

Abstract
We report a simple, efficient, and stereoselective Mukaiyama aldol approach to install the key hydroxymethyl moiety into the benzazocane framework of FR900482. Synthetic investigations revealed that the reaction is highly dependent upon the electronics of the aromatic ring. This approach enabled the economical introduction of a [13C]-label to study the biosynthesis of these structurally and biogenetically related natural products. Epimerization of the initially formed β-hydroxy ketone may enable access to mitomycin C or FR900482 biosynthetic congeners.
FR900482 (2) and congeners FR66979 (3), FK973 (4), and FK317 (5) have enormous potential as anticancer agents (Figure 1).1 These new prospective drugs are cytotoxic to tumors, but do not appear to exhibit the deleterious side effects characteristic of the structurally related drug mitomycin C (MMC, 1).2 For example, myelo-suppression and hemolytic uremic syndrome have plagued the broad clinical utility of MMC since 1974.3 The allure of less toxic and more active chemotherapeutic agents has spawned numerous total and formal syntheses of FR900482 and its congeners.4 In particular, we recently described a stereoselective, convergent total synthesis of FR900482 (2)5 as well as an improved synthesis of the benzazocane core of both FR900482 and mitomycin C.4 Although our thirty-three-step asymmetric route is among the most concise syntheses of FR900482 (2) extant, our interest in exploring the biogenesis of 1 and 2 has mandated that we improve the unselective hydroxymethylation aldol condensation reaction (dr ≈ 1:1).5
Figure 1.

Structures of Mitomycin C, FR900482, and congeners.
Throughout the course of our studies towards the synthesis of putative biosynthetic intermediates of both FR900482 and the mitomycins, it became necessary to investigate the hydroxymethylation of substrates such as 6 (Scheme 1).6 Hydroxymethylation of benzazocane intermediates en route to FR900482 under basic conditions often resulted in undesired elimination of the initially formed hydroxymethylation product. In conjuction with their synthetic studies, Rapoport7 and Danishefsky8 demonstrated that epoxidation and reductive ring opening of the exo-methylene in their respective eight- or six-membered ring substrates afforded the requisite hydroxymethyl compound indirectly. In a recent full paper describing his total synthesis of FR-900482 (2), Fukuyama reported a one-step, stereoselective, base-catalyzed hydroxymethylation requiring 115 equivalents of aqueous formaldehyde.9 Such a large molar excess is impractical in the context of the preparation of putative biosynthetic intermediates where an expensive isotopically labeled source of formaldehyde would be employed.10
Scheme 1.

Hydroxymethylation of Benzazocane 6.
With benzazocane 6 in hand, we explored alternate hydroxymethylation conditions. We were especially intrigued by the lanthanide triflate-catalyzed Mukaiyama aldol reaction of enoxysilanes developed by Kobayashi.11 Accordingly, treatment of benzazocane 6 with trimethylsilyl triflate provided the silyl enol ether which was used without purification in the hydroxymethylation step. Exposing the enoxysilane to a catalytic amount of ytterbium(III) triflate and five equivalents of formaldehyde over 48 hours provided alcohol 7 in 70% yield as a 84:16 ratio of diastereomers. Scandium(III) triflate was a superior catalyst and furnished the alcohol 7 in 82% yield (75% over two steps) as a 91:9 ratio of diastereomers in only three hours (Scheme 1). The major diastereomer in the aldol reaction was determined to be the 7S-stereoisomer by X-ray crystallographic analysis of a derivative, diol 8. This derivative was prepared by stereoselective carbonyl reduction and removal of the N-nosyl residue of the initial hydroxymethylation product, alcohol 7.12
Having developed an efficient method for the hydroxymethylation of benzazocane 6, we were interested in exploring the scope and generality of this protocol with more substrates. Specifically, we were interested to see the effect, if any, different protecting groups on the benzazocane nitrogen might have on the selectivity and general outcome of the aldol reaction. Consequently, N-alloc benzazocane 10a was prepared from compound 94 (Scheme 2). Treatment of the N-alloc benzazocane 10a to identical hydroxymethylation conditions described earlier to prepare compound 7 provided the 7S-stereoisomer of alcohol 11 in 71% yield over two steps with 94:6 dr. We also attempted the same aldol reaction of 10a with five equivalents of aqueous 13C-labeled formaldehyde to furnish labeled alcohol 13C-11 in slightly lower yield (45% for two steps, 78% brsm, unoptimized). Somewhat surprisingly, treatment of N-pMB benzazocane 10b5 to the same conditions did not provide any of the desired alcohol. Even more startling was that N-Nvoc (Nvoc = 6-nitroveratryloxycarbonyl or 3,4-dimethoxy-6-nitrobenzyloxycarbonyl) benzazocane 10c13 did not undergo hydroxymethylation under these conditions. Attempted hydroxymethylation of 10b and 10c was apparently unsuccessful since, in each case, the requisite enoxysilane intermediate could not be formed under a variety of conditions.
Scheme 2.

Hydroxymethylation of Benzazocanes 10a-c.
With these interesting results in hand for benzazocanes constituted with the FR900482 framework, we were intrigued whether this methodology could be useful in our studies towards the asymmetric total synthesis of the mitomycins and their corresponding biosynthetic precursors. As shown in Scheme 3, employing identical hydroxymethylation conditions as before failed to provide the desired alcohol from benzazocane 12.14 In tandem with unsuccessful results observed for FR900482-based benzazocanes 10b and 10c, our lack of success in forming the enoxysilane intermediate likely hindered hydroxymethylation of these substrates. Alternatively, attempts to engender enoxysilane formation under basic conditions (LHMDS, Me3SiCl) also failed. Carbamates other than N-Alloc (N-Boc and methyl) were prepared, but also failed to undergo hydroxymethylation. Curiously, attempted hydroxymethylation of 12 using conditions developed by Fukuyama,9 as well as other conditions, only afforded enone 14 (Scheme 3).
Scheme 3.

Failed Hydroxymethylations of Benzazocane 12.
From the hydroxymethylation results for both FR900482- and mitomycin-based benzazocanes, we surmise that the electronics of the substrate play a crucial role in the success of this reaction. This observation is not without precedent for this family of molecules.
Illustrated in Scheme 4 are hydroxymethylation conditions used by Fukuyama9 and Rapoport7 in their total syntheses of FR900482, respectively. The major differences among these substrates that may affect the hydroxymethylation reaction are the substituents on the arene ring and the protecting group on the benzazocane nitrogen. From the examples shown in Scheme 4, as well as our own, we may conclude that the more electron-rich the substrate, the more prone it is toward elimination. Curiously, formation of an enoxysilane intermediate from an electron-rich arene appears to be considerably more difficult for application in the scandium(III)triflate-mediated hydroxymethylation reaction which we describe.
Scheme 4.

Electronic Effects on Hydroxymethylation.
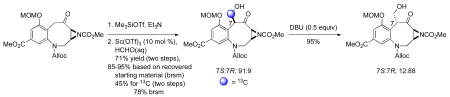
One powerful and unanticipated consequence of the Lewis acid-catalyzed Mukaiyama aldol approach is that it may be used for the construction of either the FR900482 or the MMC scaffold. Because the hydroxymethylation reaction is stereoselective and kinetically controlled, the initially formed 7S-stereoisomer of compound 11 matches the C-9 stereochemistry of MMC (1). On the other hand, equilibration of compound 11 to the thermodynamically favored 7R-isomer 16 under carefully controlled, base-catalyzed conditions should allow access to FR900482, potential late-stage biosynthetic intermediates and their congeners (Scheme 5). Unfortunately, all attempts to epimerize N-nosylsulfonamide 7S-7 gave the undesired enone 15 resulting from elimination. Successful epimerization of compound 11 (7S:7R, 91:9) to 7R-isomer 16 (7S:7R, 12:88), but not N-nosylsulfonamide 7, supports our assertion (vide infra) that electronics play a significant role in the chemistry of the benzylic position in these benzazocane architectures.
Scheme 5.

Attempts to epimerize hydroxymethylation products.
We attempted to develop a rationale to accommodate why the 7S-configuration predominates in successful hydroxymethylation reactions of FR900482-derived templates. Because reactions of medium-ring olefins are known to proceed from the periphery, the major, kinetic product of the hydroxymethylation reaction results from addition to the lowest energy conformation of the ring.15,16 The possible low-energy conformations of the intermediates in these reactions were determined and analyzed using computational methods (Figure 2).17 Conformational analysis of the likely intermediate in these reactions revealed that electrophilic addition of formaldehyde to the only available face of the electron-rich olefin in the lowest energy conformer should afford the 7S-isomer we observed. Furthermore, analysis of successful hydroxymethylation reactions from our group5,13 and results reported by Fukuyama9 suggest that electrophilic approach may proceed via a late transition state.18 In particular, the development of A1,3-strain19 between the incoming hydroxymethyl group and the aryl alkoxy group as the reaction proceeds may alter the energy barrier for each conformer in the reaction. This unfavorable steric interaction in the transition state may explain differences between experimental selectivities observed and predictions we can make on the basis of computational analysis.
Figure 2.

Comparative experimental and computational analysis of intermediates in the hydroxymethylation reaction.
In summary, we report an efficient method for the hydroxymethylation of benzazocanes en route to putative synthetic and late-stage biosynthetic intermediates of FR900482 and MMC. Electronics play a major role in this reaction, as N-Nvoc and N-PMB benzazocanes of FR900482 fail to undergo hydroxymethylation. Efforts to prepare putative biosynthetic intermediates of FR900482 and the mitomycins, as well as the asymmetric total synthesis of the mitomycins, are currently under investigation in these laboratories.
Supplementary Material
Complete experimental procedures, characterization data, and spectral data for new compounds, details of theoretical experiments, and crystallographic data in CIF format. This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgments
Dedicated to the memory of Professor Albert I. Meyers. This work was supported by the National Institutes of Health (CA51875). We also acknowledge a Graduate Student Fellowship from Eli Lilly (to D.A.G.). Mass spectra were obtained on instruments supported by the NIH Shared Instrumentation Grant GM49631.
References
- 1.(a) Boger DL, Wolkenberg SE. Chem Rev. 2002;102:2477. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]; (b) Rajski SR, Williams RM. Chem Rev. 1998;98:2723. doi: 10.1021/cr9800199. [DOI] [PubMed] [Google Scholar]
- 2.(a) Nelson SM, Ferguson LR, Denny WA. Cell & Chromosome. 2004;3:2. doi: 10.1186/1475-9268-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beckerbauer L, Tepe J, Eastman RA, Mixter PF, Williams RM, Reeves R. Chem Biol. 2002;9:427. doi: 10.1016/s1074-5521(02)00122-9. [DOI] [PubMed] [Google Scholar]; (c) Beckerbauer L, Tepe J, Cullison J, Reeves R, Williams RM. Chem Biol. 2000;7:805. doi: 10.1016/s1074-5521(00)00028-4. [DOI] [PubMed] [Google Scholar]
- 3.Bradner WT. Cancer Treat Rev. 2001;27:35. doi: 10.1053/ctrv.2000.0202. [DOI] [PubMed] [Google Scholar]
- 4.Ducept P, Gubler DA, Williams RM. Heterocycles. 2006;67:597. and references cited therein. [Google Scholar]
- 5.(a) Judd TC, Williams RM. J Org Chem. 2004;69:2825. doi: 10.1021/jo035828t. [DOI] [PubMed] [Google Scholar]; (b) Judd TC, Williams RM. Angew Chem Int Ed Engl. 2002;41:4683. doi: 10.1002/anie.200290015. [DOI] [PubMed] [Google Scholar]
- 6.Prepared in two steps (TASF-mediated desilylation, Dess-Martin oxidation) from the corresponding silyl ether 12a reported in ref. 4.
- 7.Paleo MR, Aurrecoechea N, Jung KY, Rapoport H. J Org Chem. 2003;68:130. doi: 10.1021/jo0206521. [DOI] [PubMed] [Google Scholar]
- 8.The benzylic, exocyclic olefin in Danishefsky’s synthesis of FR900482 was the product of a Heck cyclization: Schkeryantz JM, Danishefsky SJ. J Am Chem Soc. 1995;117:4722.
- 9.Suzuki M, Kambe M, Tokuyama H, Fukuyama T. J Org Chem. 2004;69:2831. doi: 10.1021/jo049862z. [DOI] [PubMed] [Google Scholar]
- 10.For example, 13CH2O solution, 20 wt. % in H2O, 99% 13C, CAS No. 3228-27-1, Aldrich Catalog No. 489417, costs US$266.00/gram.
- 11.(a) Kobayashi S, Hachiya I. J Org Chem. 1994;59:3590. [Google Scholar]; (b) Ishikawa S, Hamada T, Manabe K, Kobayashi S. J Am Chem Soc. 2004;126:12236. doi: 10.1021/ja047896i. [DOI] [PubMed] [Google Scholar]
- 12.X-ray data for compound 8 is included as Supporting Information.
- 13.Williams RM, Rollins SB, Judd TC. Tetrahedron. 2000;56:521. [Google Scholar]
- 14.Prepared in two steps (TASF-mediated desilylation, Dess-Martin oxidation) from the corresponding silyl ether 13b reported in ref. 4.
- 15.Still WC, Galynker I. Tetrahedron. 1981;37:3981. [Google Scholar]
- 16.This statement is valid if we assume that the energy barrier of reaction between both conformations of the olefin and the electrophile are nearly identical, according to Curtin-Hammett/Winstein-Holness kinetics; see: Seeman JI. J Chem Educ. 1986;63:42.
- 17.The lowest energy conformations of the eight-membered ring possessing a cis-olefin were determined by a Monte Carlo conformational search. These structures were refined using AM1, then HF/6-31G*. Lastly, the energies shown represent single-point HF/6-31G** calculations. Certain structural simplifications were made to simplify our qualitative computational experiments. These simplifications reduce computational time but do not, in our opinion, significantly alter the results. Further details and a rationale for the computational methods used are included in the Supporting Information.
- 18.Hammond GS. J Am Chem Soc. 1955;77:334. [Google Scholar]
- 19.Hoffmann RW. Chem Rev. 1989;89:1841. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete experimental procedures, characterization data, and spectral data for new compounds, details of theoretical experiments, and crystallographic data in CIF format. This material is available free of charge via the Internet at http://pubs.acs.org