cell lysates (approximately 25 μg) were incubated with 4 μg BioALVS for 2 hr at 37°. Subsequently, proteins were denatured by boiling in reducing 4 × sample buffer, separated on 12% sodium dodecyl sulphate (SDS) polyacrylamide Tris–HCl gels and electrotransferred on to polyvinylidene fluoride (PVDF) membranes. Immunoblotting was performed using streptavidin–HRP (Zymed-Invitrogen, Karlsruhe, Germany) followed by enhanced chemiluminescence (ECL, Pierce).
Flow cytometry
Flow cytometric analysis of cell surface receptors of activated or restimulated T cells was performed with standard staining and analysis procedures using FACScan and Cellquest software (BD Pharmingen). Only viable cells were gated and considered for flow cytometric analysis, except for measurement of apoptosis.
Analysis of apoptosis
Apoptotic cell death was determined using the Annexin V-FITC/propidium iodide (PI) kit from BD Biosciences, Heidelberg, Germany. Briefly, activated or resting CD4+ T cells were treated for 24 hr with the indicated concentrations of the proteasome inhibitors, washed with PBS and resuspended in binding buffer in a cell concentration of 1 × 106/ml. Annexin V-FITC and PI were added and cells were incubated for 15 min at room temperature in the dark. Apoptosis of cells was measured and quantified using flow cytometry. Percentages of specific apoptosis were calculated as follows:
 |
Analysis of T-cell proliferation
T-cell proliferation was quantified by measuring [3H]thymidine (Amersham, Munich, Germany) incorporation. Activated CD4+ T cells were incubated on day 4 for the last 24 hr with the indicated concentrations of proteasome inhibitors or DMSO controls and pulsed with 5 μCi/ml [3H]thymidine for the last 18 hr.
Restimulated T cells were treated as described in the Restimulation of T cells section and pulsed with 5 μCi/ml [3H]thymidine for the last 18 hr of the restimulation culture, but without further addition of proteasome inhibitors. Incorporation of [3H]thymidine was quantified using a beta-counter (Inotech, Wohlen, Switzerland).
Cell-cycle analysis
Cell-cycle analysis was performed as described previously.16,28 Briefly, after treatment of activated T cells with proteasome inhibitors, cells were washed in PBS, resuspended in 400 μl staining solution containing 50 μg/ml PI, 0·1% sodium citrate-2-hydrate and 0·1% Triton X-100 (Sigma) and incubated for 30 min at 4° in the dark. DNA content of the cells was analysed using a FACScan flow cytometer equipped with the Cellquest software (Becton Dickinson, Heidelberg, Germany).
Immunoblotting
For isolation of whole cell lysates, cells were treated for the indicated time with proteasome inhibitors or DMSO as control. Activated and resting CD4+ T cells were washed with ice-cold PBS, and the resulting pellets were lysed for 30 min in ice-cold radioimmunoprecipitation assay (RIPA) buffer. Homogenates were centrifuged at 10 000 g for 10 min at 4°, and the supernatant was used for further analysis. For analysis of nuclear translocation of NFATc2, nuclear and cytoplasmatic proteins were sampled using a nuclear/cytoplasmic protein extraction kit (Pierce) according to the manufacturer's instructions. Equal amounts of protein were separated by SDS–polyacrylamide gel electrophoresis on 7·5% or 12% Tris–HCl gels and electrotransferred on PVDF membranes. After blocking with a solution of PBS containing 5% bovine serum albumin and 0·1% Tween-20 for at least 1 hr at room temperature, membranes were probed with primary antibodies followed by HRP-conjugated secondary antibodies. Washing steps following incubation with antibodies were performed in PBS + 0·1% Tween-20. Immunoreactive bands were visualized by ECL using Supersignal West Femto Chemiluminescence Reagent (Pierce).
Quantification of cytokine production
To quantify the amounts of cytokine production in activated and restimulated CD4+ cells, IL-2, IL-2sRα, IL-4, IL-5, tumour necrosis factor-α (TNF-α) (R&D Systems, Wiesbaden, Germany) and IFN-γ (Biozol, Eching, Germany) enzyme-linked immunosorbent assay kits were used. Supernatants of restimulated activated CD4+ T cells were collected, and the amounts of cytokines were measured according to the manufacturer's instructions.
Statistical analyses
Statistical analyses were performed using Excel software. Differences between mean values were assessed using two-tailed paired Student's t-test. Statistical significance was set at P < 0·05.
Results
The proteasome inhibitors bortezomib and lactacystin inhibit proteasomal proteolytic subunits and activity in activated CD4+ T cells
The inhibitory profiles of bortezomib and lactacystin towards proteasomal proteolytic subunits were investigated in activated CD4+ T cells by affinity labelling, using the proteasome-specific affinity probe BioALVS.30 CD4+ T cells activated for 5 days with allogeneic DCs express all of the proteasomal proteolytic subunits: β1, β1i, β2, β2i, β5 and β5i (Fig. 1a). Affinity labelling using BioALVS, which targets these proteolytic subunits,30 reveals that bortezomib (Bor) at 10 nm and lactacystin (LC) at 5 μm and 10 μm completely occupy and inhibit β1, β1i, β2, β2i, β5 and β5i subunits (Fig. 1b). Accumulation of polyubiquitinated proteins is a functional consequence of inhibition of proteasomal protein degradation.32 To investigate the extent of proteasome inhibition in functional terms, we performed immunoblot analysis of polyubiquitinated proteins in activated CD4+ T cells exposed to bortezomib or lactacystin. Figure 1(c) shows a rapid accumulation of polyubiquitinated proteins in CD4+ T cells exposed to 10 nm bortezomib or 10 μm lactacystin, thus demonstrating inhibition of proteasomal proteolytic activity by the proteasome inhibitors.
Figure 1.
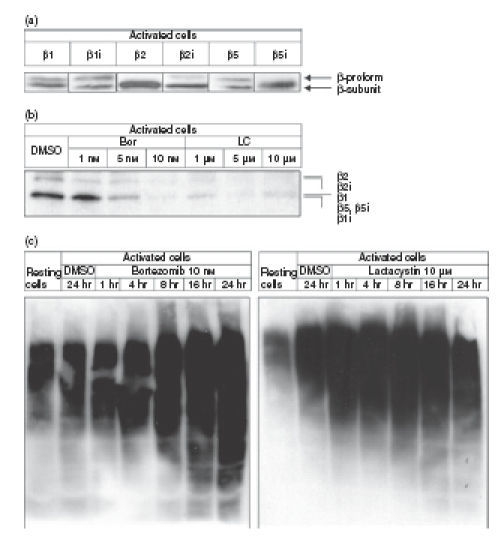
Inhibition of proteasomal proteolytic subunits and proteasomal protein degradation by bortezomib and lactacystin in activated CD4+ T cells. (a) Immunoblot analysis of expression of the proteasomal proteolytic subunits β1, β1i, β2, β2i, β5 and β5i in CD4+ T cells activated for 5 days with allogeneic DCs. (b) Immunoblot analysis of affinity labelling of the proteasomal proteolytic subunits β1, β1i, β2, β2i, β5 and β5i in activated CD4+ T cells using the specific affinity probe BioALVS. Cells were exposed for 1 hr to the indicated concentrations of bortezomib (Bor) or lactacystin (LC). (c) Immunoblot analysis of accumulation of polyubiquitinated proteins in activated CD4+ T cells using the FK2 monoclonal antibody which detects mono- and polyubiquitinated proteins. Immunoblots were performed in triplicate with similar results.
Proteasome inhibition induces apoptosis accompanied by accumulation and stabilization of p53 in activated CD4+ T cells
Flow cytometric analysis of annexin V binding to plasma membrane externalization of phosphatidylserine revealed that activated, and to a lesser extent resting, CD4+ T cells underwent apoptosis in response to treatment with bortezomib or lactacystin (Fig. 2a). As shown in Fig. 2(b), bortezomib and lactacystin were able to induce apoptosis in a dose-dependent manner in activated and, to a lesser extent, in resting CD4+ T cells. As determined by immunoblotting, induction of apoptosis by bortezomib in activated CD4+ T cells was accompanied by rapid accumulation and stabilization of pro-apoptotic p53 protein (Fig. 2c), a molecular event known to mediate the apoptosis induced by proteasome inhibitors in various cells other than T cells.33–37 We also detected accumulation of poyubiquitinated species of p53 in response to treatment of the cells with bortezomib (p53-Ubn, Fig. 2c), indicating that accumulation of p53 proteins, which become substrates for proteasomal degradation after polyubiquitination,38 was a direct consequence of proteasome inhibition by bortezomib. In all coculture experiments of DC-mediated CD4+ T-cell activation, the purity of CD4+ T cells was higher than 93% and the DC ratio was lower than 1% after coculture for 5 days (Fig. 2d). Therefore, the contribution of the DCs to the results obtained in the present study could be neglected.
Figure 2.
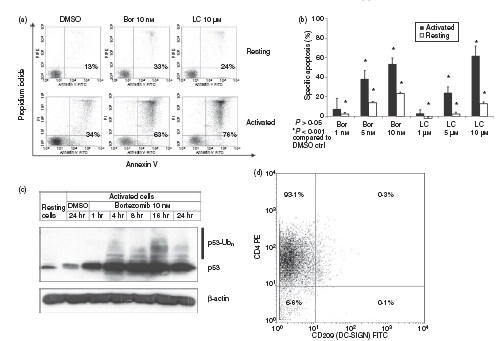
Apoptosis induced by proteasome inhibition in activated CD4+ T cells. (a) Flow cytometric analysis of cytoplasmic uptake of propidium iodide (PI) and binding of annexin V to the plasma membrane externalization of phosphatidylserine of resting CD4+ T cells and CD4+ T cells activated for 4 days with allogeneic dendritic cells (DCs) at a DC : T-cell ratio of 1 : 10. Before flow cytometric analysis, cells were exposed for 24 hr to dimethylsulphoxide (DMSO), bortezomib (Bor) or lactacystin (LC). (b) Specific apoptosis of resting and activated CD4+ T cells exposed for 24 hr to bortezomib or lactacystin. Specific apoptosis was determined by flow cytometry and calculated as described in the Materials and methods. Data are given as mean values ± SE of the mean of six independent experiments carried out in triplicate. (c) Immunoblot analyses of accumulation of p53 and polyubiquitinated p53 (p53-Ubn) in activated CD4+ T cells exposed for the indicated times to bortezomib or DMSO. Amounts of β-actin are demonstrated as a control of equal protein loading. Immunoblots were performed in triplicate with similar results. (d) Demonstration of the purity of the CD4+ T-cell population and the ratio of CD4+ T cells and DCs in the coculture experiments for CD4+ T-cell activation. Cell surface expression of CD4 and CD209 was determined by flow cytometry after 5 days of coculture. A dot plot of one representative experiment out of 10 is shown.
Proteasome inhibition suppresses proliferation of activated and restimulated CD4+ T cells
Figure 3(a) shows that proteasome inhibition by bortezomib or lactacystin strongly suppressed the proliferation of CD4+ T cells activated by allogeneic DCs. Bortezomib at 5 nm and 10 nm, and lactacystin at 5 μm and 10 μm, which are the concentrations required for efficient inhibition of proteasomal proteolytic activities (Fig. 1b,c), completely inhibited the proliferation of activated CD4+ T cells (Fig. 3a). As shown in Fig. 2(b), cells treated in this manner underwent significant apoptosis. However, the almost complete inhibition of [3H]thymidine incorporation by activated CD4+ T cells treated with 5 nm and 10 nm bortezomib or 5 μm and 10 μm lactacystin (Fig. 3a) indicated that cells surviving exposure to the proteasome inhibitors underwent inhibition of DNA synthesis and proliferation. Moreover, bortezomib was able to inhibit, in a dose-dependent manner, the proliferation of CD4+ T cells activated for 4 days with allogeneic DCs and subsequently restimulated with the same allogeneic DCs used for the initial activation, third-party allogeneic DCs or lectin (Fig. 3b).
Figure 3.
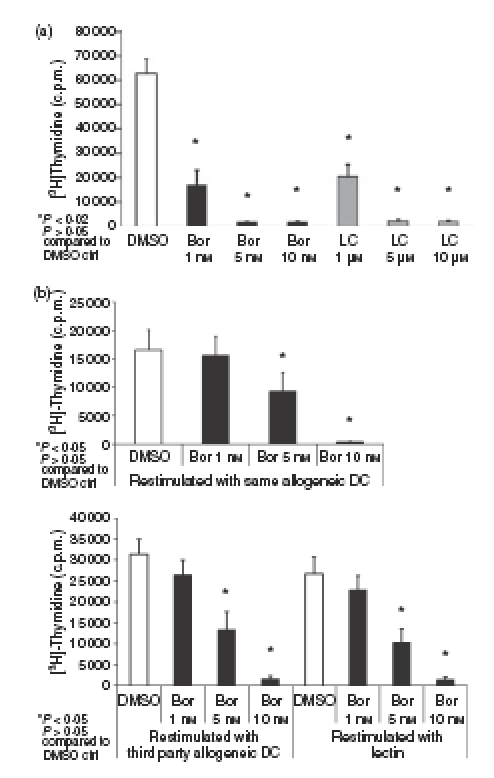
Inhibition of proliferation of activated and restimulated CD4+ T cells by proteasome inhibition. (a) Cells were activated for 4 days with allogeneic dendritic cells (DCs) at a DC : T-cell ratio of 1 : 10. Cells were subsequently exposed for 24 hr to dimethyl sulphoxide (DMSO), bortezomib (Bor) or lactacystin (LC), and incorporation of [3H]thymidine was determined as described in the Materials and methods. Data are given as mean values ± SE of the means of six independent experiments carried out in triplicate. (b) Cells were activated for 4 days with allogeneic DCs at a DC : T-cell ratio of 1 : 10, subsequently exposed for 24 hr to DMSO, Bor or LC and thereafter restimulated for 24 hr with the same allogeneic DCs used for initial activation, third-party allogeneic DCs or lectin. Data are given as mean values ± SE of the mean of six independent experiments carried out in triplicate. Incorporation of [3H]thymidine during restimulation was determined as described in the Materials and methods.
Proteasome inhibition induces G1 cell-cycle arrest, accumulation of p21WAF1/CIP1 and p27KIP1, and downregulation of PCNA, cyclin A and cyclin D2 in activated CD4+ T cells
To determine the mechanisms underlying the impaired proliferation induced by proteasome inhibition, we analysed cell-cycle progression and expression of cell-cycle-related and proliferation-related proteins in activated CD4+ T cells exposed to bortezomib or lactacystin. Flow cytometric analysis of the DNA content of viable cells exposed for 24 hr to the indicated concentrations of bortezomib or lactacystin revealed that proteasome inhibition induced G1 cell-cycle arrest in activated CD4+ T cells (Fig. 4a,b). Whereas induction of G1 cell-cycle arrest by bortezomib was dose-dependent, lactacystin showed an inverse dose dependency regarding the induction of G1 cell-cycle arrest (Fig. 4a,b). As determined by immunoblot analysis, G1 cell-cycle arrest was accompanied by accumulation of the cyclin-dependent kinase inhibitors p21WAF1/CIP1 and p27KIP1 (Fig. 4c), which are known substrates of the proteasome and the accumulation of which in response to proteasome inhibition has been shown to be associated with the induction of G1 cell-cycle arrest in different human cells.3,39–42 In addition, levels of PCNA, cyclin A and cyclin D2, proteins required for proliferation and cell-cycle progression, were found to decrease in activated CD4+ T cells treated with bortezomib (Fig. 4c). This might contribute to the induction of G1 cell-cycle arrest and inhibition of proliferation, because downregulation of PCNA, cyclin A and cyclin D2 induced by different means has previously been shown to induce G1 cell-cycle arrest and inhibition of proliferation.43–46
Figure 4.
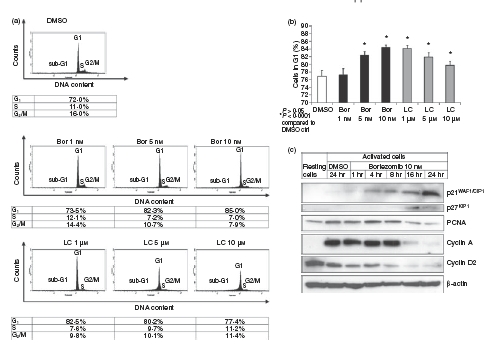
Induction of G1 cell-cycle arrest and regulation of cell cycle- and proliferation-related proteins by proteasome inhibition in activated CD4+ T cells. (a) Flow cytometric analysis of DNA content of CD4+ T cells activated for 4 days with allogeneic dendritic cells (DCs) at a DC : T-cell ratio of 1 : 10 and subsequently exposed for 24 hr to dimethyl sulphoxide (DMSO), bortezomib (Bor) or lactacystin (LC). (b) Percentages of activated CD4+ T cells in G1 cell-cycle phase after exposure of the cells for 24 hr to DMSO, Bor or LC. Data are given as mean values ± SE of the mean of six independent experiments carried out in triplicate. (c) Immunoblot analysis of intracellular amounts of p21WAF1/CIP1, p27KIP1, proliferating cell nuclear antigen (PCNA), cyclin A and cyclin D2 in activated CD4+ T cells exposed for the indicated times to Bor or for 24 hr to DMSO. Amounts of β-actin are demonstrated as a control of equal protein loading. Immunoblots were performed in triplicate with similar results.
Proteasome inhibition suppresses expression of activation-associated cell surface receptors and production of T-cell cytokines in activated and restimulated CD4+ T cells
To further investigate the functional consequences of proteasome inhibition in activated CD4+ T cells, we analysed the cell surface expression of activation-associated and functional receptors and production of T-cell-specific cytokines in viable cells exposed to bortezomib or lactacystin. As shown in Fig. 5(a), expression of CD25 (IL-2Rα-chain), CD28, CD120b (TNF-α receptor p75) and CD134 (OX40) was markedly suppressed in activated CD4+ T cells exposed for 24 hr to bortezomib or lactacystin. Expression of CD27 and CD95 (Fas) was suppressed to a lesser extent (Fig. 5a). Cells activated for 4 days with allogeneic DCs and subsequently exposed for 24 hr to DMSO or bortezomib and restimulated with the same allogeneic DCs used for initial activation, third-party allogeneic DCs or lectin, also exhibited reduced cell surface expression of CD25, CD28 and CD134 (Fig. 5b). Moreover, production of T-cell-specific cytokines, such as IFN-γ, TNF-α, IL-4 and IL-5, was suppressed in activated CD4+ T cells exposed to bortezomib and subsequently restimulated with the same allogeneic DCs used for initial activation, third-party allogeneic DCs or lectin (Fig. 6a–c). These results indicate that proteasome inhibition suppressed the basic immune functions in CD4+ T cells, such as expression of functional cell surface receptors and production of T-cell cytokines, that are required for the regulation of adaptive immune responses.
Figure 5.
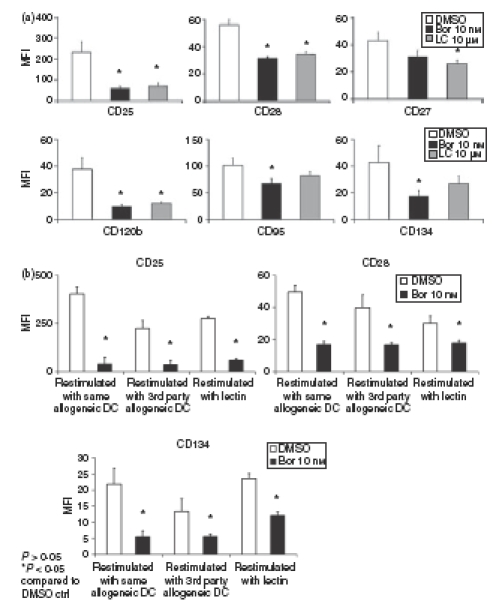
Suppression of cell surface expression of activation-associated receptors by proteasome inhibition in activated and restimulated CD4+ T cells. (a) Cell surface expression of CD25, CD28, CD27, CD120b, CD95 and CD134 in CD4+ T cells activated for 4 days with allogeneic dendritic cells (DCs) at a DC : T-cell ratio of 1 : 10 and subsequently exposed for 24 hr to dimethyl sulphoxide (DMSO), bortezomib (Bor) or lactacystin (LC). Data are given as mean values ± SE of the mean of five independent experiments carried out in triplicate. (b) Cell surface expression of CD25, CD28 and CD134 in restimulated CD4+ T cells. Cells were activated for 4 days with allogeneic DCs at a DC : T-cell ratio of 1 : 10, subsequently exposed for 24 hr to DMSO or Bor and thereafter restimulated for 24 hr with the same allogeneic DCs used for initial activation, third-party allogeneic DCs or lectin. Data are given as mean values ± SE of the mean of four independent experiments carried out in triplicate. Cell surface expression of the activation-associated receptors was determined in viable cells by flow cytometry as described in the Materials and methods.
Figure 6.
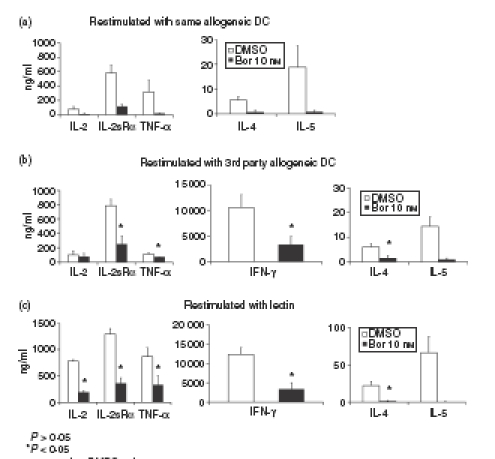
Inhibition of production of T-cell cytokines by proteasome inhibition in activated and restimulated CD4+ T cells. Inhibition of cytokine production in activated CD4+ T cells restimulated with (a) the same allogeneic dendritic cells (DCs) used for initial activation, (b) third-party allogeneic DCs, and (c) lectin. Cells were activated for 4 days with allogeneic DCs at a DC : T-cell ratio of 1 : 10, subsequently exposed for 24 hr to dimethyl sulphoxide (DMSO) or bortezomib (Bor) and thereafter restimulated for 24 hr. After restimulation for 24 hr, amounts of interleukin-2 (IL-2), soluble IL-2 receptor α (IL-2sRα), tumour necrosis factor-α (TNF-α), interferon-γ (IFN-γ), IL-4 and IL-5 were determined in the culture supernatant by enzyme-linked immunosorbent assay as described in the Materials and methods. Data are given as mean values ± SE of the mean of five independent experiments carried out in triplicate.
Proteasome inhibition suppresses nuclear translocation and abundance of NFATc2 in activated CD4+ T cells
Since transcription of genes encoding CD25, IL-2, IFN-γ, TNF-α, IL-4 and IL-5 is directly mediated by transcription factors of the NFAT family,47,48 we tested the hypothesis that activation and nuclear translocation of NFAT proteins in activated CD4+ T cells might be perturbed by proteasome inhibition. Immunoblot analysis of the amount of NFATc2 protein in the nuclear fractions of activated CD4+ T cells revealed that nuclear abundance of NFATc2, the principal NFAT member in mature T cells, was rapidly downregulated in response to proteasome inhibition (Fig. 7). Levels of cytoplasmic NFATc2 were also rapidly downregulated in response to exposure of the cells to bortezomib (Fig. 7). This strongly suggested that proteasomal activity was required for activation and nuclear translocation and abundance of NFATc2 in activated CD4+ T cells, leading to impaired expression of CD25, IL-2, IFN-γ, TNF-α, IL-4 and IL-5.
Figure 7.
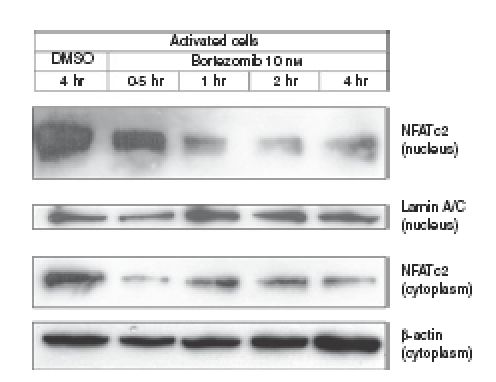
Suppression of nuclear translocation and abundance of nuclear factor of activated T cells (NFAT) c2 by proteasome inhibition in activated CD4+ T cells. Cells were activated for 4 days with allogeneic dendritic cells (DCs) at a DC : T-cell ratio of 1 : 10 and subsequently exposed for the indicated times to dimethyl sulphoxide (DMSO) or bortezomib (Bor). Amounts of NFATc2 were determined in the nuclear and cytoplasmic fractions of the cells by immunoblotting as described in the Materials and methods. Amounts of lamin A/C and β-actin are demonstrated as controls of equal protein loading of the nuclear and cytoplasmic fractions, respectively. Immunoblots were performed in triplicate with similar results.
Discussion
Previous studies in rodents revealed that proteasome inhibitors can attenuate deregulated and unwanted T-cell-mediated immune responses, which contribute to the pathogenesis of polyarthritis, psoriasis, allograft rejection and graft-versus-host disease.22–26 To provide a mechanistic and molecular explanation for these phenomena, we have investigated the effects and mechanisms induced by proteasome inhibition in human CD4+ T cells, which constitute the key regulators of adaptive immune responses.49–51
We demonstrate that human CD4+ T cells activated in a physiological manner by allogeneic DCs undergo apoptosis in response to proteasome inhibition, whereas resting CD4+ T cells display reduced but detectable susceptibility to apoptosis induced by proteasome inhibition. This might be the result of the greater propensity of activated CD4+ T cells already for spontaneous apoptosis. These results are in accordance with our previous study showing that CD4+ T cells activated and induced to proliferate by phytohaemagglutinin or anti-CD3ε IgG undergo apoptosis in response to exposure to the proteasome inhibitor lactacystin.28 Similar results have recently been shown by Blanco et al.,29 who used human CD3+ T cells activated either in the mixed lymphocyte reaction or by phytohaemagglutinin, and exposed to the proteasome inhibitor bortezomib. However, Blanco et al.29 demonstrated apoptosis induced by bortezomib selectively in activated, but not in resting, CD3+ T cells. This discrepancy to our results might be because we used pure populations of CD4+ T cells, which might be, particularly in the resting state, more susceptible to proteasome inhibitor-induced apoptosis than heterogeneous populations of CD3+ T cells. These results demonstrate that preferentially activated and proliferating T cells undergo apoptosis in response to proteasome inhibition. Interestingly, analogous results have been obtained in proliferating human myelogenic leukaemia cells. Such cells have been shown to undergo apoptosis in response to exposure to proteasome inhibitors, but become refractory to proteasome inhibitor-induced apoptosis after the induction of terminal differentiation leading to non-proliferating and mature myelogenous cells.14,15
Because tightly regulated and temporally controlled proteasomal degradation of cell-cycle regulatory proteins is essential for proliferation and cell-cycle progression,3,39 it is apparent that proteasome inhibition has deleterious effects preferentially in proliferating and cycling cells, as shown herein for activated human CD4+ T cells. Induction of apoptosis by proteasome inhibition in activated CD4+ T cells is probably mediated by the accumulation and stabilization of the pro-apoptotic protein p53, which is known to be continuously degraded by the proteasome under steady-state conditions38 and whose accumulation and stabilization in response to proteasome inhibition has been shown to induce apoptosis or G1 cell-cycle arrest in various mammalian cells other than T cells.33–37,52
We demonstrate that proteasome inhibition strongly inhibits the proliferation of CD4+ T cells activated or restimulated with allogeneic DCs or lectin. This cannot be attributed to induction of apoptosis alone because not all activated CD4+ T cells undergo apoptosis in response to proteasome inhibition. In particular, CD4+ T cells surviving exposure to the proteasome inhibitors undergo G1 cell-cycle arrest. While induction of G1 cell-cycle arrest by bortezomib is dose-dependent, lactacystin shows an inverse dose-dependency regarding the induction of G1 cell-cycle arrest. The latter unexpected results might be explained by the ability of lactacystin to induce apoptosis in a dose-dependent manner preferentially in G1 phase cells, leading to a relative decrease of viable G1 phase cells accompanied by a relative increase of S phase and G2/M phase cells. Such a phenomenon has been previously observed in HL60 leukemic cells.14
Induction of G1 cell-cycle arrest by proteasome inhibition in activated CD4+ T cells is associated with accumulation of the cyclin-dependent kinase inhibitors p21WAF1/CIP1 and p27KIP1. Both p21WAF1/CIP1 and p27KIP1 are degraded by the proteasome at G1 and early S phase of the cell cycle to allow progression of the cell cycle from G1 to S phase,3,39,53–58 and the accumulation of p21WAF1/CIP1 and p27KIP1 as a result of proteasome inhibition has been shown to be linked to the induction of G1 cell-cycle arrest and the inhibition of proliferation in different human cells.40–42 In addition, p21WAF1/CIP1 has been shown to be transcriptionally induced by p53, and to mediate p53-dependent G1 cell-cycle arrest and apoptosis.59,60
We also show that the expression of cyclin A and cyclin D2 in activated CD4+ T cells is completely abrogated after treatment of the cells for 16 hr with bortezomib. Cyclin A and cyclin D2 are cell-cycle regulators required for the entry into S phase and for S phase progression, and their abundance is regulated by proteasomal degradation.3,39 Down-regulation of cyclin A and cyclin D2 expression has previously been shown to be associated with G1 cell-cycle arrest in activated T cells.44–46 Therefore, it is likely that downregulation of cyclin A and cyclin D2 and accumulation of p21WAF1/CIP1 and p27KIP1 in response to proteasome inhibition contribute to the induction of G1 cell-cycle arrest and inhibition of proliferation in activated CD4+ T cells. In addition, the nuclear abundance of PCNA, an auxilliary protein for DNA polymerase δ required for DNA replication in proliferating cells,61,62 is shown here to decrease in response to proteasome inhibition in activated CD4+ T cells. Since induction and stabilization of p53 results in downregulation of PCNA messenger RNA and protein expression, leading to the inhibition of cell-cycle progression into S phase and the suppression of proliferation,63 the decrease of PCNA observed in activated CD4+ T cells might be a direct consequence of accumulation and stabilization of p53 induced by proteasome inhibition. Moreover, induction and stabilization of p21WAF1/CIP1 has been shown to downregulate PCNA protein expression, leading to G1 cell-cycle arrest and the suppression of DNA replication and proliferation.43,64 Inhibition of proliferation and induction of G1 cell-cycle arrest in response to proteasome inhibition in activated CD4+ T cells is probably mediated by a complex and highly-regulated interplay of p53, p27KIP1, p21WAF1/CIP1, cyclin A, cyclin D2 and PCNA.
We show that activation and restimulation of CD4+ T cells are suppressed by proteasome inhibition. In addition, we demonstrate the inhibition of cell surface expression of activation-associated and functional receptors, such as CD25 (IL-2Rα chain), CD28, CD120b (TNF-α receptor p75), and CD134 (OX40), as well as impaired production of the T-cell-specific cytokines IFN-γ, TNF-α, IL-4 and IL-5 in response to proteasome inhibition in CD4+ T cells activated or restimulated with allogeneic DCs or lectin. Interestingly, we observed that expression of activation-associated cell surface receptors and production of T-cell-specific cytokines is suppressed in response to proteasome inhibition in CD4+ T cells restimulated by both third-party allogeneic DCs and the same allogeneic DCs used for initial activation. These results are in contrast to those provided by Blanco et al.29, which demonstrated only marginal suppression of CD3+ T-cell activation after stimulation with third-party lymphocytes. This might be explained by the use of different cells (allogeneic lymphocytes) for restimulation. However, we demonstrate here that restimulation of CD4+ T cells by allogeneic DCs, which are known as professional and potent stimulators of T cells, is markedly suppressed in response to proteasome inhibition.
Transcription of genes encoding cell surface receptors and cytokines essential for T-cell activation and T-cell-mediated immune responses, including CD25, CD40L, CD95L, CTLA4, IL-2, IFN-γ, IL-4, IL-5 and TNF-α, is directly mediated by transcription factors of the NFAT family.47,48 We demonstrate that nuclear translocation and abundance of NFATc2, the principal NFAT member in mature T cells,65 is suppressed by proteasome inhibition in CD4+ T cells activated by allogeneic DCs. This apparently results in impaired gene transcription and expression of the NFAT-dependent cell surface receptors and cytokines investigated. Since nuclear translocation of NFAT transcription factors is induced by their dephosphorylation by the calcium- and calmodulin-dependent serine/threonine phosphatase calcineurin47,66,67 and proteasome inhibition is shown herein to suppress NFATc2 nuclear translocation in activated CD4+ T cells, it can be speculated that proteasome activity is required for the initiation of signalling events leading to calcium-/calmodulin-dependent calcineurin activation and/or activation and subcellular trafficking of NFAT from the cytoplasm into the nucleus. Such a proteasome-dependent mechanism is well defined for the activation and nuclear translocation of the transcription factor nuclear factor-κB,68,69 but awaits further characterization in the case of NFAT activation and nuclear translocation.
In conclusion, it is demonstrated that proteasomal proteolytic activity is required for survival, proliferation, cell-cycle progression and expression of important cell surface receptors and cytokines of activated human CD4+ T cells. The inhibitory effect of proteasome inhibition on the immune function of activated CD4+ T cells might be a summation of the effects of proteasome inhibition on p53, cell-cycle regulators, cell surface proteins, cytokines and NFAT that still awaits direct experimental evidence.
The proteasome clearly plays a critical role in the regulation of CD4+ T-cell activation and may represent a novel molecular target for the treatment of deregulated and unwanted T-cell-mediated immune responses.
Abbreviations
- BioALVS
AdaK(Bio)AhX3L3VS
- Bor
bortezomib
- CM
culture medium
- DCs
dendritic cells
- DMSO
dimethylsulphoxide
- ECL
enhanced chemiluminescence
- FCS
fetal calf serum
- FITC
fluorescein isothiocyanate
- GM-CSF
granulocyte–macrophage colony-stimulating factor
- HRP
horseradish peroxidase
- iDCs
immature DCs
- IFN-γ
interferon-γ
- IgG
immunoglobulin G
- IL-4
interleukin-4
- LC
lactacystin
- LPS
lipopolysaccharide
- mAb
monoclonal antibody
- mDCs
mature DCs
- MFI
mean fluorescence intensities
- NFAT
nuclear factor of activated T cells
- pAb
polyclonal antibody
- PBS
phosphate-buffered saline
- PVDF
polyvinylidene difluoride
- rh
recombinant human
- PCNA
proliferating cell nuclear antigen
- PE
phycoerythrin
- PI
propidium iodide
- RIPA
radioimmunoprecipitation assay
- SDS
sodium dodecyl sulphate
- TNF-α
tumour necrosis factor-α
- UPS
ubiquitin–proteasome system
References
- 1.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 2.Glickman MH, Ciechanover A. The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 3.Naujokat C, Hoffmann S. Role and function of the 26S proteasome in proliferation and apoptosis. Lab Invest. 2002;82:965–80. doi: 10.1097/01.lab.0000022226.23741.37. [DOI] [PubMed] [Google Scholar]
- 4.Ciechanover A. The ubiquitin proteolytic system. Neurology. 2006;66(Suppl. 1):S7–19. doi: 10.1212/01.wnl.0000192261.02023.b8. [DOI] [PubMed] [Google Scholar]
- 5.Baumeister W, Walz J, Zühl F, Seemüller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–80. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 6.Voges D, Zwickl P, Baumeister W. The proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–68. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 7.Kloetzel PM. Antigen processing by the proteasome. Nat Rev Mol Cell Biol. 2001;2:179–87. doi: 10.1038/35056572. [DOI] [PubMed] [Google Scholar]
- 8.Dick TP, Nussbaum AK, Deeg M, et al. Contribution of proteasomal beta-subunits to the cleavage of peptide substrates analyzed with yeast mutants. J Biol Chem. 1998;273:25637–46. doi: 10.1074/jbc.273.40.25637. [DOI] [PubMed] [Google Scholar]
- 9.Kisselev AF, Akopian TN, Castillo V, Goldberg AL. Proteasome active sites allosterically regulate each other, suggesting a cyclical bite–chew mechanism for protein breakdown. Mol Cell. 1999;4:395–402. doi: 10.1016/s1097-2765(00)80341-x. [DOI] [PubMed] [Google Scholar]
- 10.Groll M, Huber R. Inhibitors of the eukaryotic 20S proteasome core particle: a structural approach. Biochim Biophys Acta. 2004;1695:33–44. doi: 10.1016/j.bbamcr.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 11.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (bortezomib) Cancer Invest. 2004;22:304–11. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 12.Gaczynska M, Osmulski PA. Small-molecule inhibitors of proteasome activity. Methods Mol Biol. 2005;301:3–22. doi: 10.1385/1-59259-895-1:003. [DOI] [PubMed] [Google Scholar]
- 13.Naujokat C, Fuchs D, Berges C. Adaptive modification and flexibility of the proteasome system in response to proteasome inhibition. Biochim Biophys Acta. 2007;1773:1389–97. doi: 10.1016/j.bbamcr.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 14.Drexler HCA. Activation of the cell death program by inhibition of proteasome function. Proc Natl Acad Sci USA. 1997;94:855–60. doi: 10.1073/pnas.94.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C, Lin H, Karanes C, Pettit GR, Chen BD. Human THP-1 monocytic leukemic cells induced to undergo monocytic differentiation by bryostatin 1 are refractory to proteasome inhibitor-induced apoptosis. Cancer Res. 2000;60:4377–85. [PubMed] [Google Scholar]
- 16.Naujokat C, Sezer O, Zinke H, Leclere A, Hauptmann S, Possinger K. Proteasome inhibitors induce caspase-dependent apoptosis and accumulation of p21 in human immature leukemic cells. Eur J Haematol. 2000;65:221–36. doi: 10.1034/j.1600-0609.2000.065004221.x. [DOI] [PubMed] [Google Scholar]
- 17.Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol. 2005;23:630–9. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 18.Mitsiades CS, Mitsiades N, Hideshima T, Richardson PG, Anderson KC. Proteasome inhibitors as therapeutics. Essays Biochem. 2005;41:205–18. doi: 10.1042/EB0410205. [DOI] [PubMed] [Google Scholar]
- 19.Voorhees PM, Orlowski RZ. The proteasome and proteasome inhibitors in cancer therapy. Annu Rev Pharmacol Toxicol. 2006;46:189–213. doi: 10.1146/annurev.pharmtox.46.120604.141300. [DOI] [PubMed] [Google Scholar]
- 20.Nencioni A, Schwarzenberg K, Brauer KM, et al. Proteasome inhibitor bortezomib modulates TLR4-induced dendritic cell activation. Blood. 2006;108:551–8. doi: 10.1182/blood-2005-08-3494. [DOI] [PubMed] [Google Scholar]
- 21.Naujokat C, Berges C, Höh A, et al. Proteasomal chymotrypsin-like peptidase activity is required for essential functions of human monocyte-derived dendritic cells. Immunology. 2007;120:120–32. doi: 10.1111/j.1365-2567.2006.02487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palombella VJ, Conner EM, Fuseler J W, et al. Role of the proteasome and NF-κB in streptococcal cell wall-induced polyarthritis. Proc Natl Acad Sci USA. 1998;95:15671–6. doi: 10.1073/pnas.95.26.15671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zollner TM, Podda M, Pien C, Elliot PJ, Kaufmann R, Bohncke WH. Proteasome inhibition reduces superantigen-mediated T cell activation and the severity of psoriasis in a SCID-hu model. J Clin Invest. 2002;109:671–9. doi: 10.1172/JCI12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo H, Wu Y, Qi S, Wan X, Chen H, Wu J. A proteasome inhibitor prevents mouse heart allograft rejection. Transplantation. 2001;72:196–202. doi: 10.1097/00007890-200107270-00005. [DOI] [PubMed] [Google Scholar]
- 25.Sun K, Welniak LA, Panoskaltsis-Mortari A, et al. Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. Proc Natl Acad Sci USA. 2004;101:8120–5. doi: 10.1073/pnas.0401563101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Y, Han B, Luo H, Shi G, Wu J. Dipeptide boronic acid, a novel proteasome inhibitor, prevents islet-allograft rejection. Transplantation. 2004;78:360–6. doi: 10.1097/01.tp.0000128855.10397.db. [DOI] [PubMed] [Google Scholar]
- 27.Mattingly LH, Gault RA, Murphy WJ. Use of systemic proteasome inhibition as an immune-modulating agent in disease. Endocr Metab Immune Disord Drug Targets. 2007;7:29–34. doi: 10.2174/187153007780059397. [DOI] [PubMed] [Google Scholar]
- 28.Naujokat C, Daniel V, Bauer TM, Sadeghi M, Opelz G. Cell cycle- and activation-dependent regulation of cyclosporin A-induced T cell apoptosis. Biochem Biophys Res Commun. 2003;310:347–54. doi: 10.1016/j.bbrc.2003.08.141. [DOI] [PubMed] [Google Scholar]
- 29.Blanco B, Perez-Simon JA, Sanchez-Abarca LI, et al. Bortezomib induces depletion of alloreactive T lymphocytes and decreases the production of Th1 cytokines. Blood. 2006;107:3575–83. doi: 10.1182/blood-2005-05-2118. [DOI] [PubMed] [Google Scholar]
- 30.Kessler BM, Tortorella D, Altun M, et al. Extended peptide-based inhibitors efficiently target the proteasome and reveal overlapping specificities of the catalytic beta-subunits. Chem Biol. 2001;8:913–29. doi: 10.1016/s1074-5521(01)00069-2. [DOI] [PubMed] [Google Scholar]
- 31.Berkers CR, Verdoes M, Lichtman E, et al. Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nature Methods. 2005;2:357–62. doi: 10.1038/nmeth759. [DOI] [PubMed] [Google Scholar]
- 32.Princiotta MF, Schubert U, Chen W, et al. Cell adapted to the proteasome inhibitor 4-hydroxy-5-iodo-3-nitrophenylacetyl-Leu-Leu-leucinal-vinyl sulfone require enzymatically active proteasomes for continued survival. Proc Natl Acad Sci USA. 2001;98:513–8. doi: 10.1073/pnas.021132398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopes UG, Erhardt P, Yao R, Cooper GM. P53-dependent induction of apoptosis by proteasome inhibitors. J Biol Chem. 1997;272:12893–6. doi: 10.1074/jbc.272.20.12893. [DOI] [PubMed] [Google Scholar]
- 34.Chen F, Chang D, Goh M, Klibanov SA, Ljungman M. Role of p53 in cell cycle regulation and apoptosis following exposure to proteasome inhibitors. Cell Growth Differ. 2000;11:239–46. [PubMed] [Google Scholar]
- 35.Kim SC, Rho MC, Lee HS, Kim YK, Kim K. Caspase-3-dependent apoptosis in vascular smooth muscle cell by proteasome inhibition. J Cardiovasc Pharmacol. 2003;42:554–60. doi: 10.1097/00005344-200310000-00014. [DOI] [PubMed] [Google Scholar]
- 36.Nair VD, McNaught KS, Gonzales-Maeso J, Sealfon SC, Olanow CW. p53 mediates nontranscriptional cell death in dopaminergic cells in response to proteasome inhibition. J Biol Chem. 2006;281:39550–60. doi: 10.1074/jbc.M603950200. [DOI] [PubMed] [Google Scholar]
- 37.Concannon CG, Koehler BF, Reimertz C, et al. Apoptosis induced by proteasome inhibition in cancer cells: predominant role of the p53/PUMA pathway. Oncogene. 2007;26:1681–92. doi: 10.1038/sj.onc.1209974. [DOI] [PubMed] [Google Scholar]
- 38.Maki CG, Huibregtse M, Howley PM. In vivo ubiquitination and proteasome-mediated degradation of p53. Cancer Res. 1996;56:2649–54. [PubMed] [Google Scholar]
- 39.Yew PR. Ubiquitin-mediated proteolysis of vertebrate G1- and S-phase regulators. J Cell Physiol. 2001;187:1–10. doi: 10.1002/1097-4652(2001)9999:9999<1::AID-JCP1049>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 40.Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc Natl Acad Sci USA. 1999;96:7797–802. doi: 10.1073/pnas.96.14.7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumeda SI, Deguchi A, Toi M, Omura S, Umezawa K. Induction of G1 arrest and selective growth inhibition by lactacystin in human umbilical vein endothelial cells. Anticancer Res. 1999;19:3961–8. [PubMed] [Google Scholar]
- 42.Chen WJ, Lin JK. Induction of G1 arrest and apoptosis in human Jurkat T cells by pentagalloylglucose through inhibiting proteasome activity and elevating p27Kip1, p21WAF1/Cip1, and Bax proteins. J Biol Chem. 2004;279:13496–505. doi: 10.1074/jbc.M212390200. [DOI] [PubMed] [Google Scholar]
- 43.Gehen SC, Vitiello PF, Bambara RA, Kang PC, O'Reilly MA. Downregulation of PCNA potentiates p21-mediated growth inhibition in response to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L716–24. doi: 10.1152/ajplung.00135.2006. [DOI] [PubMed] [Google Scholar]
- 44.Kirkham PA, Lam EWF, Takamatsu HH, Parkhouse RME. Transcription factor E2F controls the reversible γδ T cell growth arrest mediated through WC1. J Immunol. 1998;161:1630–6. [PubMed] [Google Scholar]
- 45.Laliberté J, Yee A, Xiong Y, Mitchell BS. Effects of guanine nucleotide depletion on cell cycle progression in human T lymphocytes. Blood. 1998;91:2896–904. [PubMed] [Google Scholar]
- 46.Ma A, Qi S, Xu D, Daloze P, Chen H. Baohuside-1 inhibits activated T cell proliferation at G1–S phase transition. Transpl Immunol. 2005;15:55–62. doi: 10.1016/j.trim.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 47.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–47. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 48.Schuh K, Twardzik T, Kneitz B, Heyer J, Schimpl A, Serfling E. The interleukin 2 receptor α chain/CD25 promoter is a target for nuclear factor of activated T cells. J Exp Med. 1998;188:1369–73. doi: 10.1084/jem.188.7.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frasca L, Piazza C, Piccolella E. CD4+ T cells orchestrate amplification and deletion of CD8+ T cells. Crit Rev Immunol. 1998;18:569–94. doi: 10.1615/critrevimmunol.v18.i6.50. [DOI] [PubMed] [Google Scholar]
- 50.Mills DM, Cambier JC. B lymphocyte activation during cognate interactions with CD4+ T lymphocytes: molecular dynamics and immunologic consequences. Semin Immunol. 2003;15:325–9. doi: 10.1016/j.smim.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 51.Castellino F, Germain RN. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu Rev Immunol. 2006;24:519–40. doi: 10.1146/annurev.immunol.23.021704.115825. [DOI] [PubMed] [Google Scholar]
- 52.Dietrich C, Bartsch T, Schanz F, Oesch F, Wieser RJ. p53-dependent cell cycle arrest induced by N-acetyl-l-leucinyl-l-leucinyl-l-norleucinal in platelet-derived growth factor-stimulated human fibroblasts. Proc Natl Acad Sci USA. 1996;93:10815–9. doi: 10.1073/pnas.93.20.10815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo Y, Hurwitz J, Massague J. Cell cycle inhibition by independent CDK and PCNA domains in p21Cip1. Nature. 1995;375:159–61. doi: 10.1038/375159a0. [DOI] [PubMed] [Google Scholar]
- 54.Blagosklonny MV, Wu GS, Omura S, el-Deiry WS. Proteasome-dependent regulation of p21WAF1/Cip1 expression. Biochem Biophys Res Commun. 1996;227:564–9. doi: 10.1006/bbrc.1996.1546. [DOI] [PubMed] [Google Scholar]
- 55.Cayrol C, Knibiehler M, Ducommun B. p21 binding to PCNA causes G1 and G2 cell cycle arrest in p53-deficient cells. Oncogene. 1998;16:311–20. doi: 10.1038/sj.onc.1201543. [DOI] [PubMed] [Google Scholar]
- 56.Cayrol C, Ducommun B. Interaction with cyclin-dependent kinases and PCNA modulates proteasome-dependent degradation of p21. Oncogene. 1998;17:2437–44. doi: 10.1038/sj.onc.1202189. [DOI] [PubMed] [Google Scholar]
- 57.Pagano M, Tam SW, Theodoras AM, et al. Role of the ubiquitin–proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–5. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 58.Sutterluty H, Chatelain E, Marti A, et al. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999;1:207–14. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- 59.El-Deiry WS, Harper JW, O′Connor PM, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–74. [PubMed] [Google Scholar]
- 60.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–90. [PubMed] [Google Scholar]
- 61.Kurki P, Lotz M, Ogata K, Tan EM. Proliferating cell nuclear antigen (PCNA)/cyclin in activated human T lymphocytes. J Immunol. 1987;138:4114–20. [PubMed] [Google Scholar]
- 62.Jaskulski D, deRiel JK, Mercer WE, Calabretta B, Baserga R. Inhibition of cellular proliferation by antisense oligodeoxynucleotides to PCNA cyclin. Science. 1988;240:1544–6. doi: 10.1126/science.2897717. [DOI] [PubMed] [Google Scholar]
- 63.Mercer WE, Shields MT, Lin D, Appella E, Ullrich SJ. Growth suppression induced by wild-type p53 is accompanied by selective down-regulation of proliferating-cell nuclear antigen expression. Proc Natl Acad Sci USA. 1991;88:1958–62. doi: 10.1073/pnas.88.5.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994;369:574–8. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 65.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–84. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 66.Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483–521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 67.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–32. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 68.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin–proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell. 1994;78:773–85. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 69.Chen ZJ, Hahler J, Palombella V, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin–proteasome pathway. Genes Dev. 1995;9:1586–97. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]