

Abstract
Aspartoacylase catalyzes the deacetylation of N-acetylaspartic acid (NAA) to produce acetate and L-aspartate, and is the only brain enzyme that has been shown to effectively metabolize NAA. Although the exact role of this enzymatic reaction has not yet been completely elucidated, the metabolism of NAA appears to be necessary in the formation of myelin lipids and defects in this enzyme lead to Canavan disease, a fatal neurological disorder. The low catalytic activity and inherent instability observed with the Escherichia coli-expressed form of aspartoacylase suggested the need for a suitable eukaryotic expression system that would be capable of producing a fully functional, mature enzyme. Human aspartoacylase has now been successfully expressed in Pichia pastoris. While the expression yields are lower than in E. coli, the purified enzyme is significantly more stable. This enzyme form has the same substrate specificity, but is 150-fold more active than the E. coli-expressed enzyme. The molecular weight of the purified enzyme, measured by mass spectrometry, is higher than predicted, suggesting the presence of some posttranslational modifications. Deglycosylation of aspartoacylase or mutation at the glycosylation site causes decreased enzyme stability and diminished catalytic activity. A carbohydrate component has been removed and characterized by mass spectrometry. In addition to this carbohydrate moiety, the enzyme has also been shown to contain one zinc atom per subunit. Chelation studies to remove the zinc results in a reversible loss of catalytic activity, thus establishing aspartoacylase as a zinc metalloenzyme.
N-acetyl-L-aspartate (NAA)1 is the second most abundant amino acid in mammalian brain, yet its precise role has not been definitively established. The levels of NAA in the brain must be tightly regulated, with abnormally low levels detected in multiple sclerosis (1) and Alzheimer’s disease (2) and abnormally high levels measured in Canavan disease (3). A defect in the human aspA gene that encodes for Aspartoacylase, the enzyme responsible for metabolizing NAA, has been identified as the cause of Canavan disease (3), a fatal neurodegenerative disorder for which there is currently no effective treatment. Canavan disease is the only identified genetic disorder that is caused by a defect in the processing of a metabolite that is produced exclusively in the brain. The enzyme aspartoacylase plays a critical role in brain metabolism, the deacetylation of NAA to produce acetate and aspartate, and appears to be the only brain enzyme that can effectively metabolize NAA. Aspartoacylase is encoded by six exons, and over 50 different mutations including numerous deletions, missense mutations, and premature terminations, have been reported from Canavan patients with defects spanning all six exons (4). In most cases the missense mutations cause non-conservative amino acid substitutions, resulting in an altered enzyme that is either not expressed or is expressed but has little or no catalytic activity (5).
Aspartoacylase was first partially purified from rat brain (6), and was then subsequently purified to homogeneity from bovine brain (7) to allow the study the isolated enzyme. Immunostaining techniques had originally suggested that aspartoacylase may be a membrane-bound enzyme (7), and soluble preparations have been obtained in the presence of low levels of detergent. However, subsequent work has shown that immunoreactivity to aspartoacylase antibodies is seen in the cytosol but not in membrane fractions of rat brain tissue, demonstrating that aspartoacylase is not primarily membrane-associated (8) as was initially hypothesized. Recently studies have shown that aspartoacylase is distributed primarily in the oligodendrocytes, with antibodies that were generated from purified aspartoacylase becoming localized in these cells (9).
Based on the inactivation of aspartoacylase by diisopropylfluorophosphate, a classic inactivator of enzymes with an “active” serine, this hydrolytic enzyme was suggested to belong to an esterase family, and a catalytic serine, histidine, glutamate triad was postulated (10). However, alignment studies showed few detectable similarities between aspartoacylase and these esterases (11). Instead, sequence alignments with the zinc-carboxypeptidase family led to the suggestion that aspartoacylase is a zinc-dependent peptidase (12). The overall sequence identity between the aspartoacylases and the carboxypeptidases is 10% or less, however, the essential metal ion ligands and active site functional groups of the carboxypeptidases are conserved in the aspartoacylases. Divalent cations are reported to activate the enzyme, but the addition of these cations lead to only modest increases in catalytic activity (7). In contrast, assays conducted in the presence of metal chelators did not result in a loss in activity. From these results it was concluded that aspartoacylase is not a metalloenzyme (7). However, the metal ion content of purified aspartoacylase, and any correlation between metal ion content and catalytic activity, has not been examined. In addition to the unanswered question of metal ion regulation, the enzyme activity has been hypothesized to be regulated both by glycosylation and by phosphorylation/dephosphorylation (10). A putative N-glycosylation site (Asn117) contained in an asn-x-thr/ser recognition sequence has been suggested (13), and this site is located adjacent to one of the proposed histidine metal ion ligands.
We have now cloned and expressed human aspartoacylase in Pichia pastoris and have examined the properties of this highly purified and fully active enzyme. The metal ion and carbohydrate content were characterized and roles have been proposed for these entities in the functioning of this enzyme.
MATERIALS AND METHODS
Gene Cloning
The gene encoding for human aspartoacylase, aspA, was amplified by PCR with an attached C-terminal polyhistidine-tag. The amplified gene was ligated into the pPICZ A vector, using an EcoR I/Xho I insert. Plasmid DNA was transformed into XL10 Escherichia coli cells for plasmid amplification, with the cells plated onto low salt LB medium with 50 μg/mL of zeocin. The plasmid construct was linearized with Sac I to insert the gene into Pichia pastoris genomic DNA by homologous recombination. Several yeast strains, X-33, GS115 and KM71H, were examined for chemical transformation, and the KM71H strain was selected for its optimal expression.
Enzyme Expression
The enzyme was expressed in the KM71H P. pastoris cell line following the guidelines of the Easy Select™ Pichia Expression Kit manual (Invitrogen). Colonies were grown on yeast extract-peptone-dextrose-sorbitol plates (30°C, 2–3 days) with the antibiotic zeocin included for colony selection (100 μg/mL). Colonies picked from these plates were used to inoculate 10 mL of minimal glycerol media and the cells were grown at 30°C until reaching an OD600 of ~ 4. One liter of minimal glycerol media was inoculated with this cell culture and the cells were grown until an OD600 of ~ 4. The cells were centrifuged and resuspended in minimal methanol (1% methanol) for protein expression, and the media was supplemented with 1% methanol every 24 hours. After 3–4 days the cells were harvested and the cell paste was stored at −80°C prior to purification.
Enzyme Purification
The cell paste was resuspended into 20 mM potassium phosphate, pH 7.4, containing 500 mM NaCl, 20 mM imidazole, and 5% glycerol (buffer A) with 1 mM PMSF. The cells were lysed using a Bead Beater, and the soluble lysate was loaded onto a 5 mL HiTrap Chelating HP column (Amersham Biosciences) equilibrated with buffer A using an Äkta Explorer 100 chromatography system for immobilized metal affinity chromatography (IMAC) purification. The enzyme was eluted with a linear gradient with buffer A plus 500 mM imidazole. The active fractions from the purification were combined and dialyzed against buffer B (50 mM Hepes, pH 7.4, 1 mM DTT). The final polishing step was a 10 mL anion exchange Source 15Q (Amersham Biosciences) column, with the pure enzyme eluted by a linear gradient using buffer B containing 500 mM NaCl. The purity of the enzyme at each step was analysed by SDS-PAGE (Figure 1) and the protein concentration was determined by the Bradford assay. The active fractions were then dialyzed into 50 mM Hepes, pH 7.4, containing 100 mM NaCl and 1 mM DTT, for long term storage. Typical purifications yield 10 mg of highly purified aspartoacylase from 1 L of minimal methanol-cell culture with a specific activity of 10–15 units/mg.
Figure 1.
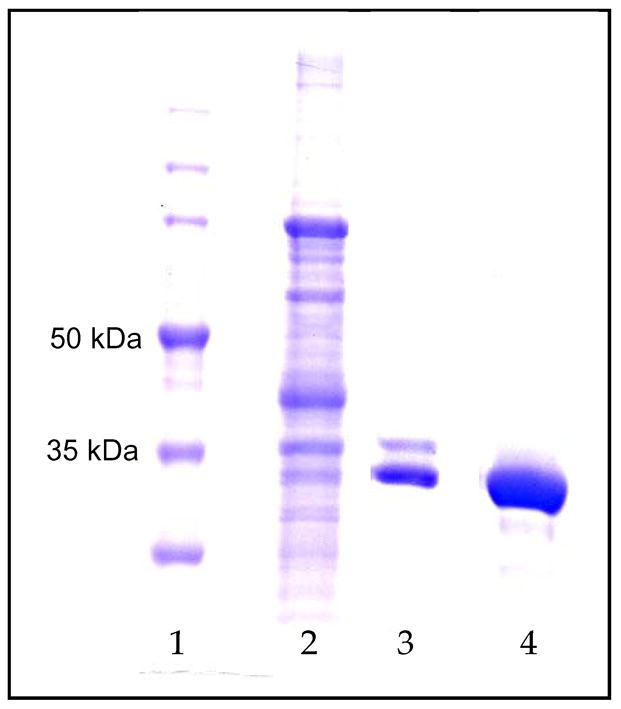
SDS-PAGE gel: Lane 1: M.W. markers; Lane 2: soluble P. pastoris cell lysate; Lane 3: IMAC purified aspartoacylase; Lane 4: purified aspartoacylase after ion exchange chromatography.
Kinetic Assay
The enzyme activity was assessed by a newly developed coupled assay in which the aspartic acid product from the aspartoacylase reaction is deaminated by L-aspartase (14). Assays were performed on a Cary 50 UV-visible spectrophotometer by monitoring the increasing concentration of fumarate at 240 nm (ε = 2.53 mM−1cm−1) in an assay buffer of 25 mM Hepes, pH 7.2, containing 1 mM Mg(OAc)2, 0.1 mM β-mercaptoethanol, varying levels of NAA, and excess L-aspartase, with the reaction initiated by the addition of aspartoacylase. During the protein purification the aspartoacylase activity was followed with the NAA concentration fixed at 0.75 mM.
Deglycosylation
The enzyme was deglycosylated under native conditions by treating 20 μg of aspartoacylase with 2500 units of protein N-glycosidase (PNGase F, New England Biosciences) in 0.5 M sodium phosphate buffer, pH 7.5, containing 1 mM DTT. ZnCl2 was added to the reaction mixture in equimolar ratio to the EDTA which is present in the storage buffer of PNGase F to eliminate any inhibitory effects of this metal chelator on enzyme activity. The reaction was incubated for 1 hour at 37°C, with a control reaction incubated under the same conditions in the absence of PNGase F.
For mass spectral studies aspartoacylase was deglycosylated under denaturing conditions by dissolving in 100 μL of a digestion buffer (100 mM sodium phosphate, pH 7.6, containing 5 mM DTT) and heating in boiling water for 10 minutes. After cooling at room temperature, 0.1% Triton X-100 and PNGase F (1μL, ~10U) were added, and the mixture was incubated at 37 °C for 17 hours. The digestion mixture was then boiled for 3 min to complete the deactivation of PNGase F and then applied to PGC-SPE column to remove salts and buffer.
The enzyme was also deglycosylated using two different glycosidases: neuraminidase (New England Biosciences) and endoglycosidase H (Endo H, New England Biosciences). For neuraminidase, 0.18 nmol of aspartoacylase and 100 units of neuraminidase were mixed in 50 mM sodium citrate buffer, pH 6. The reaction was incubated for up to 1 hr at 37 °C, with a control reaction incubated under the same conditions in the absence of neuraminidase. Aspartoacylase was also treated deglycosylated under native conditions by treating 20 μg of aspartoacylase with 2500 units of Endo H in 50 mM sodium citrate buffer, pH 5.5. The reaction was incubated for up to 8 hr at 37 °C, with a control reaction incubated under the same conditions in the absence of Endo H.
Carbohydrate Characterization
Digested oligosaccharides released by PNGase F were purified by PGC-SPE. The PGC cartridge was washed with nanopure water at a flow rate of 1 mL/min to remove salts and the glycans were eluted by washing with 10% acetonitrile in water (v/v), 20% acetonitrile in water (v/v), and then 40% acetonitrile in 0.05% TFA/water (v/v). Each fraction was collected and evaporated in vacuo prior to MALDI analysis. Mass spectra were recorded on an external source HiResMALDI (IonSpec Corporation, Irvine, CA) equipped with a 7.0 Tesla magnet and a pulsed YAG laser (266 nm). 2,5-Dihydroxybenzoic acid and 2,5-dihydroxyacetophenone were used as a matrix (5 mg/100 μL in ethanol). For the positive mode, one microliter of 0.01 M NaCl in acetonitrile/water solution (50:50) was added to the probe tip to enrich the Na+ concentration and produce primarily sodiated species. Matrix solution (1 μL) was followed to the MALDI probe. The sample was dried under a stream of air and subjected to mass spectrometric analysis.
Mutation at the N-glycosylation Site
The mutation N117Q was constructed to study the unglycosylated form of human aspartoacylase with the aim of understanding the role of the carbohydrate component. The QuickChange II Site Directed Mutagenesis Kit (Stratagene) was used to construct the mutant, and the mutation was confirmed by sequencing. The vector containing the mutated gene was linearized and inserted into the yeast genome of the KM71H cell line, and was then expressed and purified under the same conditions as the native enzyme.
Metal Content Determination
Metal chelation studies were conducted by dialyzing purified human aspartoacylase against 50 mM Hepes, pH 7.4, containing 100 mM NaCl, 1 mM DTT, 5% glycerol and 5 mM orthophenantroline (OP) at 4°C. Aliquots were removed at various times to assay for catalytic activity and metal ion content. As a control the enzyme was dialyzed into the same buffer but without OP. The metal content of the enzyme was determined before and after dialysis by inductively coupled plasma (ICP) emission spectroscopy. Different concentrations of ZnCl2 solutions were used to construct a standard curve to determine zinc levels within the enzyme. Zinc analysis was conducted at 213 nm, and the buffer solution was analysed as the background.
Quaternary Structure
Solutions of purified aspartoacylase (0.7 – 1.0 mg/ml) were examined by dynamic light scattering (DynaPro Titan DLS, Wyatt Technologies) at 20°C to determine the average particle size in solution and the corresponding molecular weight.
RESULTS
Aspartoacylase Stability
Aspartoacylase had previously been overexpressed to relatively high levels in E. coli, but was produced primarily as inclusion bodies by these cells (14). A refolding protocol was developed to obtain active and soluble enzyme, however this enzyme form is quite unstable, losing a significant fraction of its catalytic activity within 24 hours. The presence of a putative N-glycosylation site in human aspartoacylase (10) suggests that some type of posttranslational modification may be required to produce a stable and fully active enzyme. Since bacterial cells do not have the capability to correctly glycosylate mammalian proteins, human aspartoacylase has now been expressed in a Pichia pastoris cell line which can support many of these glycosylation reactions. The purified enzyme expressed in P. pastoris (Figure 1) is significantly more stable than the E. coli-expressed enzyme, losing less than 10% of its catalytic activity after two weeks when stored under conditions where the E. coli-expressed enzyme becomes completely inactivated within 24 hours. The P. pastoris expressed aspartoacylase shows sensitivity to oxidation overtime and will precipitate if not kept under the proper reducing conditions. However, the addition of DTT (1mM) is sufficient to reverse the precipitated state of the enzyme with no significant loss of activity.
Aspartoacylase Activity
The aspartoacylase expressed in P. pastoris is significantly more active as compared with the enzyme that was expressed in E. coli. Kinetic studies were conducted with both the natural substrate, NAA, and with N-trifluoroacetyl-L-aspartate (trifluoro-NAA), an alternative substrate with the highest activity (14), to examine the substrate specificity of these enzyme forms. Comparable Km values were measured between the E. coli and P. pastoris enzyme forms for each substrate, but a substantially higher kcat is observed for each with the P. pastoris-expressed enzyme (Table 1).
Table 1.
Kinetics of aspartoacylases expressed in E. coli and P. pastoris
| substratea | parameters | E. coli-expressed murine aspartoacylase | P. pastoris-expressed human aspartoacylase |
|---|---|---|---|
| X = –CH3 | kcat (s−1) | 0.083 ± 0.006 | 12. 7 ± 0.05 |
| Km (mM) | 0.36 ± 0.06 | 0.12 ± 0.03 | |
| X = –CF3 | kcat (s−1) | 1.2 ± 0.09 | 116 ± 13 |
| Km (mM) | 0.21 ± 0.04 | 0.15 ± 0.05 |
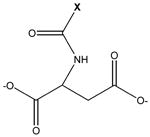
parent compound:
While the yeast-expressed enzyme has higher catalytic activity, some unusual kinetic properties have been revealed for this enzyme form. At low NAA concentrations aspartoacylase shows the classical sigmoidal behavior that is generally ascribed to subunit cooperativity (Figure 2). At higher NAA concentrations substantial substrate inhibition is observed, an indicator of the presence of an additional site on the enzyme where substrate binding can occur. Similar sigmoidal behavior and substrate inhibition are also seen with the more efficient trifluoro-NAA, in the same concentration range as with the physiological substrate.
Figure 2.
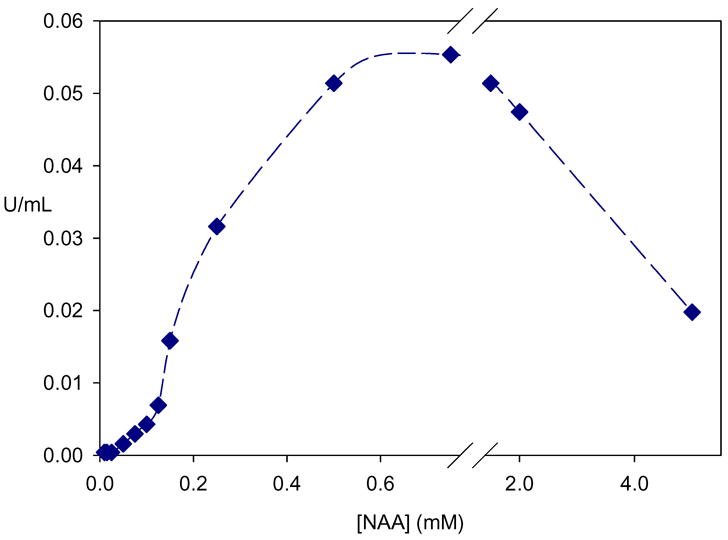
Kinetics of aspartoacylase. At low substrate concentration the enzyme shows sigmoidal behavior which is characteristic of cooperative kinetics. At higher substrate concentration pronounced substrate inhibition is observed.
Metal Ion Content of Aspartoacylase
Earlier studies had suggested that aspartoacylase could be a metalloenzyme. However, the addition of metal chelators, such as EDTA or OP, to purified aspartoacylase has no inhibitory effect on the catalytic activity of this enzyme. These results suggest that either metal ions are not required or that they are present and essential but are not easily removed. Subsequent dialysis of the enzyme against a buffer containing 5 mM OP leads to a substantial loss of catalytic activity in 24 to 72 hours (Table 2), demonstrating that more aggressive treatment is required for removal of metal ions from aspartoacylase. There is no loss of activity in the control experiment in which the enzyme is dialyzed against the same buffer but without added OP. Direct addition of low levels of ZnCl2 (<3 μM) to the OP-dialyzed enzyme leads to an increase in the zinc content of the enzyme and to a nearly complete recovery of catalytic activity. The addition of higher levels of zinc cause enzyme inhibition (Figure 3), and similar inhibition of activity is observed upon addition of zinc to the native enzyme that had not been treated with OP.
Table 2.
Effect of Metal Chelation on Aspartoacylase
| Enzyme form | Metal content a | S.A. (U/mg) | S.A. (percent) |
|---|---|---|---|
| Native enzyme | 1.30 ± 0.04 | 8.9 | 100 % |
| Treatment with OP for 48 hrs | 0.31 ± 0.02 | 2.0 | 22 % |
| Treatment with OP for 72 hrs | 0.23 ± 0.01 | 1.2 | 14 % |
atoms of Zn bound per 36 kDa
Figure 3.

Effect of added zinc on the kinetics of aspartoacylase after the enzyme was dialysed against 5 mM OP for 72 hours to remove metal ions.
In order to correlate these changes in catalytic activity with the metal ion content of aspartoacylase the enzyme was examined by ICP emission spectroscopy. The ICP measurements showed a metal content for the native enzyme of 1.3 atoms of zinc per 36 kDa (Table 2), which can be approximated to one zinc atom per enzyme subunit. Upon extensive dialysis against a buffer containing OP the metal content decreases to ~0.2 zinc atoms per subunit, and the catalytic activity shows a corresponding decrease to less than 20% of the native aspartoacylase. Addition of even low levels of ZnCl2 (0.1 μM) increases the zinc content of aspartoacylase to 0.42 ± 0.03 atoms per subunit, with a corresponding increase to 47% of the specific activity of the native enzyme.
Deglycosylation of Aspartoacylase
The higher stability and catalytic activity of the P. pastoris-expressed enzyme is hypothesized to be the result of the posttranslational glycosylations that can occur in this yeast strain. The presence of a carbohydrate component was established by treating aspartoacylase with a deglycosylating enzyme. The addition of PNGase F, an enzyme that selectively cleaves N-glycosidic bonds, to native aspartoacylase has no immediate effect on catalytic activity (Figure 4, curve A). However, after incubation in the presence of PNGase F for 1 hour at 37°C, more than 80% of the aspartoacylase activity is lost, and a substantial lag is also observed in the time course of the reaction (Figure 4, curve B). A control sample incubated under the same conditions but in the absence of PNGase F shows no change in catalytic activity (Figure 4, curve C), suggesting that the removal of a carbohydrate component is responsible for this loss of activity. To determine the effect of selective removal of carbohydrates on the activity of aspartoacylase the enzyme was treated with either neuraminidase or endoglycosidase H. Neuraminidase catalyses the hydrolysis of N-acetyl-neuraminic acid residues from glycoproteins and oligosaccharides, while Endo H cleaves within the chitobiose core of high mannose and some hybrid oligosaccarides from N-linked glycoproteins. In each case the enzyme showed no measurable loss of activity when incubated with high levels of these glycosidases for extended periods of time.
Figure 4.
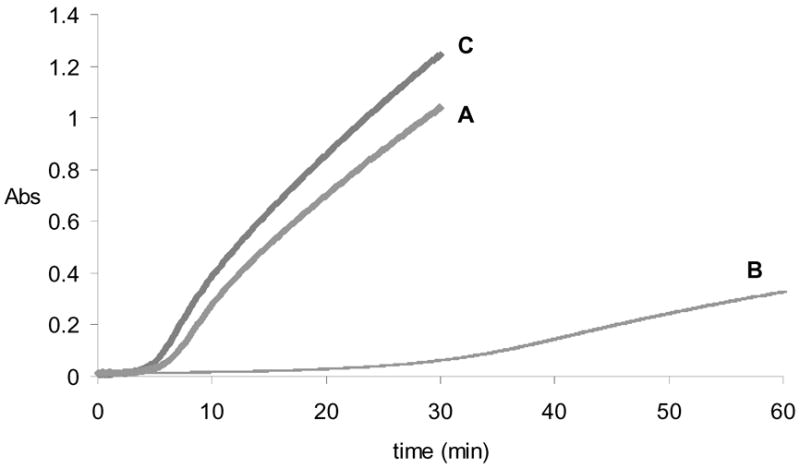
Time course of aspartoacylase activity after treatment with PNGase F to remove N-glycosyl groups. Curve A: rate of aspartic acid production after addition of PNGase F to aspartoacylase at time = 0. Curve B: rate of aspartic acid production one hour after addition of PNGase F, showing an extended lag and decreased catalytic activity. Curve C: control rate of aspartic acid production after one hour incubation of aspartoacylase in the absence of PNGase F.
Carbohydrate Content of Aspartoacylase
The nature of the carbohydrate component of native aspartoacylase was examined by subjecting the glycan released by PNGase F cleavage to mass spectral analysis. A MALDI-MS of this carbohydrate fraction shows one major component at m/z = 2544.836, and several minor components at much lower abundances in this mass range (Figure 5a). This mass corresponds to an oligosaccharide with six hexoses, six N-acetylhexosamines, and one N-acetylneuraminic acid (sialic acid), which gives an excellent fit to the quasimolecular ion [M-H+2Na]+ with a calculated mass of m/z of 2544.871. The putative structure of this oligosaccharide (OS1) is provided in Figure 5b. This oligosaccharide is monosialylated, which upon ionization with MALDI coordinates to a sodium atom and replaces the carboxylic acid proton with a second sodium. A number of lower mass components are also found, and are likely fragmentation products from the major peak. Minor components corresponding to less than 10% of the base peak were also found in this higher mass range. These peaks at m/z 2503.805 and m/z 2758.948 have putative structures which are also shown in Figure 5b (OS2 and OS3, respectively). All three of these carbohydrate components correspond to complex-type oligosaccharides.
Figure 5.
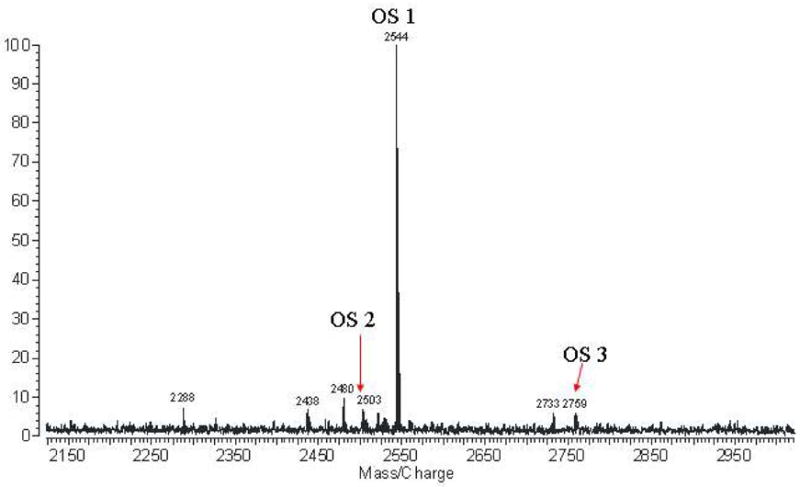
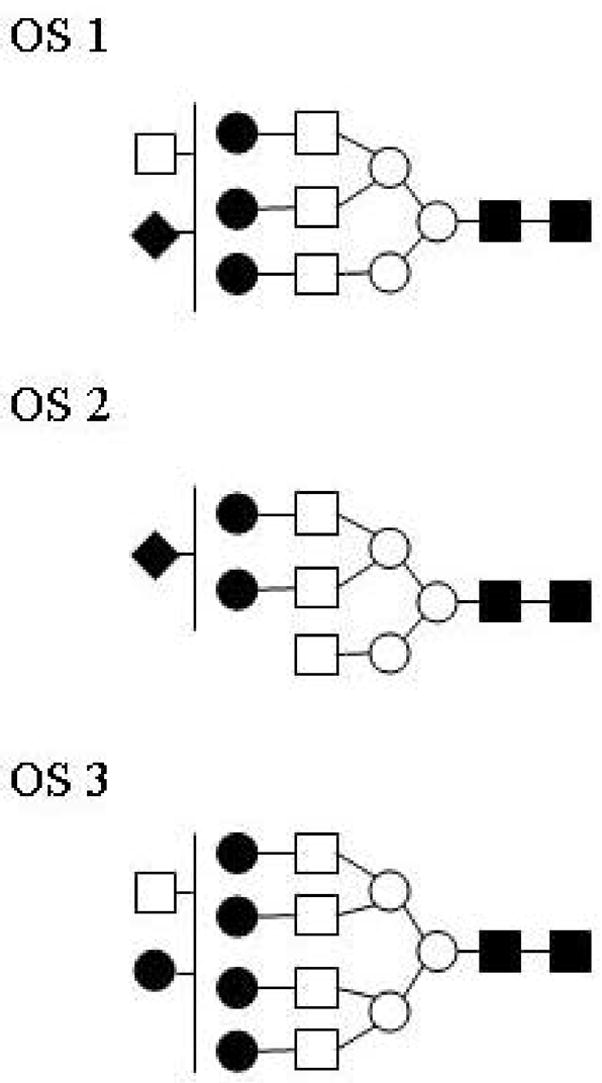
Characterization of the carbohydrate component of aspartoacylase. (a) MALDI-MS of the carbohydrate fraction of human aspartoacylase, with the spectrum expanded over the region from m/z = 2150 to 2950. (b) The putative structure of the major oligosaccharide (OS1) with best fit to the major peak at m/z = 2544.836 obtained with a parent ion containing six hexoses (○, ●), six N-acetylhexosamines (□, ■), one N-acetylneuraminic acid (◆), and two sodium ions (theoretical m/z = 2544.871). Other minor components (OS 2 and OS 3) are also found in this mass range, each corresponding to complex-type oligosaccharides with these proposed structures.
Mutation at the Putative N-glycosylation Site
Now that a single, major carbohydrate component has been identified, it remains to be established that Asn117 is the correct and unique N-glycosylation site. A conservative N117Q mutant was prepared to eliminate the possibility of glycosylation at this site while retaining the same side chain functionality. The fully purified N117Q mutant has a comparable catalytic behavior to that of the deglycosylated native enzyme in terms of reaction time course lag and catalytic activity. This unglycosylated N117Q enzyme also shows substrate inhibition (data not shown), with a maximum activity within the same range of NAA concentration as the native enzyme. Sigmoidal kinetics is still observed at low NAA concentrations with the N117Q mutant, however the mutant retains less than 10% of its catalytic activity when stored for one week at 4 °C whereas the native enzyme shows no loss of activity under these storage conditions.
To establish whether aspartoacylase contains only a single N-glycosylation site the N117Q mutant was treated with PNGase F under the same condition as the native enzyme. The activity of the unglycosylated mutant enzyme is unaffected by incubation with PNGase F at 37 °C. The calculated mass of the histidine-tagged N117Q mutant is 36,435, while the measured mass by MALDI-mass spectrometry is 36,412. This slight difference confirms that the mutation has eliminated glycosylation at this site, and also demonstrates the absence of any additional glycosylation sites in aspartoacylase. These results support the hypothesis that the absence of the N-glycosylated moiety at Asn117 is responsible for the substantial loss of activity and decrease in stability of aspartoacylase.
DISCUSSION
Aspartoacylase is an essential enzyme whose catalytic activity is necessary for proper brain functioning, however neither its precise role nor that of its substrate, NAA, have been definitively established. Aspartoacylase was expressed in a yeast strain that was selected for its capacity to carry out posttranslational glycosylations that are similar to those found in mammalian cells, while avoiding the hyperglycosylations seen in other yeast strains. The P. pastoris-expressed enzyme is stable and fully soluble, confirming a recent study that located aspartoacylase in the cytosol of oligodendrocytes cells (9) rather than as a membrane-bound enzyme as had been previously speculated (7).
Aspartoacylase Activity and Regulation by Substrate
Mammalian aspartoacylase had been previously expressed in bacterial cells, however both the activity and the stability of this enzyme form are dramatically impaired (14) since E. coli cannot do the necessary posttranslational modifications to produce a fully mature enzyme. A change in expression system seemed necessary and indeed the activity of human aspartoacylase expressed in yeast is 150-fold higher than the E. coli-expressed enzyme when examined with its physiological substrate NAA. In fact, the specific activity of the yeast-expressed human enzyme (S.A. ~ 10–15 U/mg) is within the same range as that reported for the mammalian enzyme isolated and purified from bovine brain (S.A. ~ 20 U/mg) (7).
The activity of human aspartoacylase is found to be exquisitely sensitive to the levels of its substrate, NAA. At low substrate concentrations (0.05 – 0.2 mM) the enzyme shows pronounced sigmoidal behavior that is indicative of positive cooperativity in substrate binding, suggesting the presence of at least a functionally active dimer. Dynamic light scattering (DLS) studies of the purified enzyme show a primary solution component with a hydrodynamic radius of a spherical particle that corresponds to a molecular mass of ~73 kDa, the expected mass of an aspartoacylase dimer. At substrate concentrations above 0.75 mM significant substrate inhibition is observed, which is indicative of additional NAA binding sites that adversely affect catalysis upon binding of the substrate at those sites. This same effect on enzyme activity is also observed with the alternative substrate trifluoro-NAA. The mechanism of regulation of NAA levels has not yet been established, but this brain metabolite must be maintained within fairly narrow limits. Low levels of NAA have been observed in cases of multiple sclerosis, Alzheimer’s disease and epilepsy (1,15–17), while high levels of NAA are correlated with Canavan disease (3). The cooperative kinetics and substrate inhibition shown by aspartoacylase are likely involved in the regulation of NAA metabolism in the brain.
Carbohydrate Content and its Structural Role
Glycoproteins with N-linked carbohydrates have a core structure which typically consists of β-(1–>4) linked N-acetylglucosamines (18) coupled to the amide side chain of an asparagine that is located in a consensus N-glycosylation sequence. This core structure is then extended through a mannose branch and additional mannose units (Scheme 1), with each mannose providing an opportunity for further branching through extension with additional mannoses (19). Each mannose branch can then be terminated by the addition of an N-acetylglucosamine or a D-galactosyl-N-acetylglucosamine unit in various combinations along with further structural variations (13,20,21). After accounting for this core structure (Scheme 1), linking the remaining four N-acetylglucosamines, three hexoses (mannose or galactose), and one N-acetylnuraminic acid of the fitted oligosaccharide will complete the glycan structure. The putative structure of the major oligosaccharide component of aspartoacylase is given in Figure 5b, along with the proposed structures of two minor carbohydrate components.
Scheme 1.
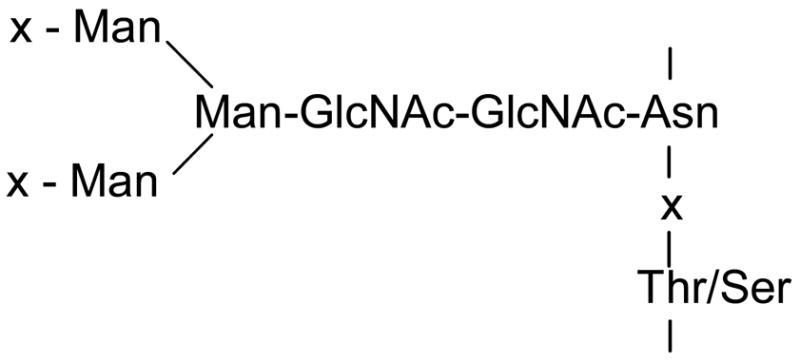
Typical Core N-linked Oligosaccharide Structure
Aspartoacylase loses a substantial fraction (80%) of its catalytic activity upon removal of the carbohydrate at the N-glycosylation site by treatment with PNGase F under native conditions. The N117Q mutant, in which this site is altered to prevent glycosylation, has a similar activity (1–2 U/mg) as the deglycosylated enzyme and also shows low stability. ICP measurement of the Zn2+ content gives 1.1 ± 0.06 metal atoms per monomer, showing that the absence of the carbohydrate portion does not affect metal ion binding. Thus removal of the carbohydrate moiety, either by PNGase F treatment or through mutagenesis, leads to a less active and unstable enzyme. Selective removal of portions of the carbohydrate had no effective on catalytic activity, suggesting that the presence of some glycan at this position is more important than the exact carbohydrate composition. Exposure of a hydrophobic region of the enzyme that is normally shielded by glycosylation could lead to protein aggregation and precipitation. A DLS analysis of the N117Q mutant gives a hydrodynamic radius for the major component consistent with the molecular mass of an aspartoacylase dimer, but also shows a significant fraction of this mutated protein in higher aggregation states. These results support the idea that the low activity and poor stability of the E. coli-expressed enzyme, which is primarily expressed in inclusion bodies (14) is related to the inability of this bacterial host to carry out the posttranslational glycosylation of aspartoacylase.
A similar loss of activity has been observed in the case of a related enzyme, glutamate carboxypeptidase II, which is expressed in the central nervous system. The enzyme catalyzes the cleavage of the neurotransmitter N-acetyl-L-aspartyl-L-glutamate releasing glutamate and NAA (22). This enzyme is N-glycosylated at ten different sites and mutation of any of these glycosylation sites drastically impairs the activity of the enzyme. In addition, when the native enzyme was treated with PNGase F to remove all of the carbohydrates the resulting enzyme is unstable and precipitates. The same behavior is observed in the case of native aspartoacylase, with the deglycosylated enzyme undergoing a slow aggregation over several days when stored at 4°C.
Metal Ion Content and its Role in Catalysis
The deacetylation reaction catalyzed by aspartoacylase is similar to that of the broader spectrum aminoacylase family. However, previous studies have proposed a relationship between aspartoacylase and the serine proteases. Later sequence alignments suggested that aspartoacylase might be a member of a zinc metalloenzyme family (12). The experiments reported here support this latter hypothesis by showing that human aspartoacylase contains one bound Zn2+ ion per enzyme subunit, and that the presence of this metal ion is essential. Removal of the zinc atom results in a loss of catalytic activity, which can be restored by replacement with added zinc. The putative zinc ligands, His-21, Glu-24, and His-116, are fully conserved in the aspartoacylase family and can be aligned with the well-established zinc ligands in the carboxypeptidase family. A more complete analysis of the primary sequence places the aspartoacylases in the carboxypeptidase H subfamily (23), based on the conserved consensus sequence –HGNE- which contains two of the zinc binding ligands. By analogy with the carboxypeptidases, this zinc ion in aspartoacylase is proposed to function in binding and promoting the ionization of a water molecule to generate an active site hydroxide nucleophile. A dual functional carboxypeptidase/aminoacylase has recently been characterized from Pyrococcus horikoshii (24), suggesting that carboxypeptidases and aminoacylases have a common origin and catalyzes a similar hydrolytic reaction mechanism.
Acknowledgments
The murine and human AspA genes were generously provided by Dr. M. A. Namboodiri (Uniformed Military Services University; Bethesda, MD). Protein mass spectroscopic studies were conducted at the Ohio State University Mass Spectrometry facility under the direction of Dr. Kari Green-Church. The inductively coupled plasma spectroscopic studies were conducted with the assistance of Dr. Pannee Burckel in the Instrumentation Center at the University of Toledo.
Footnotes
Supported by a grant to R.E.V. from the National Institutes of Health (NS45664).
Abbreviations: DLS, dynamic light scattering; DTT, dithiothreitol; endo H, endoglycosidase H; ICP, inductively coupled plasma; IMAC, immobilized metal affinity chromatography; MALDI, matrix assisted laser desorption ionization; NAA, N-acetyl-L-aspartate; OP, orthophenantroline; PGC-SPE, porous graphitized column-solid phase extraction; PMSF, Phenylmethanesulfonyl-fluoride; PNGase F, protein N-glycosidase; trifluoro-NAA, N-trifluoroacetyl-L-aspartate.
References
- 1.Richards TL. Proton MR spectroscopy in multiple sclerosis: value in establishing diagnosis, monitoring progression, and evaluating therapy. Am J Neuroradiol. 1991;157:1073–1078. doi: 10.2214/ajr.157.5.1927795. [DOI] [PubMed] [Google Scholar]
- 2.Klunk WE, Moossy J, McClure RJ, Pettegrew JW. N-acetyl-L-aspartate and other amino acid metabolites in Alzheimer’s disease brain. Neurology. 1992;42:1578–1585. doi: 10.1212/wnl.42.8.1578. [DOI] [PubMed] [Google Scholar]
- 3.Matalon R, Michals-Matalon K, Sebesta M, Deanching M, Gashkoff P, Casanova J. Aspartoacylase Deficiency and N-acetylaspartic aciduria in Patients with Canavan Disease. Am J Med Genet. 1988;29:463–471. doi: 10.1002/ajmg.1320290234. [DOI] [PubMed] [Google Scholar]
- 4.Zeng BJ, Wang ZH, Ribeiro LA, Leone P, De Gasperi R, Kim SJ, Raghavan S, Ong E, Pastores GM, Kolodny EH. Identification and characterization of novel mutations of the aspartoacylase gene in non-Jewish patients with Canavan disease. J Inher Metabol Dis. 2002;25:557–570. doi: 10.1023/a:1022091223498. [DOI] [PubMed] [Google Scholar]
- 5.Matalon R, Michals-Matalon K. Recent Advances in Canavan Disease. Adv Pediat. 1999;46:493–506. [PubMed] [Google Scholar]
- 6.D’Adamo AF, Peisach J, Manner G, Weiler CT. N-Acetyl-Aspartate Amidohydrolase: Purification and Properties. J Neurochem. 1977;28:739–744. doi: 10.1111/j.1471-4159.1977.tb10621.x. [DOI] [PubMed] [Google Scholar]
- 7.Kaul R, Casanova J, Johnson AB, Tang P, Matalon R. Purification, Characterization, and Localization of Aspartoacylase from Bovine Brain. J Neurochem. 1991;56:129–135. doi: 10.1111/j.1471-4159.1991.tb02571.x. [DOI] [PubMed] [Google Scholar]
- 8.Klugmann M, Symes CW, Klaussner BK, Leichtlein CB, Serikawa T, Young D, During MJ. Identification and distribution of aspartoacylase in the postnatal rat brain. Neuroreport. 2003;14:1837–1840. doi: 10.1097/00001756-200310060-00016. [DOI] [PubMed] [Google Scholar]
- 9.Madhavarao CN, Moffett JR, Moore RA, Viola RE, Namboodiri MA, Jacobowitz DM. Immunohistochemical Localization of Aspartoacylase in the Rat Central Nervous System. J Compar Neurol. 2004;472:318–329. doi: 10.1002/cne.20080. [DOI] [PubMed] [Google Scholar]
- 10.Kaul R, Gao GP, Balamurugan K, Matalon R. Cloning of the human aspartoacylase cDNA and a common missense mutation in Canavan disease. Nature Genet. 1993;5:118–123. doi: 10.1038/ng1093-118. [DOI] [PubMed] [Google Scholar]
- 11.Cygler M, Schrag JD, Sussman JL, Harel M, Silman I, Gentry MK, Doctor BP. Relationship between sequence conservation and three-dimensional structure in a large family of esterases, lipases, and related proteins. Protein Sci. 1993;2:366–382. doi: 10.1002/pro.5560020309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makarova KS, Grishin NV. The Zn-peptidase Superfamily: Functional Convergence After Evolutionary Divergence. J Molec Biol. 1999;292:11–17. doi: 10.1006/jmbi.1999.3059. [DOI] [PubMed] [Google Scholar]
- 13.An HJ, Peavy TR, Hedrick JL, Lebrilla CB. Determination of N-Glycosylation Sites and Site Heterogeneity in Glycoproteins. Anal Chem. 2003;75:5628–5637. doi: 10.1021/ac034414x. [DOI] [PubMed] [Google Scholar]
- 14.Moore RA, Le Coq J, Faehnle CR, Viola RE. Purification and Preliminary Characterization of Brain Aspartoacylase. Arch Biochem Biophys. 2003;413:1–8. doi: 10.1016/s0003-9861(03)00055-9. [DOI] [PubMed] [Google Scholar]
- 15.Hugg JW, Laxer KD, Matson GB, Maudsley AA, Weiner MW. Neuron loss localizes human temporal lobe epilepsy by in vivo proton magnetic resonance. Ann Neurol. 1993;34:788–794. doi: 10.1002/ana.410340606. [DOI] [PubMed] [Google Scholar]
- 16.Plaitakis A, Constantakakis E. Altered metabolism of excitatory amino acids, N-acetyl-aspartate and N-acetyl-aspartyl-glutamate in myotrophic lateral sclerosis. Brain Res Bull. 1993;30:381–386. doi: 10.1016/0361-9230(93)90269-h. [DOI] [PubMed] [Google Scholar]
- 17.Klunk WE, Panchalingam K, Moossy J, McClure RJ, Pettegrew JW. N-acetyl-L-aspartate and other amino acid metabolites in Alzheimer’s disease brain: A preliminary proton nuclear magnetic resonance study. Neurology. 1992;42:1578–1585. doi: 10.1212/wnl.42.8.1578. [DOI] [PubMed] [Google Scholar]
- 18.Lee YC, Scocca JR. A Common Structural Unit in Asparagine-Oligosaccharides of Several Glycoproteins from Different Sources. J Biol Chem. 1972;247:5753–5758. [PubMed] [Google Scholar]
- 19.Trimble RB, Atkinson PH, Tschopp JF, Townsend RR, Maley F. Structure of oligosaccharides on Saccharomyces SUC2 invertase secreted by the methylotrophic yeast Pichia pastoris. J Biol Chem. 1991;266:22807–22817. [PubMed] [Google Scholar]
- 20.Kuster B, Wheeler SF, Hunter AP, Dwek RA, Harvey DJ. Sequencing of N-Linked Oligosaccharides Directly from Protein Gels: In-Gel Deglycosylation Followed by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry and Normal-Phase High-Performance Liquid Chromatography. Anal Biochem. 1997;250:82–101. doi: 10.1006/abio.1997.2199. [DOI] [PubMed] [Google Scholar]
- 21.Charlwood J, Skehel JM, Camilleri P. Analysis of N-linked Oligosaccharides Released from Glycoproteins Separated by Two-Dimensional Gel Electrophoresis. Anal Biochem. 2000;284:49–59. doi: 10.1006/abio.2000.4687. [DOI] [PubMed] [Google Scholar]
- 22.Barinka C, Sacha P, Sklenar J, Man P, Bezouska K, Slusher BS, Konvalinka J. Identification of the N-glycosylation sites on glutamate carboxypeptidase II necessary for proteolytic activity. Protein Sci. 2004;13:1627–1635. doi: 10.1110/ps.04622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rawlings ND, Barrett AJ. Evolutionary families of metallopeptidases. Methods Enzymol. 1995;248:183–228. doi: 10.1016/0076-6879(95)48015-3. [DOI] [PubMed] [Google Scholar]
- 24.Ishikawa K, Ishida H, Matsui I, Kawarabayasi Y, Kikuchi H. Novel Bifunctional Hyperthermostable Carboxypeptidase/Aminoacylase from Pyrococcus horikoshii OT3. Appl Environ Microbiol. 2001;67:673–679. doi: 10.1128/AEM.67.2.673-679.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]