

Abstract
The total synthesis of cytostatin, an antitumor agent belonging to the fostriecin family of natural products is described in full detail. The convergent approach relied on a key epoxide opening reaction to join the two stereotriad units and a single-step late stage, stereoselective installation of the sensitive (Z,Z,E)-triene through a β-chelation controlled nucleophilic addition. The synthetic route provided rapid access to the C4–C6 stereoisomers of the cytostatin lactone, which were prepared and used to define the C4–C6 relative stereochemistry of the natural product. In addition to the natural product, each of the C10–C11 diastereomers of cytostatin was divergently prepared (11 steps from key convergence step) by this route and used to unequivocally confirm the relative and absolute stereochemistry of cytostatin. Each of the cytostatin diastereomers exhibited a reduced activity towards inhibition of PP2A (>100-fold), demonstrating the importance of the presence and stereochemistry of the C10-methyl and C11-hydroxy groups for potent PP2A inhibition. Extensions of the studies provided dephosphocytostatin (40), sulfocytostatin (67, a key analogue related to the natural product sultriecin), 11-deshydroxycytostatin (78), and 72 lacking the entire C12–C18 (Z,Z,E)-triene segment and were used to define the magnitude of the C9-phosphate (>4000-fold), C11-alcohol (250-fold), and triene (220-fold) contribution to PP2A inhibition. A model of cytostatin bound to the active site of PP2A is presented, compared to that of fostriecin which is also presented in detail for the first time, and used to provide insights into the role of the key substituents. Notably, the α,β-unsaturated lactone of cytostatin, like that of fostriecin, is projected to serve as a key electrophile providing a covalent adduct with Cys269 unique to PP2A contributing to its potency (≥200-fold for fostriecin) and accounting for its selectivity.
Introduction
Cytostatin (1),1 an antitumor agent isolated from Streptomyces sp. MJ654-Nf4 belonging to the fostriecin family of natural products,2 displays potent cytotoxic activity towards leukemia and melanoma cancer cell lines (IC50 100 nM), induces apoptosis,3 and inhibits lung tumor metastasis.4 Like the parent of its class, fostriecin (2), cytostatin is a potent and selective inhibitor of protein phosphatases 2A and 4 (PP2A IC50 = 210 nM, PP2A/PP1 >1000)5 and it shares the distinctive phosphate monoester, Z,Z,E-triene, and α,β-unsaturated δ-lactone structural units. Additional related natural products include sultriecin (3), phospholine (4), the leustroducsins (5), and the phoslactomycins (6) (Figure 1).6
Figure 1.
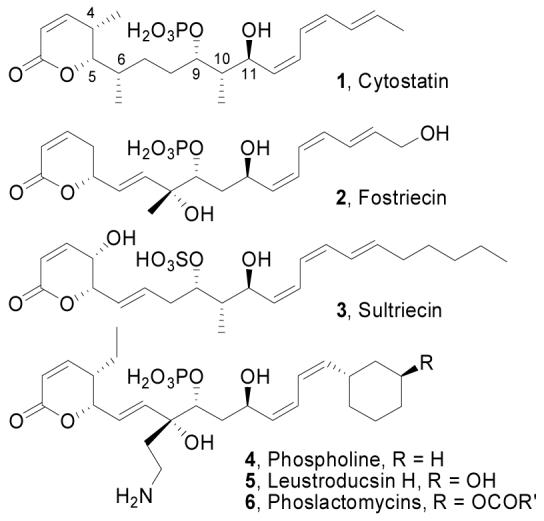
Cytostatin and related natural products.
The natural products 4–6 display different biological profiles than 1 and 2 as they are only weakly cytotoxic, but they exhibit significant broad spectrum antifungal activity. Based on reported PP2A inhibition values, fostriecin (2, IC50 = 3.2 nM)7 is apparently considerably more potent than cytostatin (1, IC50 = 210 nM), phospholine (4, IC50 = 5.8 μM),8 leustroducsin H (5, IC50 = 130 nM),4b and the phoslactomycins (6, IC50 = 3.7–4.9 μM).8 However, no side-by-side comparisons have been conducted between fostriecin and the other members of its class, limiting interpretations of the potentially useful structure–function information embedded in the natural products. Cross-assay comparisons are particularly interesting since IC50 values for fostriecin inhibition of PP2A are not only dependent on the assay enzyme concentration, but also the phosphorylated substrate used in the assay.7,9
In preceding efforts related to the fostriecin family of natural products, we determined the stereochemical configuration of fostriecin (2) and reported its first total synthesis.10 Subsequent to this work, a number of additional synthetic efforts have been described including eight total or formal syntheses of 2, each highlighting the utility of alternative asymmetric synthetic methods for the construction of the 4 chiral centers of 2 in a stereoselective manner.11 A total synthesis of leustroducsin B (phospholine)12 and a recent total synthesis of phoslactomycin B13 have also been reported. In 2002, Waldmann reported the total synthesis of 1 and several analogues,14 and Marshall has since disclosed an alternative route to an advanced intermediate of the Waldmann synthesis.15 The reported syntheses of 1 and 2, including our own initial fostriecin total synthesis, typically have been conducted in a linear fashion and have relied on cross-coupling strategies to install the triene in a multi-step, segmental manner that contrasts the approach to cytostatin described herein.
In efforts aimed at identifying the features of fostriecin (2) required for its potent and selective PP2A inhibition, we prepared analogues of 2 that were used to demonstrate the importance of the phosphate monoester and that defined the role of the unsaturated lactone (Figure 2).16 In these studies, we provided evidence that the unsaturated lactone serves as a critical electrophile that reacts with C269 of PP2A that is not present in PP1, accounting for the PP2A potency and selectivity of 2,16 and this has now been confirmed in studies of PP2A inhibition by phoslactomycin (6).17 Docking fostriecin into a PP2A homology model2,18 revealed additional potential active site interactions including a hydrogen bond between the C11-hydroxy and Arg214 and the penetration of the triene unit into a hydrophobic cleft, both of which are conserved binding features of the non-selective PP1 inhibition pharmacophore.19 The importance of the extended hydrophobic segment for PP2A binding has been confirmed with a cytostatin analogue lacking part of the triene unit that displayed reduced potency.14 In addition, the putative role of the C11-hydroxy is supported by the lack of activity of both a fostriecin analogue and a cytostatin analogue acetylated at C11.14,16 However, both of these acetylated analogues as well as that which probed the triene contribution were otherwise altered in ways that might significantly attenuate their PP2A inhibition, leaving the true magnitude of their contributions in question.
Figure 2.
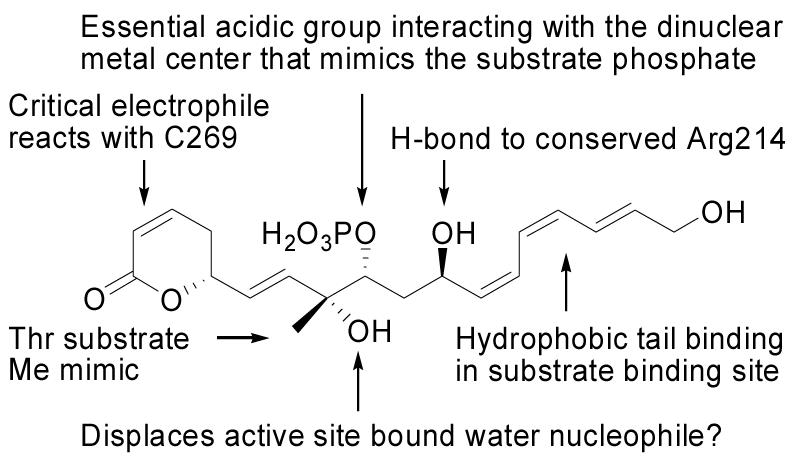
Key elements of fostriecin's PP2A activity.16
An additional conserved feature of fostriecin, cytostatin, and other PP inhibitors is a methyl group proximal to the acidic metal binding moiety, which has been proposed to mimic the methyl group of phosphothreonine.18 This is a particularly compelling hypothesis given that PP2A displays a substrate preference for phosphothreonine peptides over phosphoserine peptides. Although it is unclear whether the conserved methyl group is beneficial for inhibition, it has been proposed that it may contribute to PP binding either through hydrophobic contact with a protein residue or by restricting the conformation of the natural product to one favorable for PP binding.18 The stereochemical integrity of fostriecin's C8 center is crucial for PP2A activity,9 but whether this is due to effects related to the C8-methyl or C8-hydroxy or both has not yet been determined.
Herein, we provide full details of a total synthesis of cytostatin that is complementary to previous syntheses of 1 and 2, and by design provides rapid access to all of the stereoisomers of cytostatin.20 Accordingly, its extension to the preparation of each of the three additional C10–C11 stereoisomers of cytostatin is also described, and these compounds in conjunction with four diastereomeric lactone partial structures were used to unequivocally confirm the relative and absolute stereochemistry of cytostatin. The comparative biological assessment of cytostatin and its diastereomers is reported and demonstrates the importance of the conserved C10-methyl group and the C11-hydroxy for potent PP2A inhibition. Further extensions of the approach provided dephosphocytostatin (40), 11-deshydroxycytostatin (78), and 72 lacking the entire C12–C18 triene subunit and their comparative biological evaluation provided direct assessments of the contribution that the cytostatin C9-phosphate, C11-alcohol, and C12–C18 terminal triene make toward protein phosphatase inhibition. Removal of the C9-phosphate resulted in a complete loss in PP2A inhibition (>104-fold loss in activity) as anticipated, removal of the C11-alcohol resulted in a 250-fold loss in PP2A inhibition, and removal of the terminal C12–C18 triene resulted in a 220-fold loss in PP2A inhibition. Insights into the origin of these contributions as well as the behavior of the cytostatin diastereomers were retrospectively derived from the modeling of its binding to a PP2A homology model18 and comparison to our earlier fostriecin model.2,16 Finally and as a consequence of cytostatin's structural similarity to sultriecin (3), the corresponding cytostatin sulfate was prepared for preliminary comparison.
Chemistry
Stereochemical Assignment
The structure of cytostatin was disclosed without a definition of its relative or absolute stereochemistry, prompting us to secure a stereochemical assignment prior to initiating the synthetic work. In previous efforts, we defined the (5S,9S,11S)-stereochemistry for fostriecin,10a and this assignment was extended to cytostatin based on its structural and functional similarity (Figure 3a). In these studies, we observed an intramolecular hydrogen bond between the C9-phosphate and C11-hydroxy group of fostriecin and demonstrated that the resulting cyclic structure exists in a rigid twist boat conformation (Figure 3b) that gives rise to distinct 1H–1H coupling constants between H11 and H10a (syn) or H10b (anti) (J10a,11 = 3.7 Hz, J10b,11 = 9.6 Hz). Based on a well-founded assumption of a similar cyclic structure for cytostatin, its reported H10–H11 coupling constant (J10,11 = 9.4 Hz)1a indicated a 10,11-anti configuration for the natural product. Alternatively, assumption of the adoption of one of two possible hydrogen bonded chair conformations for the C9–C11 structure of cytostatin gives rise to a similar anti H10–H11 coupling constant and led Waldmann14 to the same (10S)-stereochemical assignment (Figure 3c).
Figure 3.
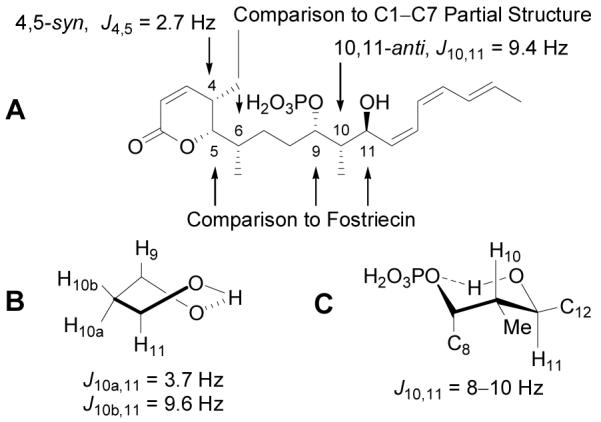
Stereochemical assignment for cytostatin.
To decipher the C4–C6 relative stereochemistry, four diastereomeric lactones (C1–C7 partial structures) were synthesized for spectral comparison (Scheme 1). Construction of lactone 12 began with silylation (TBDPSCl, imidazole, DMF, 25 °C, 2 h, 99%) of methyl (S)-3-hydroxy-2-methylpropionate (7) followed by conversion of the ester to the corresponding aldehyde 9 (DIBAL-H, toluene, −78 °C, 1 h; TPAP, NMO, CH2Cl2, 0 °C, 30 min, 82%, 2 steps).21 Crotylation of 9 (cis-2-butenyldiisopinocampheylborane, THF, ether,−78 °C, 12 h; NaOH, H2O2, 70 °C, 5 h, 63%, 8:1 dr)22 gave alcohol 10 which was converted to the α,β-unsaturated lactone 12 via acylation (acryloyl chloride,i-Pr2NEt, CH2Cl2, 0 °C, 2 h, 91%) and subsequent ring closing metathesis (Grubbs' I catalyst, CH2Cl2, 40 °C, 12 h, 89%).23 This sequence was used to access lactones 13, 14, and 15 in a stereodivergent manner by treating (R)- or (S)-9 with the appropriate crotylboration reagent followed by acylation and ring closing metathesis (Scheme 1).
Scheme 1.
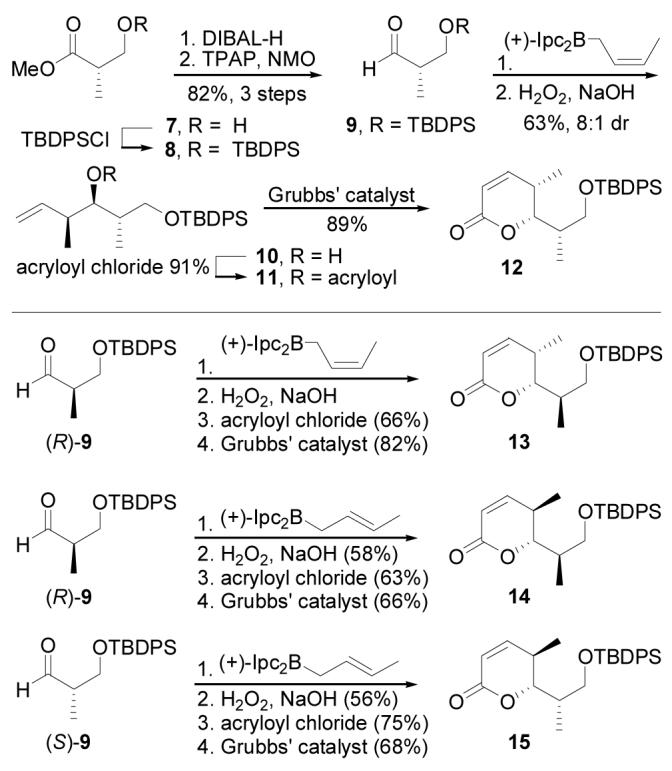
Synthesis of 12–15.
Upon examination of the 1H NMR spectra of 12–15, it became apparent that the 4,5-syn and 4,5-anti configurations exhibit distinct coupling constants (Figure 4).24 Thus, the coupling constants observed for cytostatin were indicative of a 4,5-syn relative configuration.25 Of the two 4,5-syn lactones, the 1H NMR spectrum of 12 was closer to that of cytostatin as exemplified by an average absolute difference in chemical shift values (for H2–H6) of only 0.08 ppm, suggesting a 5,6-anti relative configuration for the natural product. Most notably, the chemical shifts (CD3OD) of 13 for H4, H5, 4-CH3 (δ 1.18) and especially the key 6-CH3 (δ 0.88) were considerably distinct from those of 1 (4-CH3 at δ 1.00, 6-CH3 at δ 0.98) and 12 (4-CH3 at δ 1.08, 6-CH3 at δ 0.99). Based on this analysis, the (4S,5S,6S,9S,10S,11S) configuration was assigned to cytostatin. Independent of our efforts, Waldmann14 conducted and has disclosed an analogous set of studies using a different lactone series and a similar retrospective interpretation of the cytostatin spectroscopic properties to arrive at the same stereochemical assignments for which our relative and absolute stereochemical assignments of the fostriecin distal C4 and C9/C11 centers served as the foundation.10
Figure 4.
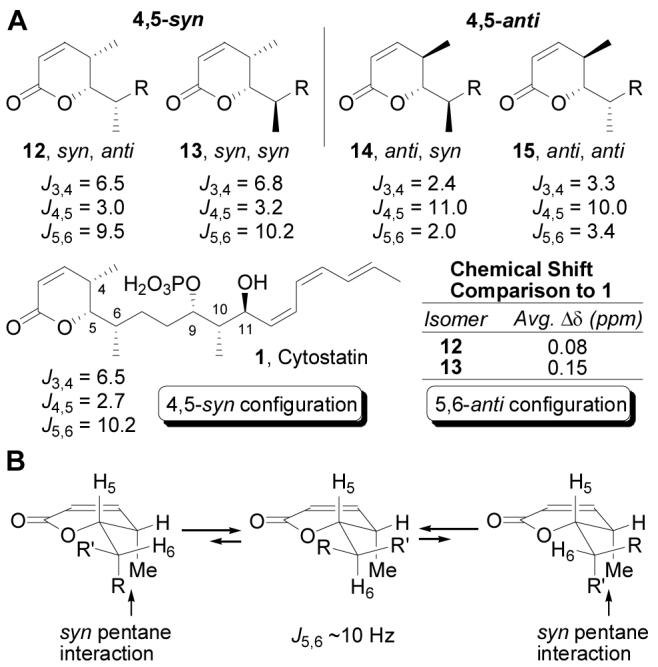
1H NMR comparison for lactones 12–15.
In digesting the conformational features embedded in this segment of cytostatin, we came to recognize that the distinguishing H5–H6 coupling constant was also diagnostic of the side chain adopting a single, preferred conformation (Figure 4b). This single side chain orientation is adopted to avoid the syn pentane interactions of the two alternatives (ΔE ca. 3.7 kcal/mol). In addition to accounting for the large H5–H6 coupling constant (J = 9.5–10.2 Hz) characteristic of their anti relationship in this conformation, this also suggests that the role of the cytostatin C4 and C6 methyl groups is to confine the side chain to a single orientation facilitating binding to PP2A, a potential role that is reinforced in our retrospective modeling of the cytostatin binding to a PP2A homology model.
Synthetic Plan
We designed a synthetic route to cytostatin that would confirm its stereochemical assignment and provide rapid access to the C10–C11 stereoisomers of 1 that could be used to define the role of the C11-hydroxy and C10-methyl group in PP2A binding. The convergent approach relied on installation of the triene at a late stage and in a single-step by addition of 18 to a C11 aldehyde, enlisting substrate control to set the C11 stereochemistry. Assembly of the C7–C8 bond by coupling a cuprate derived from iodide 16 with epoxide 17 isolates the two stereotriads of cytostatin, allowing for independent adjustment of their stereochemistry. In addition, this bond construction provided the opportunity to utilize lactones 12–15 as intermediates in the total synthesis of the natural product and its C4–C6 diastereomers. A Sharpless epoxidation served to set the C9 and C10 stereochemistry, and the triene was synthesized in a short, stereospecific approach relying on an electrocyclic ring opening to set the geometry of the three olefins (Figure 5).
Figure 5.
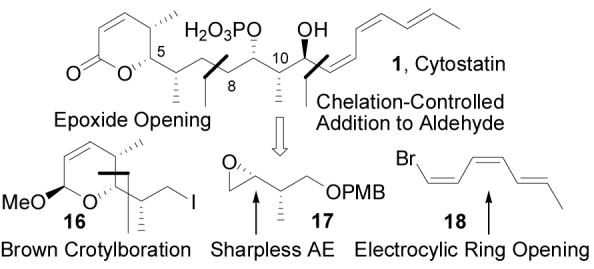
Synthetic plan for cytostatin.
Synthesis of C1–C7
Lactone 12, developed during our work to secure a stereochemical assignment for cytostatin, served as the starting point for synthesis of the C1–C7 fragment of cytostatin (Scheme 2). The electrophilic α,β-unsaturated lactone was masked by carbonyl reduction (DIBAL-H, CH2Cl2, −78 °C, 30 min) and conversion of the intermediate lactol to the methyl acetal 19 (PPTS, MeOH, 25 °C, 10 min, 82%, 2 steps). Acetal 19 formed as a single isomer26 under these conditions, but equilibrated to a 2:1 mixture of anomers when subjected to MeOH/PPTS treatment for longer reaction times. Although the C1 stereocenter would eventually be removed, selective formation of (1R)-19 allowed us to carry forward a single diastereomer, simplifying the isolation and characterization of intermediates in the synthesis. Interestingly, the alternative and more direct generation of 19 employing the corresponding methoxy acetal precursor in the Grubbs' metathesis cyclization proved less satisfactory with the substrate being more challenging to effectively prepare from 10 (30–50%) and the product being a mixture of C1 diastereomers. After desilylation of 19 (Bu4NF, THF, 25 °C, 12 h) to give 20, efforts to directly convert the alcohol to the corresponding halide proved problematic as 20 either remained unchanged (PPh3, NIS; PPh3, CBr4) or decomposed (PPh3, I2) under standard conditions. However, 20 could be converted to iodide 16 through a 2-step process of tosylation (p-TsCl, NaH, benzene, 25 °C, 4 h, 81%, 2 steps) followed by iodide displacement (NaI, acetone, 56 °C, 12 h, 90%). The C1–C7 precursor 16 contains the C4–C6 stereotriad of cytostatin, installed in a stereochemically-tunable sequence through Brown crotylboration of chiral building block 9.
Scheme 2.
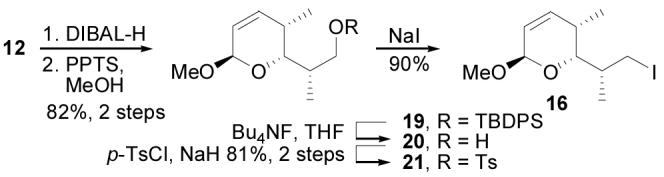
Synthesis of C1–C7.
Synthesis of C8–C11
Assembly of the C8–C11 epoxide unit (Scheme 3) began with 22, which was prepared on a multigram scale by a known procedure27 that relies on a Sharpless asymmetric epoxidation reaction to set the chirality of the epoxy alcohol. Epoxide opening using a procedure developed by Tius (MeMgI, CuI, Et2O, THF, −40 °C, 3 h, 69%)28 provided a 3:1 mixture of 23 and its regioisomer (1,2-diol), which was removed by periodate treatment (NaIO4, H2O, Et2O, 25 °C, 1.5 h, 91%) followed by chromatography. After debenzylation (H2, Pd/C, MeOH, 25 °C, 2h, 99%), triol 2429 was transformed to epoxide 17 in a sequence that mirrors a reported conversion of epi-(3R)-24 into its corresponding epoxide.30 Thus, 1,2-diol protection (3,3-dimethoxypentane, p-TsOH, DMF, 25 °C, 12 h, 68%) followed by PMB ether formation (NaH, PMBCl, Bu4NI, THF, 0 to 20 °C , 20 h, 80%) gave 26. After acetal removal (p-TsOH, MeOH, 25 °C, 7 h, 89%), diol 27 was transformed to epoxide 17 through tosylation of the primary alcohol followed by its intramolecular displacement (NaH, pTs-imidazole, THF, −78 to 25 °C, 4 h, 82%). The C8–C11 unit bears the C9 and C10 stereocenters of cytostatin that contain the subtituents proposed to affect PP2A active site binding. Alternative C9–C10 stereochemical configurations of cytostatin are accessible by transforming the stereoisomers of triol 24 to their corresponding epoxides through this simple and reliable synthesis.
Scheme 3.
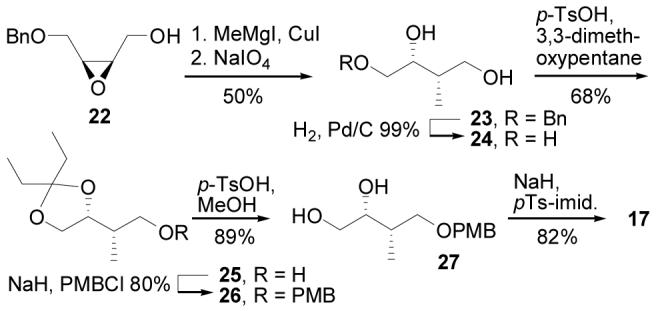
Synthesis of C8–C11.
Synthesis of C12–C18.
Crucial to the implementation of the convergent synthetic plan set forth herein was the ability to supply and successfully manipulate the potentially labile bromotriene 18. In a modification of Taylor's synthesis of Z,E-dienals,31 addition of MeLi (THF, −78 °C, 2 h) to pyrylium tetrafluoroborate followed by electrocyclic ring opening provided 28 in 66% as a single isomer (Scheme 4). Transformation of 28 to dibromoolefin 29 (CBr4, PPh3, Et3N, CH2Cl2, 0 °C, 10 min, 97%)32 and selective (E)-bromide reduction (Bu3SnH, Pd(PPh3)4, ether, 0 °C, 15 min, 95% conversion)33 gave 18 stereoselectively. Isolation of bromide 18 was complicated by its volatility and tendency to polymerize during distillation or concentration. However, chromatographic purification (silica gel, pentane) followed by careful concentration in the presence of Et3N to prevent trace acid-induced polymerization allowed isolation of 18 in superb yields (73%) considering its lability. Storage of bromide 18 proved more problematic, but it was readily generated immediately prior to use from the dibromide 29 which was stable for months when stored as a refrigerated benzene solution. The C12–C18 precursor 18 contains the (Z,Z,E)-conjugated triene, ready for incorporation into cytostatin as a single unit.
Scheme 4.
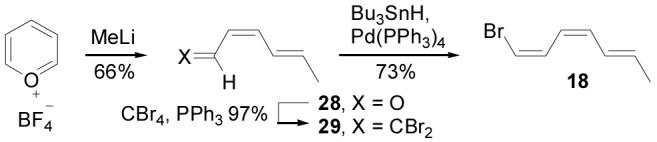
Synthesis of C12–C18.
Synthesis of Cytostatin.
Assembly of 1 (Scheme 5) was initiated by converting iodide 16 into the corresponding cuprate (t-BuLi, ether, −78 °C, 5 min; then (2-Th)CuCNLi, THF, −78 to 0 °C, 5 min)34 followed by addition of 17 at 0 °C to give 30 in 84%. The coupling of 16 and 17 was slow when alternative metalated forms of 16 were used (i.e. R2CuLi, RMgI/cat. CuI, R2CuCNLi2, RLi/BF3), and the success of the transformation was dependent on the use of the higher order cuprate at 0 °C and at concentrations of 50 mM or greater. Acetal formation (ethyl vinyl ether, PPTS, CH2Cl2, 25 °C, 30 min, 90%) gave 31 as a 1.2:1 mixture of diastereomers which, for ease of isolation and characterization, were separated after PMB removal (DDQ, CH2Cl2, H2O, 25 °C, 30 min, 85%) and carried through the subsequent 4 steps of the synthesis separately. Oxidation of 32 (DMP, CH2Cl2, 25 °C, 15 min, 91%)35 provided aldehyde 33 setting the stage for installation of the triene. Bromine/lithium exchange on 18 followed by addition of 33 gave 34 as a 2:1 mixture of diastereomers favoring the undesired 10,11-syn isomer (Felkin product). Following Still's precedent,36 the diastereoselectivity of the addition could be modulated by using a copper-based nucleophile, which promotes addition to a chelated β-alkoxy aldehyde. Thus, conversion of 18 to its Bu3P-stabilized cuprate (t-BuLi, ether, −78 °C, 1.5 h; CuI–PBu3, 10 min) prior to addition of 33 (−78 °C, 30 min, 76%) produced 34 as a pair of chromatographically-separable diastereomers in a ratio of 7:1 favoring the desired 10,11-anti product. As anticipated, the diastereoselectivity of the triene addition was also highly dependent on the C9 protecting group (Figure 6). When the non-chelating TES group was used instead of EE, the Felkin product (syn) predominated when either the organolithium or organocuprate nucleophile was added to the aldehyde. This observation is consistent with the Felkin versus chelation model for the diastereoselective addition and proved useful for the synthesis of the cytostatin stereoisomers. Assignment of the C10–C11 relative stereochemistry of anti-34 was initially based on its H10–H11 coupling constant (J = 9.0 Hz), which was indicative of a 10,11-anti configuration and consistent with a C9-alkoxy/C11-hydroxy hydrogen bonded cyclic structure (Figure 3), and this stereochemical assignment was later confirmed through analysis of the 9,11-acetonide derived from a more advanced synthetic intermediate. The minor isomer (syn-34) from the β-chelation controlled triene addition exhibited an H10–H11 coupling (J = 3.6 Hz) expected to arise from a 10,11-syn configuration.
Scheme 5.
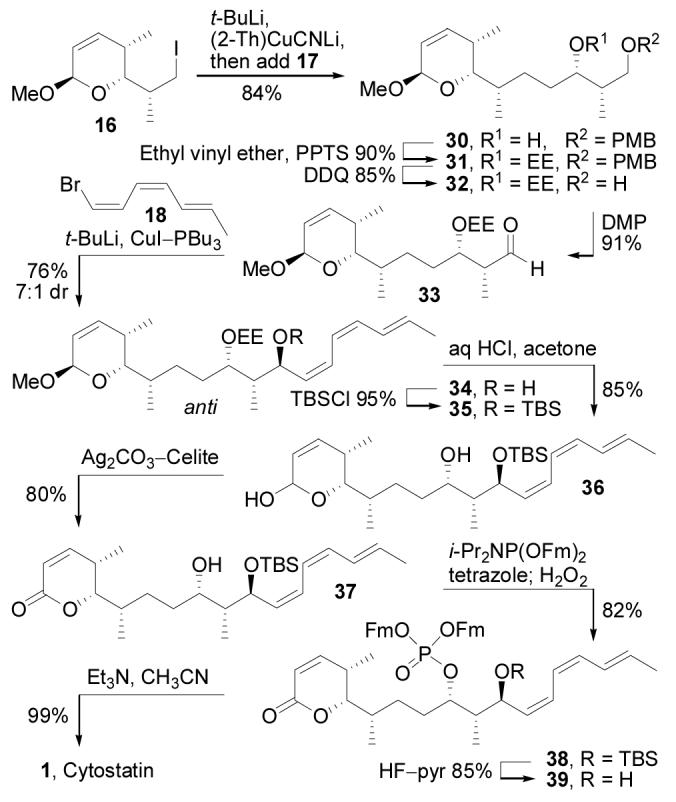
Synthesis of cytostatin.
Figure 6.
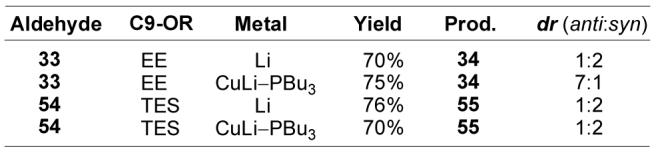
Diastereoselectivity of the triene addition.
Following silylation (TBSCl, imidazole, DMF, 25 °C, 2 h, 95%) of alcohol 34, treatment of 35 with dilute HCl for short reaction times (0.02 N HCl, acetone, water, 25 °C, 10 min, 85%) induced simultaneous C1 and C9 acetal hydrolysis without affecting the silyl ether, whereas more conventional acidic deprotection methods (0.2 N HCl–acetone; 80% aq. HOAc; 90% TFA–H2O) led to competitive global deprotection even when conducted at low temperatures. In this context, the use of the C9 EE protecting group proved valuable as it served not only to direct the formation of the C11 stereocenter (through chelation), but could be effectively removed in the presence of the labile silyl ether and conjugated triene. Selective oxidation (Ag2CO3–Celite, benzene, 80 °C, 2 h, 80%) of lactol 36 produced 37, which was phosphorylated (i-Pr2NP(OFm)2, tetrazole, CH3CN, CH2Cl2, 25 °C, 15 min; H2O2, 10 min, 82%) using the protocol introduced by Waldmann14c to give 38. Desilylation (HF–pyr, THF, pyridine, 25 °C, 4 h, 85%) followed by fluorenylmethyl removal (Et3N, CH3CN, 25 °C, 17 h, 99%) provided 1, identical to a sample of natural cytostatin (1H NMR, TLC, HPLC, HRMS).
At this stage, the C9–C11 relative stereochemistry of synthetic cytostatin was confirmed by examining the spectral properties of acetonide 41, derived from intermediate 37. Cleavage of the silyl ether of37 (HF–pyr, THF, pyridine, 25 °C, 2 h, 99%) provided dephosphocytostatin (40), which was subjected to acetal formation (2,2-dimethoxypropane, p-TsOH, THF, 25 °C, 1 h, 99%) to deliver 41 in excellent yield. The acetonide methyl groups (δ 24.6, 25.1) and ketal carbon (δ 101.2) of 41 exhibited 13C NMR chemical shifts (CD3CN, 150 MHz) characteristic of anti-1,3-diol acetonides (δ 23.6–25.6 and 100.2–101.0) and distinct from those observed for syn-1,3-diol acetonides (δ 18.6–19.9/29.8–30.2, and 98.0–99.3).37 Furthermore, the H10–H11 coupling constant (J = 8.4 Hz) confirmed the expected 10,11-anti stereochemistry. Dephosphocytostatin (40) displayed spectral properties similar to 41, and particularly striking is its H10–H11 coupling constant (J = 8.4 Hz) and C10-methyl group 13C NMR chemical shift (δ 10.8, CD3CN, 150 MHz), both of which are indistinguishable from those of 41. This comparison suggests that the C9–C11 hydrogen-bonded cyclic structure of 40 (and 1) exists in the twist boat conformation observed for 41 and fostriecin (Figure 3b) rather than the alternative chair conformation (Figure 3c) suggested by Waldmann. The latter places the C10-methyl group in an equatorial position that would result in a downfield shifted 13C NMR resonance for the C10-Me instead of the identical signal that is observed.
Synthesis of Cytostatin Diastereomers
The completion of the total synthesis of cytostatin set the stage for preparation of the C10–C11 diastereomers (42–44, Figure 8) that were used to confirm the C9–C11 stereochemical assignments and to further delineate the cytostatin–PP2A interaction. epi-(11R)-Cytostatin (42) was synthesized by advancing the 10,11-syn isomer of 34 through the final 6 steps of the cytostatin synthesis, which occurred without incident and was conducted without significant optimization of the individual steps (Scheme 6). As noted above, syn-34 was the major product of the addition of triene 18 to aldehyde 33 when the lithium deriviative of 18 was used as the nucleophile (Figure 6), and this procedural adjustment allowed accumulation of sufficient material to produce 42.
Figure 8.
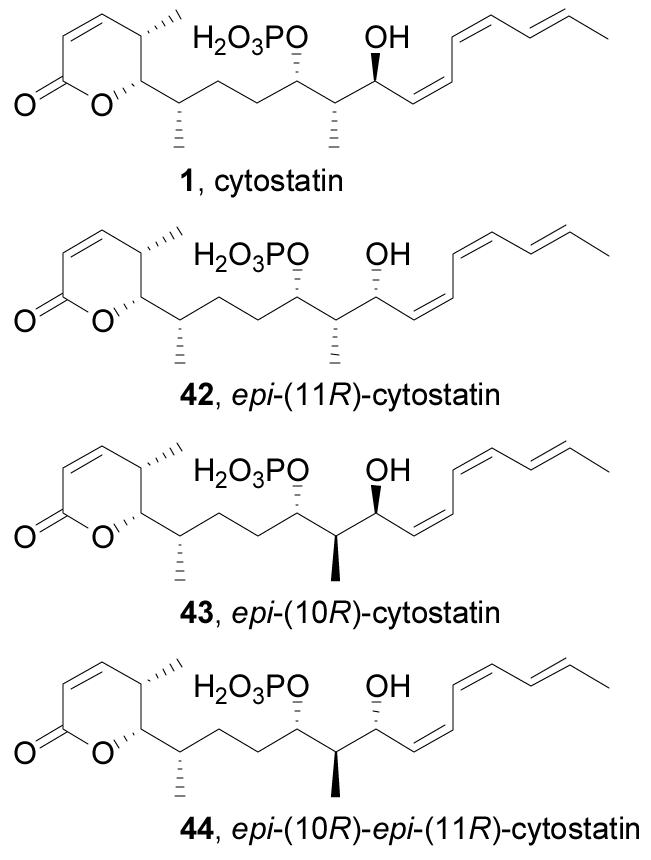
Structures of cytostatin C10–C11 diastereomers.
Scheme 6.
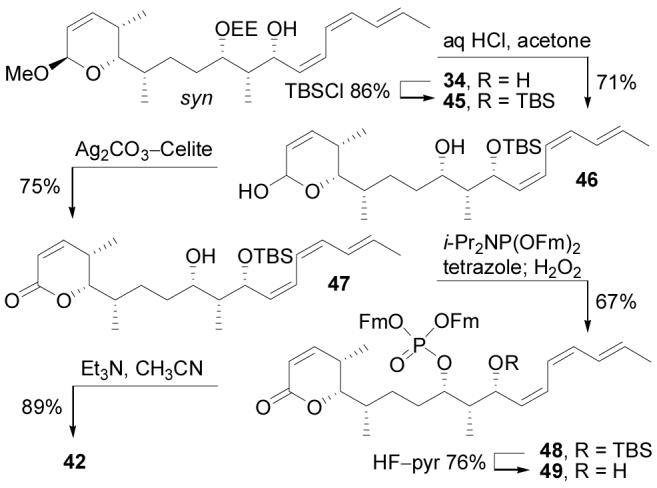
Synthesis of 42.
The synthesis of cytostatin isomers 43 and 44 bearing the (10R) configuration was achieved by utilizing the known epoxide 5030 in place of 17 in the synthesis (Scheme 7). A key element of the derivation of the cytostatin synthesis in which the two stereotriads are independently created and joined in a convergent epoxide opening followed by a single-step, late stage addition of the intact triene with diastereocontrolled introduction of the C11 center is that the approach provides rapid, divergent access (11 steps from convergence point) to each C9–C11 diastereomer. Thus, coupling of 16 and 50 proceeded as expected under the previously optimized conditions (t-BuLi, ether, −78 °C, 5 min; then (2-Th)CuCNLi, THF, −78 to 0 °C, 5 min; 50, ether, 0 °C, 1 h, 72%) to provide 51 in superb yield. A C9 TES group was selected in preference to EE for synthesis of the (10R) series since β-chelation was not required for installation of the natural (11S)-configuration and the silyl ether formation does not produce additional diastereomers. Thus, through a sequence of silylation (TESCl, imidazole, DMF, 25 °C, 1 h, 87%), PMB removal (DDQ, CH2Cl2, H2O, 25 °C, 15 min, 72%), and oxidation (DMP, CH2Cl2, 25 °C, 10 min, 90%),35 51 was converted to aldehyde 54. Addition of 54 to the lithium derivative of 18 (t-BuLi, ether, −78 °C, 1.5 h; 54, −78 °C, 1 h, 70%) produced 55 as a 2:1 mixture of diastereomers favoring the Felkin 10,11-syn product (Figure 6). The expected stereochemical assignments were confirmed based on the H10–H11 coupling for 55 (J10,11 = 9.0 for anti, J10,11 = 2.4 for syn). After chromatographic separation, syn-55 and anti-55 were converted to 43 and 44 by way of the process developed for the synthesis of 1 (Scheme 7). Of note, the TES group was also cleaved at a useful rate under the conditions (0.02 N HCl, acetone, water, 25 °C, 10 min, 61%) developed for removal of the EE group in the presence of the triene and labile allylic TBS ether. Thus, the utility of our convergent total synthesis of cytostatin is highlighted by the fact that only 23 additional steps were required to prepare its entire complement of C10–C11 diastereomers.
Scheme 7.
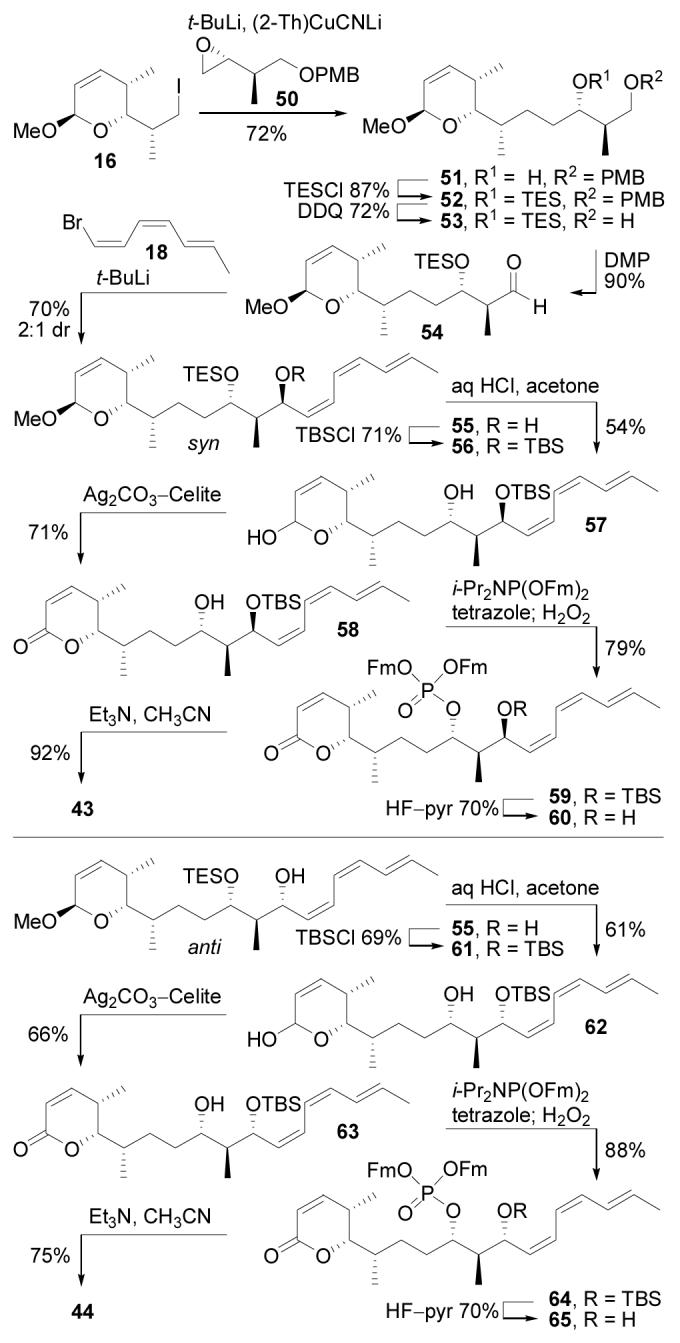
Synthesis of 43 and 44.
Confirmation of Stereochemical Assignment
The stereochemistry of the C9–C11 portion of cytostatin has thus far been assigned based on a key coupling constant (J10,11 = 9.4 Hz) that suggests a 10,11-anti relative stereochemistry and the assumption that cytostatin and fostriecin have identical absolute configurations at shared chiral centers (C9 and C11). While this is sound reasoning and synthetic 1 proved spectroscopically indistinguishable from natural cytostatin,14,20 the magnitude of the optical rotation of synthetic 1 reported both by Waldmann14c ([α]20D +46 (c 0.27, MeOH)) and found herein ([α]25D +45 (c 0.07, MeOH)) did not match that measured for natural cytostatin ([α]25D +20 (c 0.27, MeOH)) by Waldmann on an aged authentic sample,14c leaving room for doubt about the identity of natural cytostatin. Visual comparison of the 1H NMR spectra of the four C10–C11 diastereomers (1, 42, 43, 44, Figure 9) with that of natural cytostatin demonstrates that synthetic 1 matches natural cytostatin identically, while 42–44 display substantially different resonances arising from C11-H, C9-H, and C10-Me. In general, the C9 signals for the 10,11-syn isomers (42, 43) were further upfield than for the 10,11-anti isomers (1, 44), while the C11 signals in the syn compounds were further downfield than those of the anti isomers. The 10,11-anti isomer 44 most closely resembles 1 spectrally, but the large difference in the shift of their H11 protons (Δδ = 0.22 ppm) clearly distinguishes the two. The sensitivity of the 1H NMR signals to stereochemical changes in the C9–C11 region and the magnitude of the H10–H11 coupling for each diastereomer can be best interpreted by their adoption of a C9-phosphate/C11-hydroxy hydrogen bonded twist boat conformation. These results also remove any doubt about the 9,10-relative configuration since similarly large spectral changes would be observed if the C9–C11 cyclic structure were altered through that locus. In total, the dissonance of the alternative C10–C11 diastereomers with cytostatin, the comparison of the C1–C7 lactone partial structures with cytostatin, the spectral identity of synthetic 1 with natural cytostatin, the biological profile of synthetic 1, and the fact that synthetic 1 shares the same sign of optical rotation with natural cytostatin authenticate the (4S, 5S, 6S, 9S, 10S, 11S) assignment. The discrepancy in the magnitude of the optical rotation of synthetic 1 and natural cytostatin has been attributed by Waldmann to impurities in his natural sample that resulted from long-term storage.14c
Figure 9.
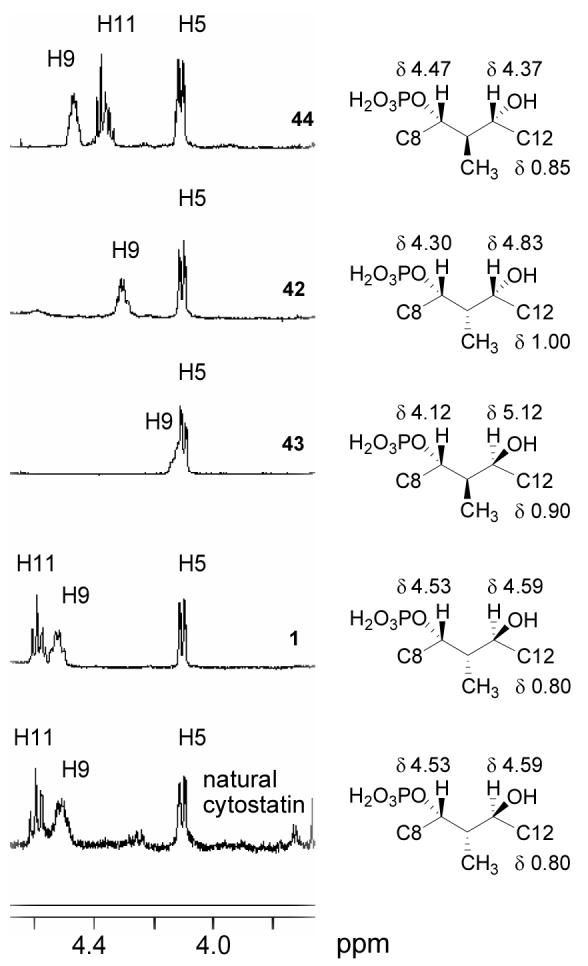
Partial 1H NMR comparison for natural cytostatin, 1, and 42–44.
Synthesis of Additional Key Analogues
In addition to the C10–C11 diastereomers, our synthetic route provided access to several other analogues useful for defining the impact of the C9–C11 substituents and the triene unit on PP2A inhibition. Intermediate 37, ideally suited for the preparation of phosphate replacements, provided not only dephosphocytostatin (40) but by sulfation (SO3–pyridine, THF, 25 °C, 30 min, 71%) followed by desilylation (HF–pyr, THF, pyridine, 25 °C, 1.5 h, 83%) also provided 67, the sulfated version of cytostatin (Scheme 8).
Scheme 8.
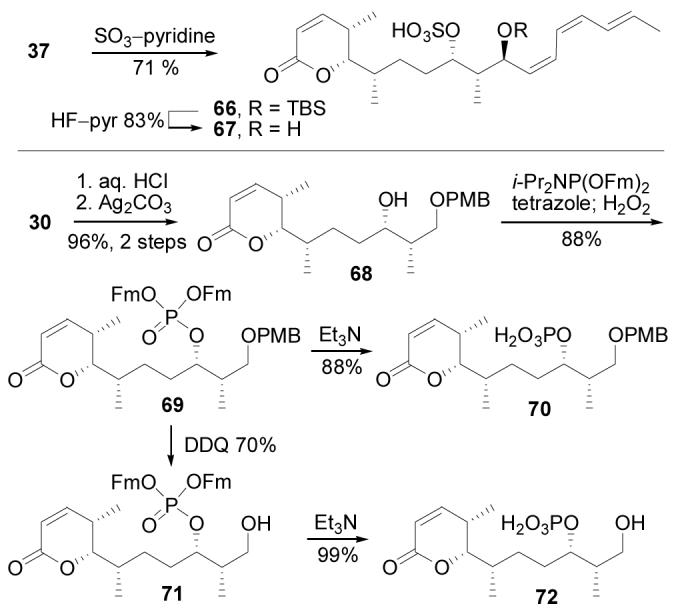
Synthesis of cytostatin analogues.
Conversion of acetal 30 to its corresponding lactone through hydrolysis (0.02 N HCl, acetone, water, 25 °C, 10 min, 99%) and oxidation (Ag2CO3–Celite, benzene, 80 °C, 2 h, 96%) gave 68. Phosphorylation (i-Pr2NP(OFm)2, tetrazole, CH3CN, CH2Cl2, 25 °C, 15 min; H2O2, 10 min, 88%) of 68 was followed by Et3N treatment (Et3N, CH3CN, 25 °C, 20 h, 88%) to provide the phosphate 70 (Scheme 8). Alternatively, cleavage of the PMB group of 69 (DDQ, CH2Cl2, H2O, 25 °C, 30 min, 70%) followed by Et3N treatment (Et3N, CH3CN, 25 °C, 17 h, 99%) produced 72, a cytostatin C1–C11 partial structure that lacks the C12–C18 triene unit.
To provide a means to quantify the impact of the C11-hydroxy on PP2A binding, 11-deshydroxy cytostatin (78) was constructed through a late-stage modification of the cytostatin total synthesis (Scheme 9). Alcohol 32 was transformed to iodide 73 (NIS, PPh3, imidazole, THF, 25 °C, 5 min, 74%), a suitable coupling partner for bromotriene 18 at the correct oxidation state to deliver 78. The union of 18 and 73 was achieved through alkylation of the cuprate derived from 18 (t-BuLi, ether, −78 °C, 1 h; CuI–PBu3, 10 min; 73, 0 °C, 1 h, 63%) to provide 74 in good yield with only minimal efforts at optimization. Notably, this cuprate coupling represents a nice example of a sp2-sp3 organometallic coupling reaction enlisting an unactivated sp3 halide complementary to the more contemporary Stille, Suzuki, and Negishi reactions of vinyl stannanes, boronic acids, or organic zinc reagents currently being explored with sp3 halides. Acetal hydrolysis (0.02 N HCl, acetone, water, 25 °C, 10 min, 77%) followed by lactol oxidation (Ag2CO3–Celite, benzene, 80 °C, 30 min, 76%) gave lactone 76, which after phosphorylation (i-Pr2NP(OFm)2, tetrazole, CH2Cl2, CH3CN, 25 °C, 30 min; H2O2, 5 min, 65%) and Et3N treatment (Et3N, CH3CN, 25 °C, 17 h, 79%) provided 78.
Scheme 9.
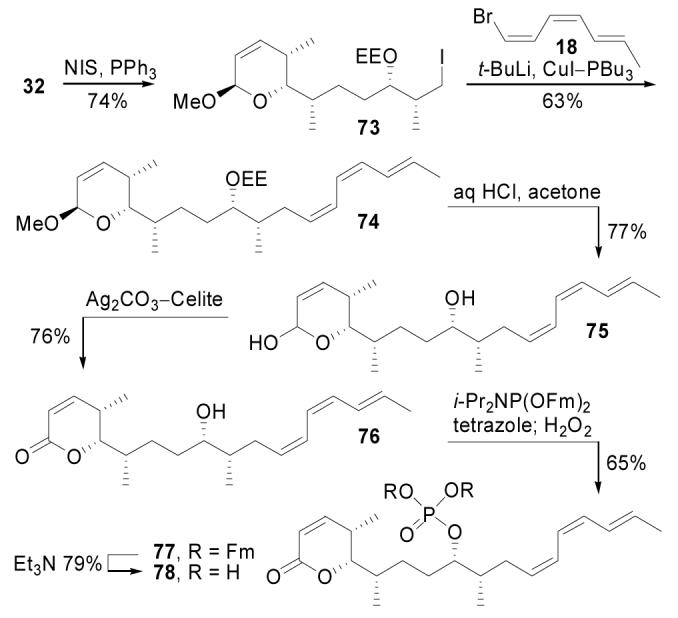
Synthesis of 78.
Biological Evaluation
Assay Substrate and Cross Assay Comparisons: Cytostatin versus Fostriecin
One of the first questions addressed was the relative PP2A inhibition activity of cytostatin versus fostriecin. Fostriecin reportedly exhibits potent PP2A inhibition enlisting phosphorylase a (IC50 = 1–3 nM, rabbit),7,16 glycogen phosphorylase (IC50 = 1.5 nM, bovine),38 or phosphohistone (IC50 = 2–5.6 nM, rabbit and human)9,39 as the substrate in assays containing roughly the same amounts of enzyme, is slightly less effective when enlisting phosphopeptide KR as the substrate (IC50 = 40 nM, human),40 but is substantially less effective when enlisting commercial phosphopeptide, phosphocasein, and p-nitrophenylphosphate as substrates (IC50 = 5–40 μM, human).9 In contrast, albeit in a much more limited series of comparisons, cytostatin displays a narrower and less potent range of PP2A inhibition depending on the substrate enlisted with reports of IC50's of 210 nM (p-nitrophenylphosphate, human at 0.05 units),5 33 nM (p-nitrophenylphosphate, bovine at 0.025 units),14 and 100 nM (phosphorylase a, rabbit at 0.01 units).41 It is clearly less potent than fostriecin using phosphorylase a, but it is less sensitive to the substrate choice in the assay and remains active enlisting p-nitrophenylphosphate as the substrate. Consequently, we conducted side-by-side comparisons of cytostatin and fostriecin enlisting both phosphohistone (a natural substrate) and 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP, a simplified artificial substrate)42 as substrates for bovine brain PP2A.7 In both assays, fostriecin was found to be more potent than cytostatin (3–11 fold), but the difference was much more significant (11-fold) in the assay using a natural substrate (phosphohistone), Figure 10. Additionally, both fostriecin (50-fold) and cytostatin (15-fold) as well as the subsequent analogue series (typically 2–7 fold) exhibited more potent activity against PP2A when a natural (phosphohistone) versus artificial (DiFMUP) substrate was employed. Both results are summarized in Figure 10 and each assay qualitatively provided identical trends in relative activity for the series examined, but those derived from the phosphohistone assay proved easier to distinguish and are probably the most relevant to discuss. Here, cytostatin proved to be ca. 10-fold less potent than fostriecin and both failed to inhibit PP1 or PP5 illustrating their exquisite selectivity (>105 fold for fostriecin and >103–104 fold for cytostatin).
Figure 10.
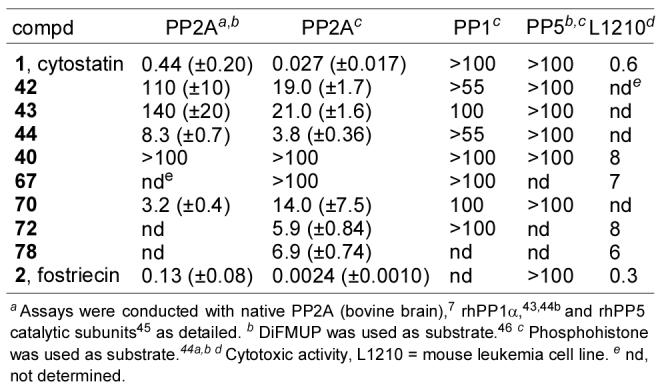
Protein phosphatase and cytotoxic activity (IC50, μM).
C9-Phosphate Modifications
Two key analogues of cytostatin containing modifications to the C9-phosphate were prepared for examination. The first, dephosphocytostatin (40), proved inactive (IC50 >100 μM) as anticipated and analogous to observations made with dephosphofostriecin16 illustrating that the phosphate is an essential component of the PP pharmacophore. The second modification, sulfocytostatin (67), replaced the phosphate with a sulfate providing a key analogue that is nearly identical in structure to sultriecin (3), a naturally occurring antitumor agent.6a Given the structural similarity of sultriecin to cytostatin and fostriecin, it may have been reasonable to suggest that its antitumor activity arises from a mechanism operative for 1 and 2 and that it would inhibit PP2A, although this has yet to be examined. Accordingly, the assessment of sulfocytostatin (67) was conducted not only to probe the importance of the C9-phosphate, but also in anticipation that it would provide insight into the mechanism of action of sultriecin itself. Remarkably, sulfocytostatin was inactive against all PP's examined but did exhibit cytotoxic acitivity (L1210 IC50 = 7 μM) at a level consistent with that observed for sultriecin (IC50 = 0.75–9 μM).6a In addition to illustrating again the essential role of the C9-phosphate, this result would suggest that the antitumor activity of sultriecin does not arise from PP inhibition.
The C11-Alcohol
Probably one of the most ambiguous features of the cytostatin and fostriecin structures has been the importance and role of the C11-hydroxy group. Both we16 and Waldmann14 have reported derivatives (–OAc) that were inactive against PP2A, but with analogues that were altered in ways that also significantly attenuated their PP2A inhibition. Thus, it was clear that the C11-alcohol plays an important role consistent with its conserved presence in all the natural products, but the magnitude of its effect could not be established in these studies. Consequently, 11-deshydroxycytostatin (78), lacking only this key C11-alcohol, was prepared for direct comparative examination (PP2A IC50 = 6.9 μM) and it was found to be 250-fold less potent than cytostatin. Consistent with this impact, the retrospective cytostatin–PP2A modeling as well as our earlier fostriecin–PP2A model2 define a key hydrogen bond between the C11-hydroxy group and a conserved Arg214. This hydrogen bond stabilizes a turn in the bound conformation of 1 or 2 orienting the terminal triene into a hydrophobic pocket where it π-stacks with the indole side chain of Trp200.
Notably, the activity of 70, which also lacks the C11-hydroxy group and replaces the sensitive triene with a p-methoxybenzyl ether, exhibited PP2A inhibition (IC50 = 14 μM) only 2-fold less potent than 11-deshydroxycytostatin suggesting that the PMB ether may serve as a simplified, stable replacement for the labile triene. The retrospective model places this PMB replacement directly over the Trp200 indole benefiting from the same π-stacking as the authentic C12–C18 triene.
The C12–C18 Triene
The examination of 72, lacking only the entire C12–C18 segment of cytostatin, permitted a direct assessment of the (Z,Z,E)-triene contribution to the magnitude of PP2A inhibition potency and selectivity. Analogue 72 inhibited PP2A (IC50 = 5.9 μM) and not PP1 or PP5 maintaining selectivity for PP2A, but it was 220-fold less active than cytostatin itself. This observation complements Waldmann's examination of a truncated C12–C13 alkyne and a C12–C13 (Z)-vinyl iodide which were found to be 10-fold less active and equipotent with cytostatin, respectively, albeit in a PP2A assay enlisting p-nitrophenylphosphate as substrate.14 Our earlier model of fostriecin bound at the PP2A active site and the retrospective modeling studies of cytostatin that follow define a beautiful π-stacking interaction of nearly all of the triene with the indole side chain of Trp200 in a hydrophobic cleft providing a compelling rationale for the (Z,Z,E)-triene enhancement of binding affinity and its unusual stereochemistry. Moreover, this model suggests that the largest share of the π-stacking interaction occurs with C13–C17 implying that the terminal (Z,E)-diene is more engaged than the internal C12–C13 (Z)-alkene.
The C10/C11 Cytostatin Diastereomers 42–44
Each of the cytostatin diastereomers 42–44 proved to be significantly or substantially less potent than the natural product itself. This satisfying result not only reinforced the natural product structural assignment, but it indicates that both the C10 and C11 substituents productively contribute to the properties of cytostatin. The C11-epimer 42, in which only the stereochemistry of the alcohol is inverted, proved to be 700-fold less potent against PP2A than cytostatin itself. This proved to be even less potent than the activity of 78 lacking the C11 alcohol as well as 72 lacking the entire triene segment consistent with a substantially disrupted binding that precludes effective accommodation of both the inverted alcohol and triene simultaneously.
The C10-epimer 43, in which only the stereochemistry of the methyl group is inverted, was even less active being ca. 780-fold less potent than cytostatin. This result indicates a prominent role for this substituent and its stereochemistry and was especially surprising since the inversion of the C10 center was expected to have the least perturbation on the conformation of free cytostatin. The retrospective modeling studies that follow did not reveal a conformational role for this substituent, but it does fit snuggly into a hydrophobic pocket defined by Tyr127 and Tyr265 and shields the active site and C9-phosphate from solvent access. Adoption of the unnatural (10R)-configuration with 43 precludes methyl binding in this pocket, but it does not introduce destabilizing interactions as the methyl group now extends out of the active site into solvent accessible space and it no longer shields the active site and C9-phosphate. Although it is not easy to define a role for this substituent that might account for a 780-fold increase in inhibitory activity, it is consistent with its proposed role18 of mimicking a phosphothreonine methyl group paralleling the substrate preference of PP2A for phosphothreonine peptides (5–100 fold) over phosphoserine peptides.46 In the retrospective modeling, the methyl group of cytostatin resides close to the phosphate tucked into a hydrophobic pocket, whereas that of 43 is distal from the phosphate incapable of serving as such a threonine methyl mimic. Moreover, in the side-by-side comparison with our fostriecin–PP2A model, we came to recognize that the fostriecin C8-methyl group and cytostatin C10-methyl group extend into the same enzyme pocket and that they are spacially much closer to one another than might be intuitively expected being displaced by only 2.85 Å. For us, this provides a compelling case for suggesting that the cytostatin C10-methyl group, like the fostreicin C8-methyl group, serves as a mimic of the phosphothreonine methyl group accounting for its prominent role.
Surprisingly and interestingly, the (10R,11R)-diastereomer 44 in which the stereochemistry of both the C10-methyl group and the C11-alcohol is inverted was the most potent of the series (IC50 = 3.8 μM) being 140-fold less active than the natural product. A cursory examination of the retrospective model did not reveal insights into why 44 might be a 5-fold more effective inhibitor than either 42 or 43, but the magnitude of the loss in activity is still so significant as to reflect a substantially disrupted binding.
The sensitivity of the cytostatin–PP2A interaction to the stereochemical orientation of the C9–C11 segment underscores the critical nature of the C9–C11 substituents and confirms the proximal methyl and C11-hydrogen bonding groups are important elements of the PP pharmacophore for cytostatin and its congeners.
Cytotoxic Activity
We also evaluated compounds 1, 2, 40, 72, and 78 for in vitro cytotoxic activity against the L1210 cell line (Figure 10). In this functional assay, fostriecin (IC50 = 0.3 μM) and cytostatin (IC50 = 0.6 μM) exhibited comparable activity, albeit with cytostatin being roughly 2-fold less potent. Similarly, dephosphocytostatin (40, IC50 = 8 μM) was 10-fold less active than the natural product and comparable in activity to dephosphofostriecin (IC50 = 20 μM),16 both exhibiting potencies that most consider as nearly inactive. Finally, both 72 and 78 were found to be roughly 10-fold less potent than cytostatin consistent with their reduced PP2A inhibition.
Models of Cytostatin and Fostriecin Bound at the PP2A Active Site
Complementary to our own earlier modeling2,16 of fostriecin bound to a homology model of PP2A,18 we examined the binding of cytostatin at the PP2A active site. The former model not only provided insights into the active site binding by fostriecin identifying key features of the interaction, but it defined the origin of the PP2A selectivity that stems from reversible conjugate addition of the active site Cys269 (unique to PP2A and PP4 and absent in PP1, PP2B, PP5, and PP7) to the α,β-unsaturated lactone. Although this covalent addition to fostriecin or cytostatin has not been experimentally verified, it was originally supported by the decreased affinity of 2,3-dihydrofostriecin (≥200-fold less potent than 2),16 and confirmed with the more recent isolation and identification of the analogous Cys269 adduct with phoslactomycin (6).17
Since the details of the fostriecin–PP2A model have not been disclosed elsewhere, it is presented herein alongside the newly generated cytostatin–PP2A model. Side-by-side overviews of the two models are presented in Figure 11 with space-filling representations of fostriecin and cytostatin. Both adopt nearly identical bent conformations with the C9-phosphate binding and bridging both active site metal cations. The lactone contacts the β12–β13 loop stabilized by hydrophobic interactions with the surrounding residues (Tyr267) and Cys269 is poised for reversible, conjugate addition to the α,β-unsaturated lactone (CysSH–C3 distance is 2.59 Å and 3.15 Å for fostriecin and cytostatin, respectively) which is activated by an Arg268 hydrogen bond to the lactone carbonyl accounting for the PP selectivity and the enhanced potency (≥200-fold for fostriecin relative to 2,3-dihydrofostriecin16 and ca. 1000-fold for an analogous pair of simplified cytostatin analogues14). This is illustrated in Figure 12 for cytostatin which also highlights a key conformational feature imposed by the interacting C4- and C6-methyl groups. The presence and stereochemistry of these two substituents restrict the side chain to a single orientation that avoids syn pentane interactions and facilitates active site binding (see also Figure 4). Fostriecin accomplishes the same side chain restriction, although with a slightly altered orientation, through preferential adoption of a C5 H-eclipsed conformation with the C6–C7 olefin.
Figure 11.
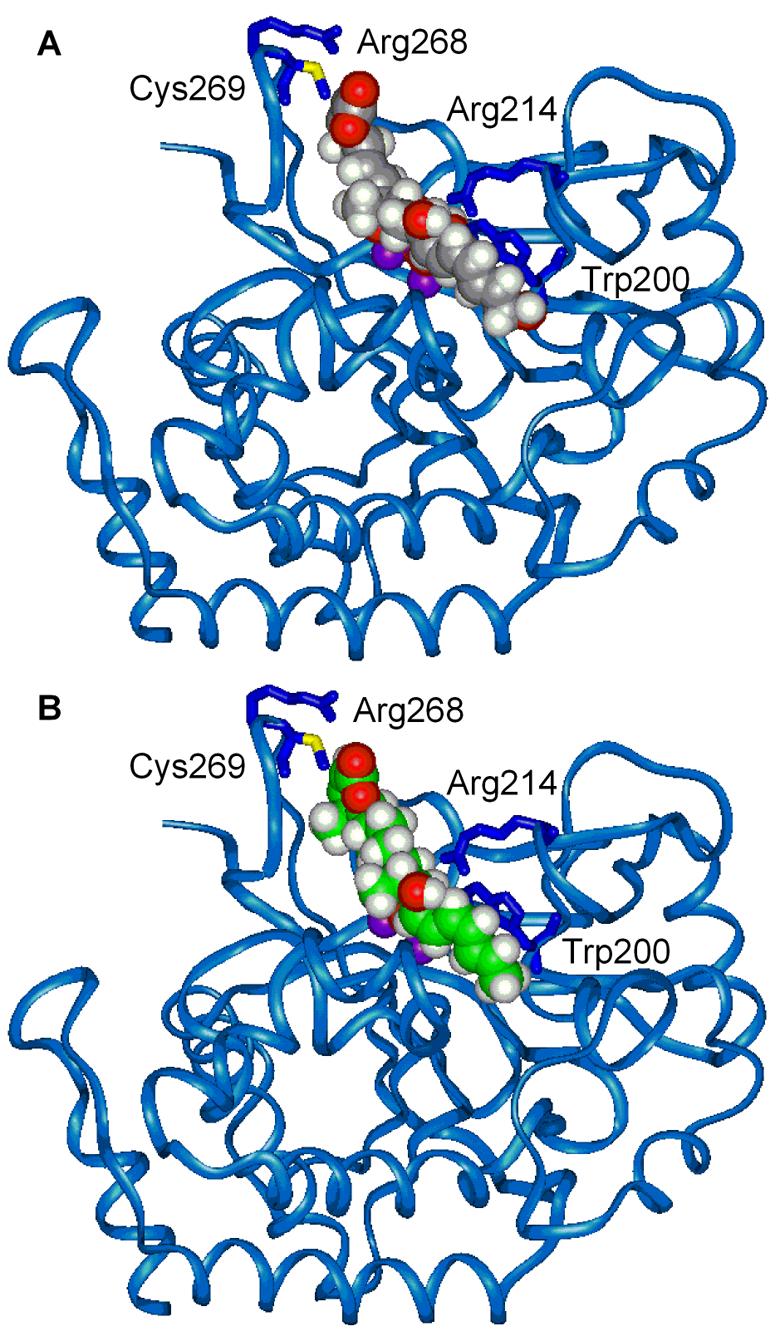
Models of fostriecin (A) and cytostatin (B) bound to PP2A.
Figure 12.
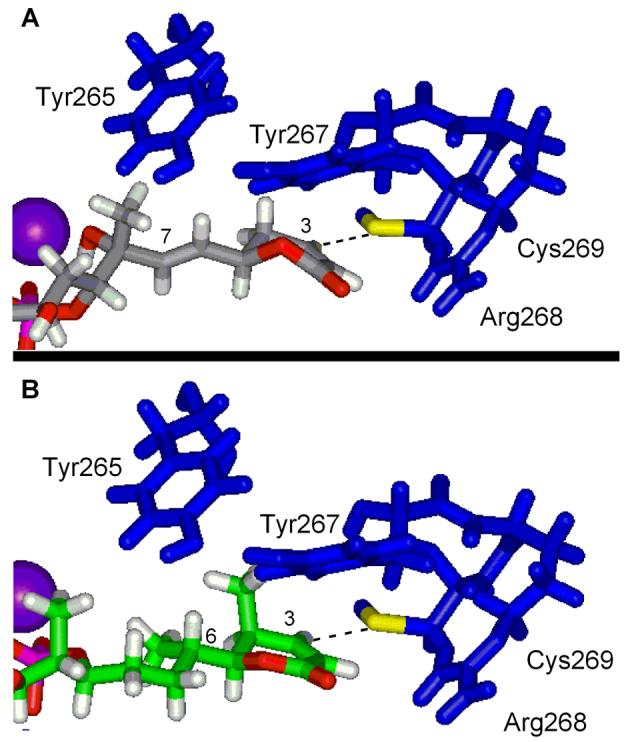
The C1–C8 region of fostriecin (A) and cytostatin (B) bound to PP2A.
The largest differences in the two models naturally emerge in the C6–C8 region with cytostatin adopting a gauche turn at C7–C8, whereas fostriecin accomplishes this at C8–C9. This latter turn in fostriecin is stablilized by a C8-OH hydrogen bond to Asp57 and its cooperative binding to the proximal metal cation potentially displacing a reactive active site water nucleophile enlisted for phosphate hydrolysis. Moreover, this places the fostriecin C8-methyl group in a hydrophobic pocket potentially reserved for the methyl group of phosphothreonine substrates. This suggests important roles for both the presence and stereochemistry of each of the fostriecin C8 substituents and it is notable that Shibasaki has reported that C8-epi-fostriecin is 2000-fold less potent than the natural product in assays enlisting a natural substrate (phosphohistone).9
The backbones for the remainder of the bound cytostatin and fostriecin are essentially identical being anchored by the C9-phosphate binding and bridging of both active site metal cations and the C11-hydroxy group forming a key hydrogen bond with the conserved Arg214 (Figure 13). The latter hydrogen bond induces, or at least stabilizes, a turn in the bound conformations orienting the C12–C18 triene into a hydrophobic cleft where it stacks with the indole side chain of Trp200. Notably, the unusual (Z,Z,E)-triene geometry allows it to π-stack across the entire length of the indole maximizing the intermolecular binding stabilization (Figure 14). The importance of the phosphate binding is clear from the reduced activity of dephosphofostriecin (>105-fold)16 and dephosphocytostatin (>103–104 fold), whereas the importance of the latter two interactions are clear from the evaluations herein of 11-deshydroxycytostatin (250-fold) and 72 lacking the entire triene segment (220-fold). No active site interaction for the fostriecin terminal alcohol was detected consistent with its lack of impact on the PP2A inhibition2,16 and its absence in all other related natural products including cytostatin. The largest and only other difference between cytostatin and fostriecin throughout this region is the cytostatin C10-methyl group. This methyl group resides close to the phosphate tucked into a hydrophobic pocket potentially mimicking the methyl group of a phosphothreonine substrate. In comparing fostriecin and cytostatin models, the cytostatin C10-methyl group was found to extend into the same hydrophobic pocket as the fostriecin C8-methyl group and the two are spacially closer to one another than one might suspect (2.85 Å) (Figure 13). The importance of the cytostatin C10-methyl (780-fold reduction for epimer) and the analogous importance of the fostriecin C8 substituents (2000-fold reduction for epimer) make a compelling case for suggesting these centers are serving similar roles in their respective natural products in which the methyl groups mimic a phosphothreonine substrate methyl group used by and sufficient for the enzyme to distinguish its substrates from phosphoserine peptides.
Figure 13.
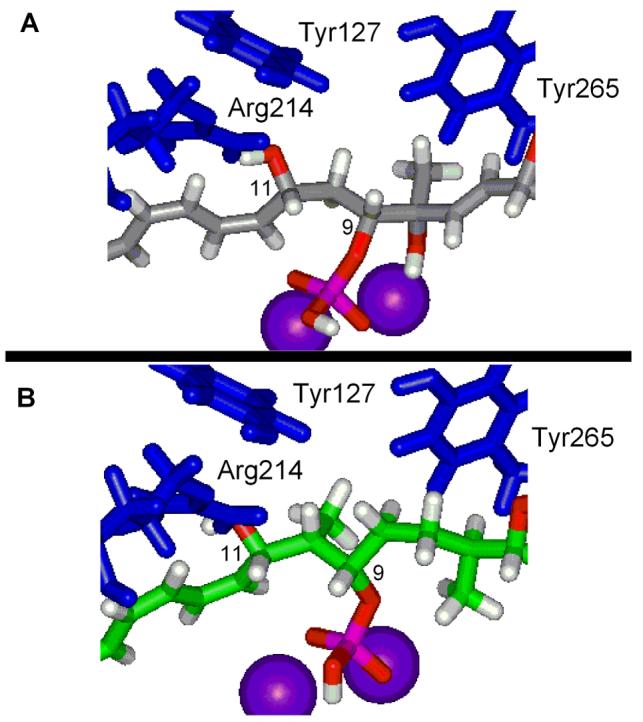
The C9–C11 region of fostriecin (A) and cytostatin (B) bound to PP2A.
Figure 14.
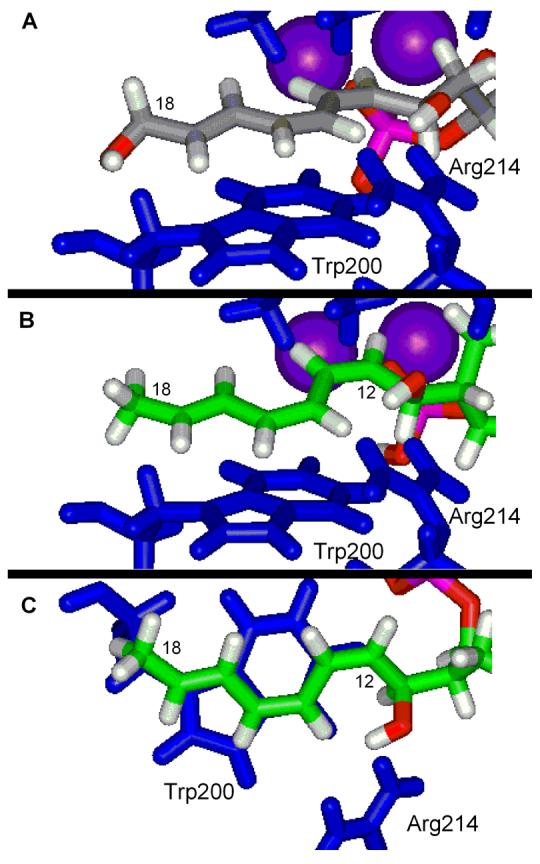
The C12–C18 region of fostriecin (A) and cytostatin (B and C) bound to PP2A.
Conclusion
Synthetic studies leading to a definitive assignment of the relative and absolute stereochemistry of cytostatin and culminating in its total synthesis and that of each of its C10–C11 stereoisomers were conducted. Key elements of the strategy include a convergent assembly of the natural product core that permits the early stage independent adjustment of the C4–C6 or C9–C10 stereochemistry coupled with a late stage single-step installation of the sensitive (Z,Z,E)-triene with a reaction that allows substrate control of the C11 stereochemistry. Extensions of the synthetic approach provided additional key analogues including dephosphocytostatin (40), sulfocytostatin (67), 11-deshydroxycytostatin (78), and 72 lacking the entire triene unit. Biological assessment of cytostatin, its diastereomers, and the key analogues demonstrated the importance and quantitated the magnitude of the presence and stereochemistry of the C10/C11 substituents (>140-fold), the C9-phosphate (>103–104 fold), C10-methyl (ca. 780-fold), C11-hydroxy (250-fold), and the C12–C18 (Z,Z,E)-triene (220-fold) contributions to the potent PP2A inhibition. Retrospective modeling of cytostatin within the active site of a PP2A catalytic subunit homology model and its comparison with an analogous fostriecin–PP2A model provided key insights into the role of each of the natural product segments, their substituents, and the accompanying stereochemistry. Remarkably, each element of the cytostatin structure plays a productive, and sometimes surprisingly prominent, role in its PP2A inhibition. Finally, indirect evidence is provided that sultriecin, despite its structural similarity to cytostatin, does not apparently derive its cytotoxic and antitumor activity through protein phosphatase inhibition.
Supplementary Material
Figure 7.
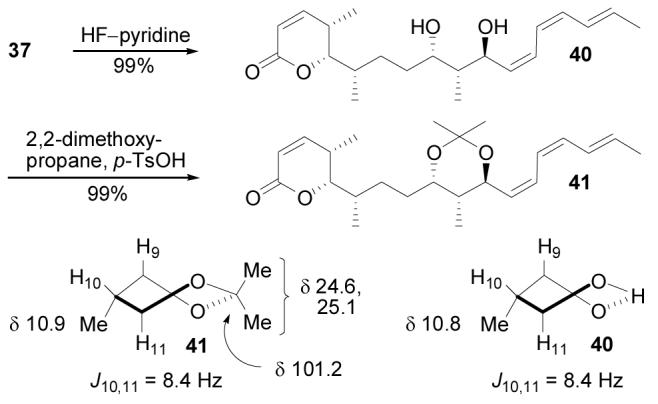
Confirmation of the C9–C11 stereochemistry.
Acknowledgments
We gratefully acknowledge the support of the NIH (CA93456 and CA42056, DLB; LA, RH). We thank Prof. Ishizuka of the Microbial Chemistry Research Foundation for an authentic sample of cytostatin and Inkyu Hwang for conducting the cytotoxic activity assays.
Footnotes
Supporting Information Available. Full experimental details.
References
- 1.(a) Amemiya M, Someno T, Sawa R, Naganawa H, Ishizuka M, Takeuchi T. J. Antibiot. 1994;47:541. doi: 10.7164/antibiotics.47.541. [DOI] [PubMed] [Google Scholar]; (b) Amemiya M, Ueno M, Osono M, Masuda T, Kinoshita N, Nishida C, Hamada M, Ishizuka M, Takeuchi T. J. Antibiot. 1994;47:536. doi: 10.7164/antibiotics.47.536. [DOI] [PubMed] [Google Scholar]
- 2.Lewy DS, Gauss CM, Soenen DR, Boger DL. Curr. Med. Chem. 2002;9:2005. doi: 10.2174/0929867023368809. [DOI] [PubMed] [Google Scholar]
- 3.(a) Yamazaki K, Amemiya M, Ishizuka M, Takeuchi T. J. Antibiot. 1995;48:1138. doi: 10.7164/antibiotics.48.1138. [DOI] [PubMed] [Google Scholar]; (b) Kawada M, Amemiya M, Ishizuka M, Takeuchi T. Jap. J. Cancer Res. 1999;90:219. doi: 10.1111/j.1349-7006.1999.tb00736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Masuda T, Watanabe S, Amemiya M, Ishizuka M, Takeuchi T. J. Antibiot. 1995;48:528. doi: 10.7164/antibiotics.48.528. [DOI] [PubMed] [Google Scholar]; (b) Kawada M, Kawatsu M, Masuda T, Ohba S, Amemiya M, Kohama T, Ishizuka M, Takeuchi T. Intl. Immunopharm. 2003;3:179. doi: 10.1016/s1567-5769(02)00231-x. [DOI] [PubMed] [Google Scholar]
- 5.Kawada M, Amemiya M, Ishizuka M, Takeuchi T. Biochim. Biophys. Acta. 1999;1452:209. doi: 10.1016/s0167-4889(99)00126-3. [DOI] [PubMed] [Google Scholar]
- 6.(a) Ohkuma H, Naruse N, Nishiyama Y, Tsuno T, Hoshino Y, Sawada Y, Konishi M, Oki T. J. Antibiot. 1992;45:1239. doi: 10.7164/antibiotics.45.1239. Sultriecin: [DOI] [PubMed] [Google Scholar]; (b) Ozasa T, Tanaka K, Sasamata M, Kaniwa H, Shimizu M, Matsumoto H, Iwanami M. J. Antibiot. 1989;42:1339. doi: 10.7164/antibiotics.42.1339. Phospholine: [DOI] [PubMed] [Google Scholar]; (c) Kohama T, Enokita R, Okazaki T, Miyaoka H, Torikata A, Inukai M, Kaneko I, Kagasaki T, Sakaida Y, Satoh A, Shiraishi A. J. Antibiot. 1993;46:1503. doi: 10.7164/antibiotics.46.1503. Leustroducsins: [DOI] [PubMed] [Google Scholar]; Kohama T, Nakamura T, Kinoshita T, Kaneko I, Shiraishi A. J. Antibiot. 1993;46:1512. doi: 10.7164/antibiotics.46.1512. [DOI] [PubMed] [Google Scholar]; (d) Fushimi S, Nishikawa S, Shimazu A, Seto H. J. Antibiot. 1989;42:1019. doi: 10.7164/antibiotics.42.1019. Phoslactomycins: [DOI] [PubMed] [Google Scholar]; (e) Tomiya T, Uramoto M, Isono K. J. Antibiot. 1990;43:118. doi: 10.7164/antibiotics.43.118. Phosphazomycins: [DOI] [PubMed] [Google Scholar]
- 7.Walsh AH, Cheng A, Honkanen RE. FEBS Lett. 1997;416:230. doi: 10.1016/s0014-5793(97)01210-6. [DOI] [PubMed] [Google Scholar]
- 8.Usui T, Marriott G, Inagaki M, Swarup G, Osada H. J. Biochem. (Tokyo) 1999;125:960. doi: 10.1093/oxfordjournals.jbchem.a022375. [DOI] [PubMed] [Google Scholar]
- 9.Maki K, Motoki R, Fujii K, Kanai M, Kobayashi T, Tamura S, Shibasaki M. J. Am. Chem. Soc. 2005;127:17111. doi: 10.1021/ja0562043. [DOI] [PubMed] [Google Scholar]
- 10.(a) Boger DL, Hikota M, Lewis BM. J. Org. Chem. 1997;62:1748. [Google Scholar]; (b) Boger DL, Ichikawa S, Zhong W. J. Am. Chem. Soc. 2001;123:4161. doi: 10.1021/ja010195q. [DOI] [PubMed] [Google Scholar]
- 11.For reviews of fostriecin total syntheses see: Shibasaki M, Kanai M. Heterocycles. 2005;66:727. and reference 2.Chavez DE, Jacobsen EN. Angew. Chem. Int. Ed. 2001;40:3667. doi: 10.1002/1521-3773(20011001)40:19<3667::aid-anie3667>3.0.co;2-6.Reddy YK, Falck JR. Org. Lett. 2002;4:969. doi: 10.1021/ol025537r.Miyashita K, Ikejiri M, Kawasaki H, Maemura S, Imanishi T. Chem. Commun. 2002:742. doi: 10.1039/b201302a.Miyashita K, Ikejiri M, Kawasaki H, Maemura S, Imanishi T. J. Am. Chem. Soc. 2003;125:8238. doi: 10.1021/ja030133v.Esumi T, Okamoto N, Hatakeyama S. Chem. Commun. 2002:3042. doi: 10.1039/b209742g.Fujii K, Maki K, Kanai M, Shibasaki M. Org. Lett. 2003;5:733. doi: 10.1021/ol027528o. and reference 9.Trost BM, Frederiksen MU, Papillon JP, Harrington PE, Shin S, Shireman BT. J. Am. Chem. Soc. 2005;127:3666. doi: 10.1021/ja042435i.Yadav JS, Prathap I, Tadi BP. Tetrahedron Lett. 2006;47:3773.
- 12.Shimada K, Kaburugi Y, Fukuyama T. J. Am. Chem. Soc. 2003;125:4048. doi: 10.1021/ja0340679. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Takeyama R, Kobayashi Y. Angew. Chem. Int. Ed. 2006;45:3320. doi: 10.1002/anie.200600458. [DOI] [PubMed] [Google Scholar]
- 14.(a) Bialy L, Waldmann H. Angew. Chem. Int. Ed. 2002;41:1748. doi: 10.1002/1521-3773(20020517)41:10<1748::aid-anie1748>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]; (b) Bialy L, Waldmann H. Chem. Commun. 2003:1872. [PubMed] [Google Scholar]; (c) Bialy L, Waldmann H. Chem. Eur. J. 2004;10:2759. doi: 10.1002/chem.200305543. [DOI] [PubMed] [Google Scholar]; (d) Bialy L, Lopez-Canet M, Waldmann H. Synthesis. 2002:2096. [Google Scholar]
- 15.Marshall JA, Ellis K. Tetrahedron Lett. 2004;45:1351. [Google Scholar]
- 16.Buck SB, Hardouin C, Ichikawa S, Soenen DR, Gauss CM, Hwang I, Swingle MR, Bonness KM, Honkanen RE, Boger DL. J. Am. Chem. Soc. 2003;125:15694. doi: 10.1021/ja038672n. [DOI] [PubMed] [Google Scholar]
- 17.Teruya T, Simizu S, Kanoh N, Osada H. FEBS Lett. 2005;579:2463. doi: 10.1016/j.febslet.2005.03.049. [DOI] [PubMed] [Google Scholar]
- 18.Gauss CM, Sheppeck JE, II, Nairn AC, Chamberlin R. Bioorg. Med. Chem. 1997;5:1751. doi: 10.1016/s0968-0896(97)00145-4. [DOI] [PubMed] [Google Scholar]
- 19.Reviews: Colby DA, Chamberlin AR. Mini-Reviews Med. Chem. 2006;6:109. doi: 10.2174/138955706777435715.Sheppeck JE, II, Gauss CM, Chamberlin AR. Bioorg. Med. Chem. 1997;5:1739. doi: 10.1016/s0968-0896(97)00146-6.
- 20.Lawhorn BG, Boga SB, Wolkenberg SE, Boger DL. Heterocycles. 2006;70 in press. [Google Scholar]
- 21.Ley SV, Anthony NJ, Armstrong A, Brasca MG, Clarke T, Culshaw D, Greck C, Grice P, Jones AB, Lygo B, Madin A, Sheppard RN, Slawin A, Williams DJ. Tetrahedron. 1989;45:7161. [Google Scholar]
- 22.Brown HC, Bhat KS, Randad RS. J. Org. Chem. 1989;54:1570. [Google Scholar]
- 23.Grubbs RH, Chang S. Tetrahedron. 1998;54:4413. [Google Scholar]
- 24.Similar coupling patterns have been reported for other 4,5-disubstituted-α,β-unsaturated lactones: Ley SV, Armstrong A, Diez-Martin D, Ford MJ, Grice P, Knight JG, Kolb HC, Madin A, Marby CA, Mukherjee S, Shaw AN, Slawin AM, Vile S, White AD, Williams DJ, Woods M. J. Chem. Soc. Perkin. 1991;1:667.Keck GE, Li X-Y, Knutson CE. Org. Lett. 1999;1:411. doi: 10.1021/ol990632u.Marshall JA, Adams ND. J. Org. Chem. 1999;64:5201. doi: 10.1021/jo9823083.Diez-Margin D, Kotecha NR, Ley SV, Mantegani S, Menendez JC, Organ HM, White AD. Tetrahedron. 1992;48:7899.
- 25.The H4–H5 coupling constant was initially reported as J4,5 = 10.4 Hz1a but our measurement on natural cytostatin indicates J4,5 = 2.7 Hz, consistent with Waldmann's report of this value.14
- 26.The trans-C1 stereochemistry was assigned based on H3–H4 coupling (J = 6.0 Hz). Similar trans-1,4-disubstituted dihydropyrans display an H3–H4 coupling (J = 5.7 Hz) distinct from that of cis-1,4-disubstituted dihydropyrans (J = 1.9 Hz), see: Valverde S, Bernabe M, Garcia-Ochoa S, Gomez AM. J. Org. Chem. 1990;55:2294.
- 27.Mori K, Seu YB. Tetrahedron. 1988;44:1035. [Google Scholar]
- 28.Tius MA, Fauq AH. J. Org. Chem. 1983;48:4131. [Google Scholar]
- 29.(a) Mori K, Nomi H, Chuman T, Kohno M, Kato K, Noguchi M. Tetrahedron. 1982;38:3705. [Google Scholar]; (b) Shimizu Y, Kiyota H, Oritani T. Tetrahedron Lett. 2000;41:3141. [Google Scholar]; (c) Kobayashi Y, Tan C, Kishi Y. J. Am. Chem. Soc. 2001;123:2076. doi: 10.1021/ja004154q. [DOI] [PubMed] [Google Scholar]
- 30.Sabes SF, Urbanek RA, Forsyth CJ. J. Am. Chem. Soc. 1998;120:2534. [Google Scholar]
- 31.(a) Belosludtsev YY, Borer BC, Taylor RJK. Synthesis. 1991:320. [Google Scholar]; (b) Furber M, Herbert JM, Taylor RJK. J. Chem. Soc., Perkin Trans. 1989;1:683. [Google Scholar]
- 32.(a) Corey EJ, Fuchs PL. Tetrahedron Lett. 1972:3769. [Google Scholar]; (b) Ramirez F, Desai NB, McKelvie N. J. Am. Chem. Soc. 1962;84:1745. [Google Scholar]
- 33.Uenishi J, Kawahama R, Yonemitsu O, Tsuji J. J. Org. Chem. 1998;63:8965. [Google Scholar]
- 34.Lipshutz BH, Moretti R, Crow R. Org. Syn., Coll. 1993;VIII:33. [Google Scholar]
- 35.Dess DB, Martin JC. J. Am. Chem. Soc. 1991;113:7277. [Google Scholar]
- 36.Still WC, Schneider JA. Tetrahedron Lett. 1980;21:1035. [Google Scholar]
- 37.(a) Rychnovsky SD, Skalitzky DJ. Tetrahedron Lett. 1990;31:945. [Google Scholar]; (b) Evans DA, Rieger DL, Gage JR. Tetrahedron Lett. 1990;49:7099. [Google Scholar]
- 38.Hastie CJ, Cohen PTW. FEBS Lett. 1998;431:357. doi: 10.1016/s0014-5793(98)00775-3. [DOI] [PubMed] [Google Scholar]
- 39.Cheng A, Balczon R, Zuo Z, Koons JS, Walsh AH, Honkanen RE. Cancer Res. 1998;58:3611. [PubMed] [Google Scholar]
- 40.Roberge M, Tudan C, Hung SMF, Harder KW, Jirik FR, Anderson H. Cancer Res. 1994;54:6115. [PubMed] [Google Scholar]
- 41.Launey T, Endo S, Sakai R, Harano J, Ito M. Proc. Natl. Acad. Sci. USA. 2004;101:676. doi: 10.1073/pnas.0302914101. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
42.Structure of DiFMUP:
- 43.rhPP1α was expressed in E. coli with the pKK-223-2 vector (accession number M77749) and purified essentially as described: Zhang L, Lee EY. Biochemistry. 1997;36:8209. doi: 10.1021/bi9704865.
- 44.(a) Honkanen RE, Zwiller J, Moore RE, Daily SL, Khatra BS, Dukelow M, Boynton AL. J. Biol. Chem. 1990;265:19401. [PubMed] [Google Scholar]; (b) Huang X, Swingle MR, Honkanen RE. Methods Enzymol. 2000;315:579. doi: 10.1016/s0076-6879(00)15869-0. [DOI] [PubMed] [Google Scholar]
- 45.Swingle MR, Honkanen RE, Ciszak EM. J. Biol. Chem. 2004;279:33992–33999. doi: 10.1074/jbc.M402855200. [DOI] [PubMed] [Google Scholar]
- 46.Gee KR, Sun W-C, Bhalgat MK, Upson RH, Klaubert DH, Latham KA, Haugland RP. Anal. Biochem. 1999;273:41–48. doi: 10.1006/abio.1999.4202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.