

Abstract
The α-helical coiled-coil motif serves as a robust scaffold for incorporating electron-transfer functionality into synthetic metalloproteins. These structures consist of a right-handed supercoiling of two or more α-helices that are formed by the self-assembly of individual polypeptide chains whose sequences contain a repeating pattern of hydrophobic and hydrophilic residues. Early work from our group attached abiotic ruthenium-based redox sites to the most surface-exposed positions of two stranded coiled-coils and used electron-pulse radiolysis to study both intra and inter-molecular electron-transfer reactions in these systems. Later work used smaller metallopeptides to investigate the effects of conformational gating within electrostatic peptide-protein complexes. We have recently designed the C16C19-GGY peptide which contains cysteine residues located at both the “a” and “d” positions of its third heptad repeat in order to construct a native-like metal-binding domain within its hydrophobic core. It was shown that the binding of both Cd(II) and Cu(I) ions induces the peptide to undergo a conformational change from a disordered random-coil to a metal-bridged coiled-coil. However, whereas the Cd(II) protein exists as a two-stranded coiled-coil, the Cu(I) derivative exists as a four-stranded coiled-coil. Upon the incorporation of other metal ions, metal-bridged peptide dimers, tetramers, and hexamers are formed. The Cu(I)-protein is of particular interest as it exhibits a long-lived (microsecond) room-temperature luminescence at 600 nm. The luminophore in this protein is thought to be a multinuclear CuI4Cys4(N/O)4 cage complex which can be quenched by exogenous electron-acceptors in solution, as shown by emission lifetime and transient absorption experiments. It is anticipated that further investigation into these systems will contribute to the expanding effort of bioinorganic chemists to prepare new kinds of functionally-active synthetic metalloproteins.
Introduction
Metalloproteins comprise approximately one-third of all structurally-characterized proteins and perform such important biochemical functions as the catalytic transformation of chemical substrates, the facilitation of redox-dependent chemical reactions, and the mediation of oxygen transport and storage. The diversity of chemical functions performed by naturally-occurring metalloproteins has inspired recent work towards the design of artificial analogs which might possess activities that mimic, enhance, or even replace those now performed by native systems.1–7 However, this goal now presents a formidable challenge to the scientific community as it requires not only the ability to construct well-defined protein structures that can bind specific kinds of metal ions, but also the ability to do so in ways that exploit the inherent chemical reactivity of these ions in order to incorporate tailorable chemical functions into these systems.
In general, two complementary approaches have been taken to synthesize new types of chemically-functional metalloproteins. The “top down” approach refers to the re-engineering of native proteins in ways that enable them to perform new chemical tasks which can be significantly different from their inherent biological functionality.2,5,8,9 Alternatively, the “bottom-up” strategy for designing functional metalloproteins refers to the rational assembly of discrete peptidic and inorganic building blocks in ways that can create synthetic protein structures whose inorganic components can perform interesting chemical reactions. To date, most work in the field of “bottom-up” metalloprotein design has concentrated on the essential, albeit preliminary, task of developing native-like peptide scaffolds that can bind specific metal ions. Notable examples of these include the designed metalloprotein in which two self-associated helix-loop-helix units are used to bind two separate iron atoms,10–13 the bridged Ni(II)-(μ2-SCys)-[Fe4S4] protein in which the helix-loop-helix motif creates a bridged metal binding site,14 several series of cysteine-containing helical bundle proteins,15–28 and the well-studied four-helix bundle heme proteins.29
Pecoraro and co-workers,15–23 have been conducting an elegant series of studies to examine the metal-binding properties of an important family of coiled-coil peptides prepared by subtle modifications of the parent peptide known as “TRI”. TRI has the sequence Ac-G(LKALEEK)4G-NH2 which places hydrophobic leucine residues at each of the heptad “a” and “d” positions of an α-helical coiled-coil (vida infra) and was found to exist predominately as a three-stranded coiled-coil at pH > 7. Importantly, it was observed that the single replacement of one leucine residue with a cysteine at either position 9 or 12 of the sequence created a metal binding site having an affinity for Hg(II) and Cd(II), and that the resulting metallopeptides existed as three-stranded coiled-coils containing a very unusual three-coordinate metal center. This unexpected assembly process was seen to occur even in the case of a truncated peptide which exists as a largely disordered coiled-coil in the absence of metal ion. The results suggested that an important relationship exists between the conformational preferences of the apopeptide backbone and the coordination chemistry of the incorporated metal ion. Careful thermodynamic studies confirmed the existence of this relationship by showing a linear free energy relationship between the self-association affinities of the TRI peptides and their ability to bind Hg(II) and Cd(II) ions in trigonal geometries.15 These studies proved that within the TRI family of metalloproteins, the conformational preferences of the protein dictate the coordination geometry of the incorporated metal ion.
Building upon recent successes in the “bottom-up” design of new metal-binding proteins, several workers have now begun to address the significant challenge of incorporating chemical functionality into these systems.7 In a notable effort, De Grado and co-workers recently prepared a computationally designed metalloprotein which has a diiron cluster, similar to those found in a variety of naturally-occurring hydrolytic enzymes, positioned in close proximity to a suitable substrate-binding domain.10–13 Significantly, this protein called DFtet was shown to catalyze the two-electron oxidation of 4-aminophenol to the corresponding quinone monoamine with a somewhat modest but distinct value of kcat/KM = 1500 M−1 min−1.13 In related efforts, workers have successfully constructed chimeric metalloproteins which contain both the helix-turn-helix DNA binding domain and either the metal-binding loop of calmodulin30,31 or an unnatural amino acid based on triazacyclononane32 in order to bind hydrolytic metal ions. Such systems have been shown to display sequence-specific nuclease activity. Most recently, Dutton and co-workers have introduced the design of new amphiphilic heme protein maquettes (minimalistic protein models) in order to facilitate their incorporation of hydrophobic cofactors in a manner that mimics native membrane proteins.33–35 Thus, significant achievements in functional metalloprotein design have begun to appear through the simultaneous incorporation of both metal-binding and substrate-binding centers into a single protein environment. It is anticipated that future study of these systems will provide considerable insight into how protein environments can be used to regulate the chemical reactivity of their incorporated transition metal ions.
Aside from catalytic functionality, redox activity is another type of chemical property that can be incorporated into designed metalloproteins. Indeed, a long-standing effort to understand how protein structures can provide pathways for long-range donor-acceptor interactions has led to the development of many peptide-based electron-transfer reagents built from the “bottom-up”. Early work in this field largely concentrated on the attachment of exogenous abiotic redox centers, such as ruthenium polypyridyl complexes, to polypeptide spacers possessing defined secondary structures such as proline helices and α-helices. Such work has been the focus of a previous review.36 However, more recent work in this field has resulted in the design of systems which can indeed be classified as examples of functionally-active synthetic proteins having well-defined tertiary and/or quarternary structures, some with more native-like redox cofactors.37–42 In a notable example of such work, Haehnel and co-workers have been studying a family of modular four-helix, template assembled synthetic proteins (TASP’s) which are amenable to the incorporation of both metalloporphyrin43 and chlorophyll44 cofactors. Recent work by this group also involved the combinatorial assembly of Cu(II)-binding sites which led to the creation of a mixed-valent dinuclear copper site reminiscent of CuA.45 The work described below will review our efforts to utilize the α-helical coiled-coil motif as a robust scaffold upon which inorganic redox centers can be either attached to their solvent-exposed surfaces, or bound to native-like binding sites designed within their hydrophobic cores.
Experimental Section
Materials
The F-moc-protected L-amino acid derivatives, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), piperidine, diisopropylcarbodiimide, and anhydrous N-hydroxybenzotriazole (HOBt) were purchased from Peptides International Inc. (Louisville, KY). The reagent tetrakis(acetonitrile)copper(I) hexafluorophosphate was purchased from the Sigma-Aldrich Company (St. Louis, MO). All reagents were used as received.
General Methods
UV-vis, circular dichroism and reverse-phase HPLC analyses were performed as previously described46 except that either a semipreparative Vydac reversed-phase 218TP™ C18 column (10 μM particle size, 10 × 250 mm) or a preparative Vydac 218T™ C18 column (10 μM particle size, 22 × 250 mm) was used. Static luminescence spectra were obtained with a single photon counting spectrofluorimeter from Edinburgh Analytical Instruments (FL/FS 900) and emission lifetime measurements were carried out using a nitrogen broadband dye laser (2–3 nm fwhm) using fundamental nitrogen excitation (337 nm) or BPBD dye (357 nm) as previously decribed.47 The emission of the Cu(I)-C16C19-GGY adduct was monitored at 600 nm and performed in argon-saturated solutions.
Synthesis of the C16C19-GGY Peptide
The 32-residue peptide C16C19-GGY having the sequence Ac-K(IEALEGK)2(CEACEGK)(IEALEGK)-GGY-NH2 was prepared and purified by reverse-phase HPLC as previously described.48 The GGY tag was attached to the peptide to allow determination of the peptide concentration by measuring the absorption of the tyrosine residue at 275 nm (ε = 1450 M−1cm −1).49
High Performance Size Exclusion Chromatography (HPSEC)
HPSEC experiments were performed using a Superdex 75 Biotech column (Amersham Biosciences) connected to a Waters Model 515 HPLC pump equipped with Waters Model 996 diode array detector. The peptide samples were eluted using 0.1 M KCl/0.05 M KH2PO4, with a 0.2–0.4 ml/min flow rate and monitored at 230 nm and their behavior was compared against those of suitable peptide standards.50
Results and Discussion
Synthetic Electron-transfer Metallproteins Based on α-helical Coiled-Coils
Our group has been preparing synthetic metalloproteins based on α-helical coiled-coils.27,28,46,48,51–53 These ubiquitous structures comprise an important dimerization domain of native proteins and are formed by the non-covalent self-assembly of α-helices to create a left-handed supercoil. Such structures are stabilized by a specific “knobs into holes” packing of regularly spaced hydrophobic residues belonging to each strand of the coiled-coil. It has been found that synthetic coiled-coils can be prepared from amino acid sequences based on a seven residue heptad repeat, (abcdefg)n, in which hydrophobic amino acids occupy positions “a” and “d” of the heptad, hydrophilic residues fill positions “b”, “c”, and “f”, and oppositely-charged residues may occupy positions “e” and “g” in order to form stabilizing inter-chain salt bridges (Figure 1).54–57
Figure 1.
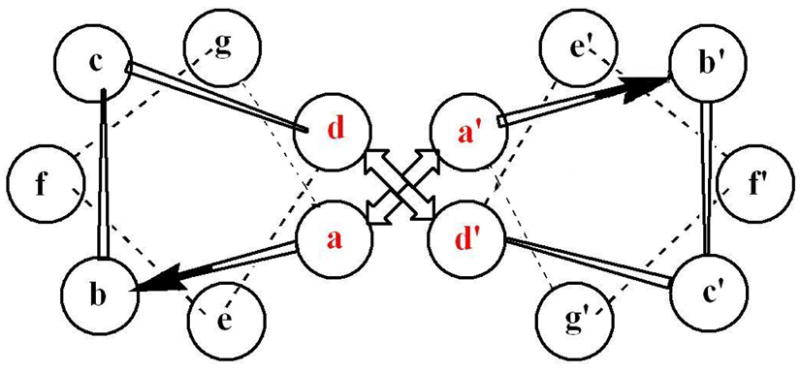
Helical wheel diagram depicting one heptad repeat of a two-stranded α-helical coiled-coil. The hydrophobic “a” and “d” positions are shown in red.
In early work, our group prepared a 30-residue polypeptide called H21(30-mer) whose sequence was designed to form two-stranded coiled-coils.46 An important feature of this Ac-K(IEALEGK)2(IEALEHK)(IEALEGK)G-NH2 H21(30-mer) sequence is that it places a single histidine residue at position 21 to provide a convenient metal-binding site at the most highly solvent-exposed “f” position of the third heptad repeat. Circular dichroism (CD) spectroscopy showed that this peptide did indeed exist as a α-helical coiled-coil in which the association process could be fit to a two-state monomer-dimer equilibrium having a value of Kd = 1.5 ± 0.4 μM and a maximum ellipticity of 69%. Treatment of the peptide with either [Ru(NH3)5(OH2)]2+ or [Ru(trpy)(bpy)(OH2)]2+ produced the corresponding metallated homodimers in which ruthenium compounds were coordinated to each of the two H21 sites of the coiled-coil. The desired ET heterodimer (Figure 2) was then prepared in a statistical distribution with the two metallated homodimers by heating an equimolar solution of the two metallated peptides to 60° C and cooling the mixture back to room temperature. These peptides were shown by a combination of analytical ultracentrifugation, SDS-PAGE, and size exclusion chromatography to exist as two-stranded coiled-coils. Importantly, EPR spin-labeling experiments were used to provide a measure of the inter-chain Cα-Cα distance of 13.5 ± 0.9 Å at position 21 of the coiled-coil, which is nearly identical to those observed for the isostructural family of bZip proteins. These results enabled computer modeling studies to estimate that the two metal centers in the ET heterodimer were separated by a metal-to-metal distance of ca. 24 Å across the non-covalent peptide interface. Oxidative pulse radioloysis experiments were used to study intramolecular electron-transfer reactions occurring from the RuII(NH3)5-H21 donor to the RuIII(trpy)(bpy)-H21′ acceptor located across the non-covalent peptide interface. The rate constant for the electron-transfer reaction was found to be kET = 380 s−1 which was independent of peptide concentration. Significantly, these experiments showed that the observed rate constant is consistent with the distance-rate behavior observed in both native58 and modified59 protein systems, and that intramolecular ET can indeed occur over long distances in this designed metalloprotein across a non-covalent peptide-peptide interface. The designed H21 metallo-heterodimer is therefore a viable model system for natural electron-transfer proteins.
Figure 2.
Computer generated model of the synthetic electron-transfer protein formed by attaching ruthenium-based redox sites to histidine residues occupying the solvent-exposed “f” positions of a two-stranded coiled-coil.
A Pathways60 analysis was conducted for the H21(30-mer) system which identified its primary coupling path (metal-to-metal) to consist of 22 covalent bonds and a critical inter-helix through-space jump of 3 Å between the Cβ of Lys22 and Cγ of Ile23′ of the next heptad repeat.61 We therefore prepared a related peptide called H18(30-mer) whose metal-coordinating histidine residues were now located at position 18 of the sequence occupying the heptad “c” positions. It was hypothesized that this change should decrease the length of the putative ET tunneling pathway by 3 covalent bonds and increase the observed rate of intra-protein ET from the relatively slow rate observed for H21(30-mer). However, pulse radiolysis experiments showed no evidence for intra-protein ET occurring in this system, and only a concentration-dependent inter-protein ET event was seen having a second-order rate constant of kET(inter) = 6 × 108 M−1 s−1. The reason for this disappointing result was explained by CD experiments which showed that placing the hydrophilic metal complexes closer to the non-covalent interface of this peptide resulted in destabilizing its coiled-coil structure. This likely led to an increased metal-metal distance in the H18(30-mer) metallopeptide to make its rate of intramolecular ET no longer competitive with that of the competing intermolecular reaction occurring between different proteins in the pulse radiolysis experiment.
Electron-transfer Along the Covalent Backbone of α-helices and Coiled-coils
Previous results by Fox and co-workers reported that the permanent dipole moment of αhelices can be used to modulate the rates of photoinduced electron-transfer occurring between organic donors and acceptors.62–64 Thus, our group designed a new peptide-based electron-transfer system to determine if this effect can also be used to regulate ET rates in metalloproteins.48,53 Two 30-residue coiled-coil apopeptides were synthesized having the following sequences: (I) Ac-K-(IEALEGK)(ICALEGK)(IEALEHK)(IEALEGK)-G-NH2, and (II) Ac-K-(IEALEGK)(IHALEGK)-(IEALECK)(IEALEGK)-G-NH2 (Figure 3). Ruthenium-based electron-donor and acceptor sites were then attached to the cysteine and histidine sites respectively, to yield the binuclear electron-transfer metallopeptides ET-I and ET-II. Photoexcitation of the ruthenium polypyridyl donor resulted in ET occurring to the pentaammine ruthenium (III) acceptor in a direction that was towards the negative end of the helix dipole in ET-I, and the positive end in ET-II. Significantly, no evidence for directional electron-transfer rates was observed in these systems in aqueous solution, the low-dielectric solvent 2,2,2-trifluoroethanol (TFE), or a 1:1 (v/v) mixture of CH2Cl2:TFE. This behavior is in marked contrast to that observed for the systems studied by Fox and co-workers.62–64 The reason for this apparent discrepancy is presently not understood. However, two important differences do exist between the two systems studied. Firstly, the earlier work examined the rates photoinduced electron-transfer occurring between non-polar redox centers, but the peptides ET-I and ET-II employ charged, divalent and trivalent, metal complexes as their donor and acceptor sites. It is possible that the electrostatic effects generated by the presence of these charged complexes may supersede the effects exerted by the helix dipole. Secondly, the metallopeptide systems use a flexible acetyl linker to attach the ruthenium polypyridyl complex to the peptide chain which can place this redox site at distances ranging from ca. 3 – 8 Å away from the helix axis (edge-to-backbone). This may also serve to reduce the effects of the helix dipole in regulating ET rates. Thus, to determine if these factors contribute to the absence of a helix dipole effect, studies are currently underway to design systems which use redox sites that consist of neutral metal complexes, which do not require the use of an acetyl linker, and which can be placed at different regions of the sequence. As discussed below, work from our group has shown that a new family of synthetic metalloproteins can be prepared in which redox active cofactors can be buried within their hydrophobic interiors. Investigations of such systems may help elucidate the necessary requirements for allowing the helix dipole moment to regulate electron-transfer rates in de novo designed metallopeptides and metalloproteins.
Figure 3.
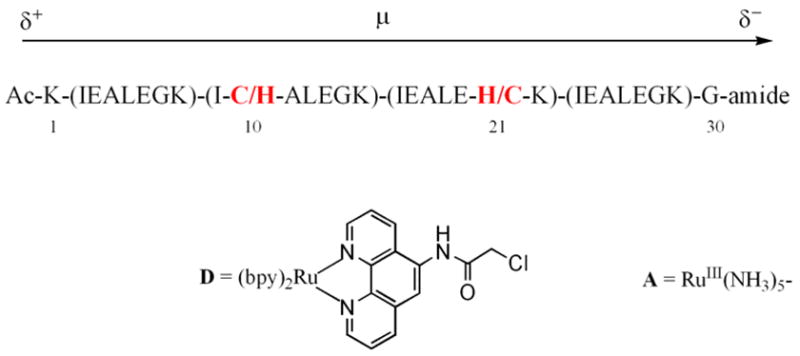
Sequence of the ET-I and ET-II in which the binuclear electron-transfer proteins were prepared by first reacting the ruthenium polypyridyl complex (D) to the cysteine residue of the appropriate peptide and then attaching the pentaammine ruthenium species (A) to the histidine residue.
Gated ET to Study the Dynamics of Peptide-Protein Complexes
Our group’s experience in preparing redox-active metallopeptides have been applied towards the study of ET processes that occur within electrostatic protein complexes. Early work in this project sought to understand how the incorporation of complementary electrostatic recognition domains onto the surface of coiled-coil metalloproteins can affect the rates of inter-molecular ET occurring between separate metalloproteins.51 Thus, inter-protein electron-transfer reactions were studied which involve a [Ru(NH3)5-H21]2+ electron-donor and a [Ru(trpy)(bpy)-H21]3+ electron-acceptor that were embedded within protein surfaces having opposite charge: a RuII(NH3)5-H21 site was placed on the positive surface of a coiled-coil peptide, and a RuIII(trpy)(bpy)-H21 site was placed on the negative surface of another peptide (Figure 4). No evidence for stable electrostatic complex formation was observed and the rates of inter-molecular electron-transfer were seen to follow bi-molecular kinetics and increase from kinter = (1.9 ± 0.4) × 107 M−1 s−1 to kinter = (3.7 ± 0.5) × 107 M−1 s−1 as the ionic strength was raised from 0.01 to 0.20 M. This somewhat unexpected result indicates that the electrostatic repulsion between the positively charged ruthenium centers dominates the kinetics of these reactions, and not the complementary surface charges of the proteins. However, analysis by two different electrostatic models indicated that the presence of the oppositely-charged protein surfaces in the coiled-coils does create an electrostatic recognition domain which substantially ameliorates the effects of this inter-metal repulsion.
Figure 4.
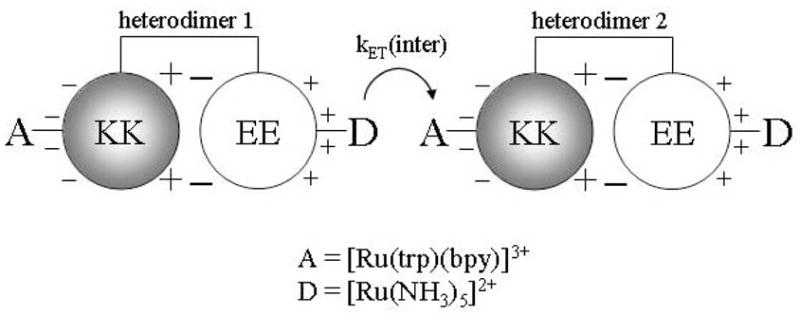
Schematic representation of the EE/KK electrostatic heterodimer emphasizing the charges on the solvent-exposed and interfacial regions of the heterodimer.
Encouraged by our ability to use electrostatic interactions to influence the rates of inter-molecular electron-transfer reactions occurring between designed metalloproteins, we designed two small negatively-charged metallopeptides capable of forming stable electrostatic complexes with ferricytochrome c. This work showed how the rates of intra-complex electron-transfer are gated by rate-limiting configurational changes occurring within the electrostatic peptide-protein complex.
Emission measurements showed that the triplet lifetime of the ruthenium metallopeptide, [Ru(bpy)2(phen-am)-Cys-(Glu)5-Gly]3− = RuCE5G, (Figure 5) is shortened and decays via biexponential kinetics when in the presence of Cyt c. These results indicate the existence of two excited-state populations of ruthenium peptides, both of which undergo photoinduced electron-transfer to the iron heme. The faster decay component displays concentration-independent kinetics demonstrating the presence of a preformed peptide-protein complex which undergoes intra-complex electron-transfer with a rate constant of . Significantly, the magnitude of decreases with increasing solvent viscosity (Figure 6) and the behavior can be fit to the expression to give α = 0.59 ± 0.05. The electron-transfer process occurring in the preformed complex is therefore gated by a rate-limiting configurational change of the complex. The slower decay component displays concentration-dependent kinetics which saturate at high concentrations of Cyt c. Analysis according to rapid equilibrium formation of an encounter complex which then undergoes unimolecular electron-transfer yields . The smaller value of suggests that a somewhat longer donor-acceptor distance exists in the encounter complex. Interestingly, the value of is also viscosity dependent showing that this reaction is also gated. However, a value of α= 0.98 ± 0.14 was observed, which emphasized the very dynamic nature of the encounter complex.
Figure 5.
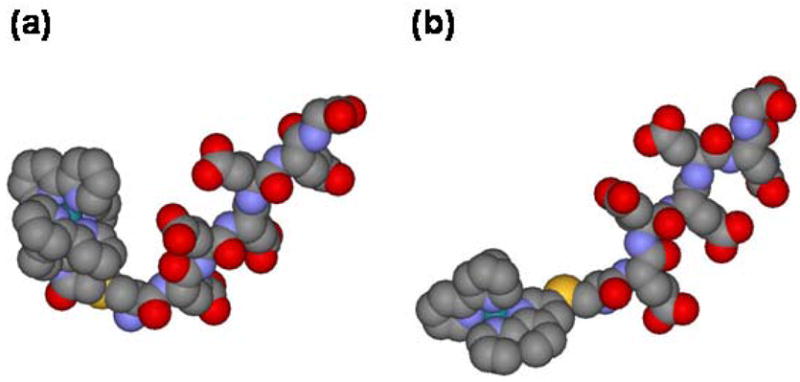
Energy-minimized structures of the metal-peptides a) RuCE5G, and b) RuCE5G-short which differ by the method of attaching the ruthenium center to the cysteine side-chain.
Figure 6.
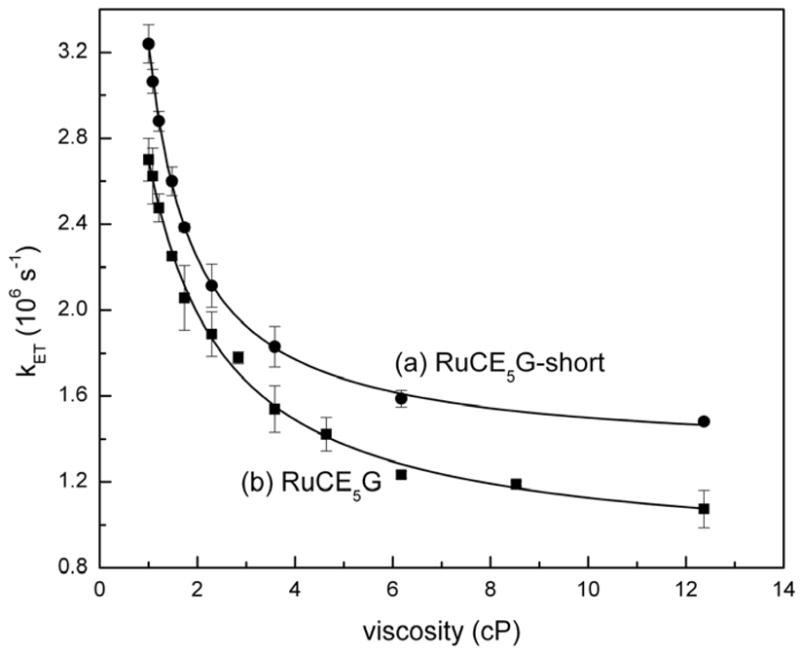
Viscosity dependence of intracomplex electron-transfer rate constants for (a) RuCE5G-short and (b) RuCE5G.
Subsequent work demonstrated how a small modification of the metal peptide could produce significant changes in the dynamics of its preformed complex.65 Thus, a new metalloprotein was prepared in which a ruthenium polypyridyl complex was coupled directly to the CE5G peptide by reacting it with [(bpy)2Ru(3-bromo-1,10-phenanthroline)](PF6)2 to yield the compound, RuCE5G-short (Figure 5). This new metallopeptide differs from the original one only by the absence of the flexible acetyl linker joining the metal complex to the cysteine side-chain of the peptide. Photoinduced electron-transfer experiments showed that RuCE5G-short also forms an electrostatic complex with Cyt c within which intra-complex electron-transfer can occur. In addition, viscosity studies showed that this process is also gated by rate-limiting configurational changes of the complex. However, it was seen that the preformed complex involving RuCE5G-short was more mobile (α = 0.97 ± 0.03) than the one involving the longer peptide (Figure 6) and had a higher binding constant. These observations were rationalized by molecular modeling studies which indicated that the two peptides likely adopt different conformations (Figure 5). Whereas the short peptide has a roughly linear rod-like geometry, the flexible acetamido linker of RuCE5G allows it to form a hairpin-like structure in which the bulky ruthenium polypyridyl cation is placed in closer proximity to the negatively-charged glutamate chain. It was speculated that the higher mobility of the RuCE5G-short:Cyt c complex may due to its rod-like conformation, and the lower binding constant of the RuCE5G complex may arise from partial charge compensation occurring between the oppositely charged portions of the metal peptide as they are brought closer together in the hairpin structure. These results demonstrated how gated electron-transfer experiments can be used to directly probe the dynamics of peptide-protein complexes, and how apparently subtle changes made to the peptide sequence may produce large changes in the dynamics of the complexes that they form.
Design of a Cd(II)-binding Site within the Interior of a Coiled-coil Protein
The work from our group described above shows how de novo designed metalloproteins and metallopeptides can be used to study various aspects of biological electron-transfer reactions. However, it is noted that the design of these systems simply appended non-native coordination complexes, such as ruthenium pentaammine and polypyridyl complexes, to the surfaces of these proteins in order to facilitate these reactions.36,66,67 Consequently, the early electron-transfer metalloproteins studied by our group can be considered to be intrinsically abiotic in that they did not contain native-like cofactors in which redox-active transition metals were directly coordinated to amino acid residues located within the hydrophobic core of these proteins, as typically observed for many native systems. Thus, consideration was given towards devising ways of preparing more native-like ET metalloproteins through the incorporation of more native-like transition metal cofactors into the interior of synthetic redox proteins. Once prepared, such functionally-active de novo designed metalloproteins will provide a useful system in which to study how protein environments can be used to tune the chemical reactivity of embedded cofactors.
Recent efforts by our group have successfully incorporated a cysteine-containing metal-binding site into the hydrophobic core of α-helical coiled-coils.27 The peptide sequence employed in this study was based on the IEALEGK heptad repeat used by our group in the mechanistic electron-transfer studies described above.36,46,48,51–53 However, here the peptide sequence was modified to contain the Cys-X-X-Cys metal binding domain of rubredoxin in which four cysteine residues create a tetrahedral coordination sphere for Fe2+. The computer model shown in Figure 7 illustrates how the appropriate incorporation of the Cys-X-X-Cys tetrad into positions 16–19 of the coiled-coil sequence place cysteine residues at the hydrophobic “a” and “d” positions of third heptad repeat which may result in the creation of a rubredoxin-like metal-binding site. Energy minimization calculations predict that binding of a Cd(II) ion to this site will result in dihedral bond angles ranging from 104.6° to 118.4°, and Cd-S distances of ca. 2.55 Å, which are consistent with those observed for cadmium substituted desulforedoxins.68 From these results, a 32-mer peptide called C16C19-GGY was prepared having the sequence:
Figure 7.
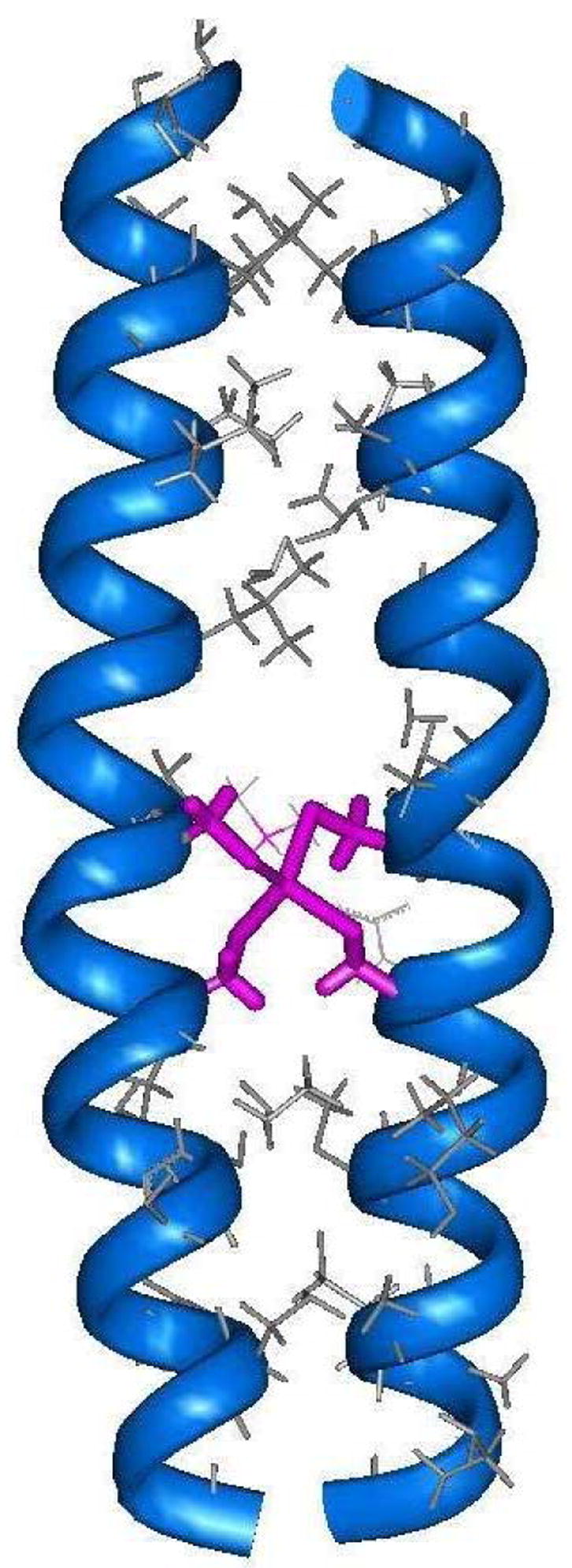
Energy minimized computer model of the Cd-bridged C16C19 peptide dimer in which cysteine residues located at postions 16 and 19 of each peptide chain bind the Cd center in a tetrahedral geometry.
| Ac-K(IEALEGK)2(CEACEGK)(IEALEGK)GGY-NH2 | C16C19-GGY |
which places cysteine residues at both the “a” and “d” positions of the third heptad repeat. The C16C19-GGY peptide was purified by reverse-phase HPLC and characterized by MALDI mass-spectroscopy. Figure 8 shows that the circular dichroism spectrum of C16C19-GGY consists of a large, negative maximum centered at 205 nm to indicate that the apopeptide exists as a disordered random coil in aqueous solution. This was an unexpected result based on our previous experience with similar peptides and shows that the substitution of cysteine residues into the hydrophobic core disrupts the coiled-coil structure. Possible reasons for this include the introduction of different side-chain packing interactions into the hydrophobic core of the putative coiled-coil, and/or the lowered hyrdophobicity of cysteinyl side-chain. Importantly, the results presented in Figure 8 also show that the CD spectrum of the reduced C16C19-GGY peptide changes substantially upon the addition of CdCl2. Negative maxima were then observed at 208 and 222 nm to indicate the presence of α-helices. Further, the intensity of these signals increased with increasing amounts of Cd(II) added to solution and yielded an ellipticity ratio of [θ222]/[ θ208] = 0.99 which falls within the range generally regarded to indicate the presence of a coiled-coil structure.57
Figure 8.
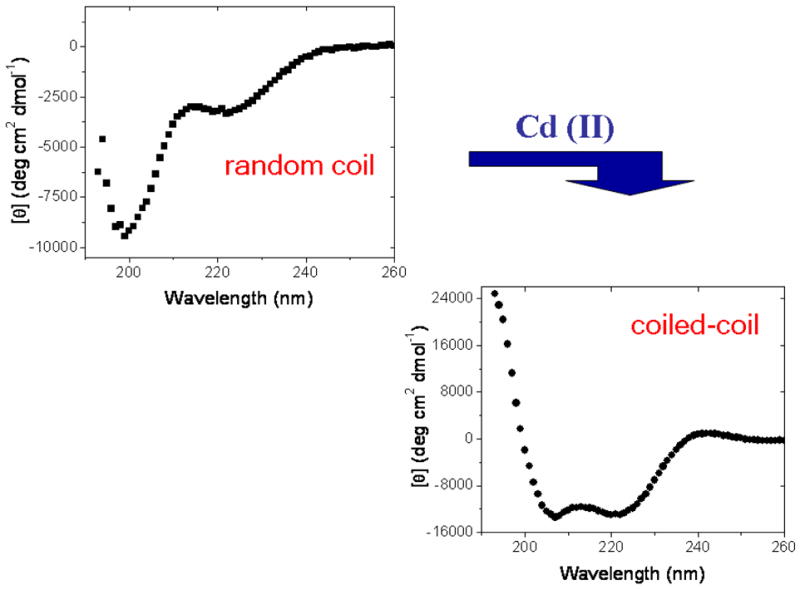
Changes in the circular dichroism spectrum of C16C19-GGY upon addition of CdCl2 to a 100 μM solution of C16C19-GGY containing the reducing agent tris-(2-carboxyethyl)phosphine (TCEP, 100–200 μM) at pH 5.3 under deaerated conditions.
The aggregation state of the metal-peptide assembly was studied by high performance size exclusion chromatography (HPSEC) which has been shown to be a reliable method for determining the oligomerization states of α-helical coiled-coils.50 In these experiments, the elution profile of C16C19-GGY was obtained in both the absence and presence of Cd(II), and then compared with those of several peptide standards. The results indicate that the apopeptide has an apparent molecular mass of 3.3 kDa which shows that it exists as a monomer. However, the behavior of the Cd-peptide yields an apparent molecular mass of 5.3 kDa which indicates that the metalloprotein exists as a peptide dimer. These results show that the C16C19-GGY peptide undergoes a metal-induced folding process from a monomeric random coil to the organized structure of a two-stranded α-helical coiled-coil upon binding Cd(II). The metal-peptide stoichiometry of the dimeric peptide was studied by UV-vis spectroscopy as a UV absorption band at 238 nm is observed upon addition of Cd(II) to a solution of C16C19. This band is assigned to the ligand-to-metal-charge-transfer (LMCT) transition of the newly formed Cd-S bond.69 A Job plot was constructed by measuring the absorbance at 238 nm as a function of the mole fraction of Cd(II) present in solution and demonstrated the existence of a 2:1 peptide:metal complex. These results were supported by spectrophotometric titrations in which the absorption intensity at 238 nm was seen to increase with successive additions of CdCl2, reaching a limiting value at higher concentrations of added Cd(II) (Figure 9). As seen, the plot has a break at 0.5 equivalents of Cd(II) added per peptide to further indicate the presence of a 2:1 peptide:metal complex.
Figure 9.
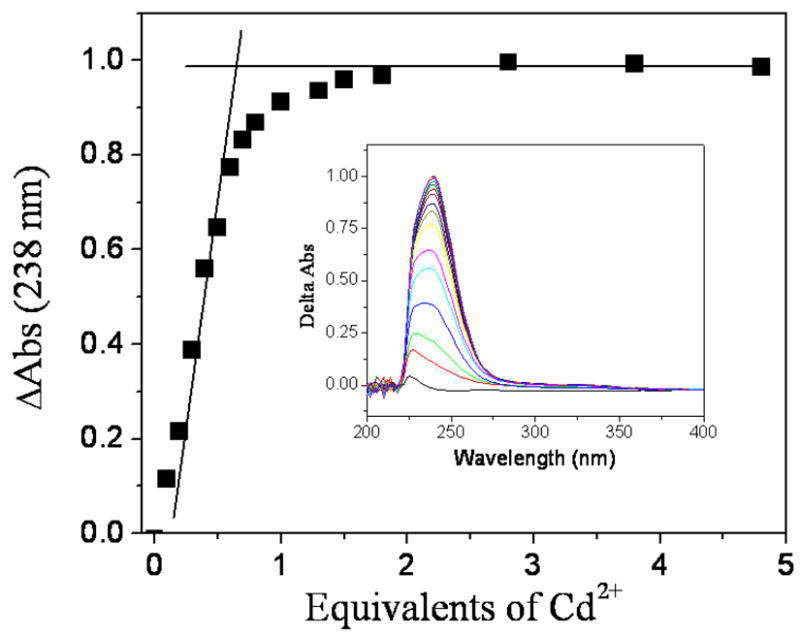
UV-vis titration in which successive additions of CdCl2 were made to a 100 μM solution of C16C19-GGY containing the reducing agent tris-(2-carboxyethyl)phosphine (TCEP, 100–200 μM) in 0.2 M acetate buffer (pH 5.4) under deaerated conditions.
In summary, the incorporation of the Cys-X-X-Cys metal-binding motif into the sequence of the C16C19-GGY apopeptide causes the peptide to exist as a monomeric random coil in free solution. However, the peptide then assembles into a metal-bridged, coiled-coil dimer upon binding Cd(II). This behavior is reminiscent of that observed for the copper chaperone HAH1 which forms a metal-bridged dimer in the presence of Cu(I),70,71 and for the related copper trafficking protein CopZ whose metal-dependent dimerization was studied by electrospray ionization mass spectrometry,72 analytical ultracentrifugation, and spectroscopic titrations.73,74
Metal-specific Protein Folding Properties of C16C19-GGY
The observation that C16C19-GGY undergoes a Cd(II)-induced conformational change from a random-coil to a two-stranded coiled-coil prompted further investigation of the metal-binding properties of this peptide.75 New CD results show that the random coil C16C19-GGY peptide monomer folds into an α-helical coiled-coil when in the presence of such soft metal ions as Hg(II), Cu(I), Au(I), and Ag(I), but continues to exist as a disordered random coil in the presence of Fe(II), Co(II), Ni(II), Zn(II), and Pb(II). The data presented in Table 1 show that binding of the various soft metal ions to C16C19-GGY results in a significant variation of the helical content of the resulting metalloproteins. Perhaps more dramatically, the oligomerization states of these different metalloproteins were determined by HPSEC and range in size from being peptide dimers for the Cd(II) and Hg(II) adducts to peptide hexamers for the Au(I) protein. The binding of Cu(I) and Ag(I) to C16C19-GGY produces the intermediate case of peptide tetramers, which in the case of the Cu(I) adduct has been verified by analytical ultracentrifugation.28 Interestingly, no obvious trend is observed as binding of the largest ion in the series (Au(I)) is seen to form the largest peptide oligomer, but the comparably-sized Ag(I) and Hg(II) ions produce peptide tetramers and dimers, respectively. Thus, the binding of different metal ions to the C16C19-GGY peptides produces significant differences in conformational properties of the resulting C16C19-GGY holoproteins. Interpreted within the Pecoraro’s TRI paradigm,15 these results indicate that the C16C19-GGY peptide does not have a strong preference for a particular coiled-coil geometry and that the coordination properties of the different metal ions strongly influence the conformation of the resulting metalloprotein.
Table 1.
Conformational properties of different metal adducts of C16C19-GGY
| Cd(II) | Hg(II) | Cu(I) | Ag(I) | Au(I) | |
|---|---|---|---|---|---|
| [θ222]/[ θ 208]a | 0.99 | 1.15 | 0.97 | 1.13 | 1.04 |
| [θ222]a (deg cm2 dmol−1 | −16,400 | −23,900 | −21,300 | −27,800 | −10,200 |
| Oligomerizationb | dimer | dimer | tetramer | tetramer | hexamer |
measured upon the addition of an equimolar amounts of metal ion (CdCl2, HgCl2, [Cu(CH3CN)4]PF6, AgNO3 and sodium aurothiomalate) and C16C19-GGY in 5 mM sodium acetate buffer (pH 5.5).
measured by HPSEC as described in the text.
Binding of Cu(I) to Create a Luminescent Coiled-coil Metalloprotein
Our studies of the Cu(I) adduct of C16C19-GGY led to the interesting observation of an intense (φ = 0.053) ambient temperature luminescence centered at 600 nm which persists upon allowing the protein to stand overnight under ambient conditions.28 It was found that this luminescence can be quenched by the addition of either ferricyanide, oxygen, or urea to respectively indicate that the emitting species is associated with the reduced Cu(I) state, has significant triplet character, and is quenched upon exposure to bulk solvent. It is noteworthy that similar photoluminescent properties have been reported for Cu(I) derivatives of the cysteine rich metal-binding protein metallothionein,76,77 as well as to those of the Cox17 copper chaperone78 and the copper responsive transcription factors ACE179 and CopY80, all of which contain polynuclear copper(I) clusters that are buried within the protein to shield them from bulk solvent.81 This is consistent with earlier studies of small molecule Cu(I)-thiolate compounds in which luminescence is observed for polynuclear metal clusters where metal-metal interactions play an important role in stabilizing the emissive photoexcited-state.82 Thus, the observation of a 600 nm luminescence from Cu(I)-C16C19-GGY suggests that it likely contains a polynuclear Cu(I) cluster.
The metal binding stoichiometry of the Cu-protein was therefore determined by monitoring the various spectral changes that can be observed upon the addition of Cu(I). The data in Figure 10 shows the emission intensity of Cu(I)-C16C19-GGY increases with increasing amounts of Cu(I) added to the solution of peptide but saturates after ca. 0.9 equivalents of metal ion have been added. As with the oligomerization state of the Cu(I) adduct of C16C19-GGY which exists as a peptide tetramer, this behavior is in marked contrast to that previously observed for the Cd(II) protein and indicates that four equivalents of Cu(I) are present in this new peptide tetramer. UV titrations were used to confirm these results, as the binding of Cu(I) to cysteine residues is known to produce both Cys-thiolate to Cu(I) ligand-to-metal-charge-transfer (LMCT), and metal-centered (MC) transitions in the UV region of the spectrum.83,84 Indeed, the addition of Cu(I) to a solution of C16C19-GGY does produce a new absorption band having a maximum at 236 nm and a shoulder at 296 nm. By analogy to studies of copper metallothionein, the lower energy absorption can be assigned to a clustered-centered transition whose intensity should be proportional to the number of bound metal ions, and the higher energy band is likely due to the LMCT transition having an extinction coefficient of ca. 5500 M−1cm−1 per metal-cysteine bond.83 The absorption at 296 nm does indeed increase with increasing amounts of added Cu(I), and saturates after one equivalent of copper has been added to confirm the 1:1 peptide:metal stoichiometry. The measured extinction coefficient at 236 nm further suggests the presence of 4.0 Cu(I)-cysteine bonds in the metal cofactor.
Figure 10.
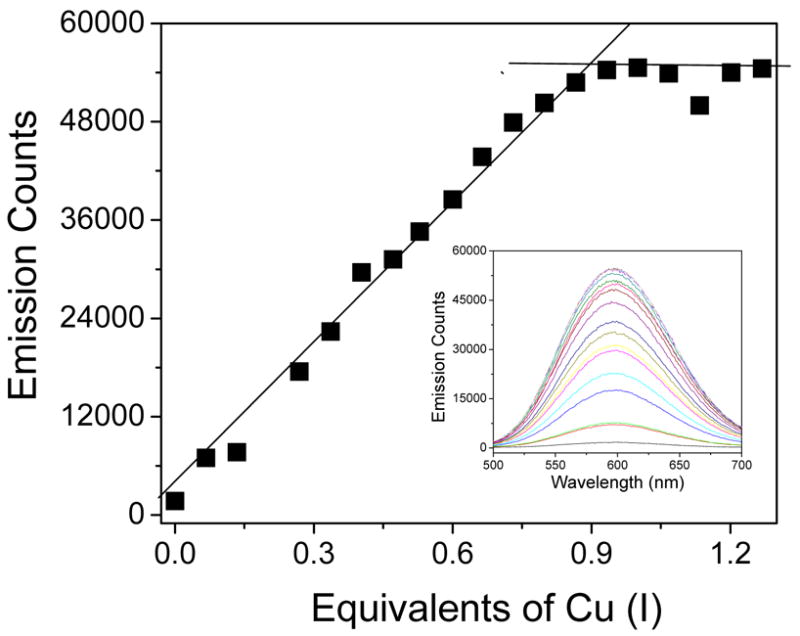
Emission titration of C16C19-GGY by [Cu(CH3CN)4]PF6. Inset: Emission spectra obtained upon addition of Cu(I) to the peptide solution. Conditions: 120 μM C16C19-GGY in 0.2 M acetate buffer (pH 5.4) containing 730 μM tris-(2-carboxyethyl)phosphine (TCEP) as a reducing agent.
The nature of the multinuclear copper center was characterized by X-ray absorption spectroscopy (XAS).28 The Cu K-edge XANES spectrum of Cu(I)-C16C19-GGY showed the existence of Cu(I) ions having a trigonal coordination geometry and EXAFS analysis suggested that each Cu(I) ion was surrounded by a ligand set consisting of one N/O and two S-donors at average distances of 1.89(2) Å and 2.22(2) Å, respectively. The data also revealed the presence of additional scatterers in the second and third coordination sphere of the Cu centers, indicating the presence of a tetranuclear Cu cluster in which adjacent Cu(I) ions are bridged by the side chains of two cysteine residues and each Cu atom also has a terminal N/O ligand. This model is consistent with the titration of free thiol groups with 5,5′-dithio-bis(2-nitrobenzoic acid) which confirmed the presence of one free thiol group per peptide chain in the metalloprotein. Figure 11 shows a likely model of Cu(I)-C16C19-GGY.
Figure 11.
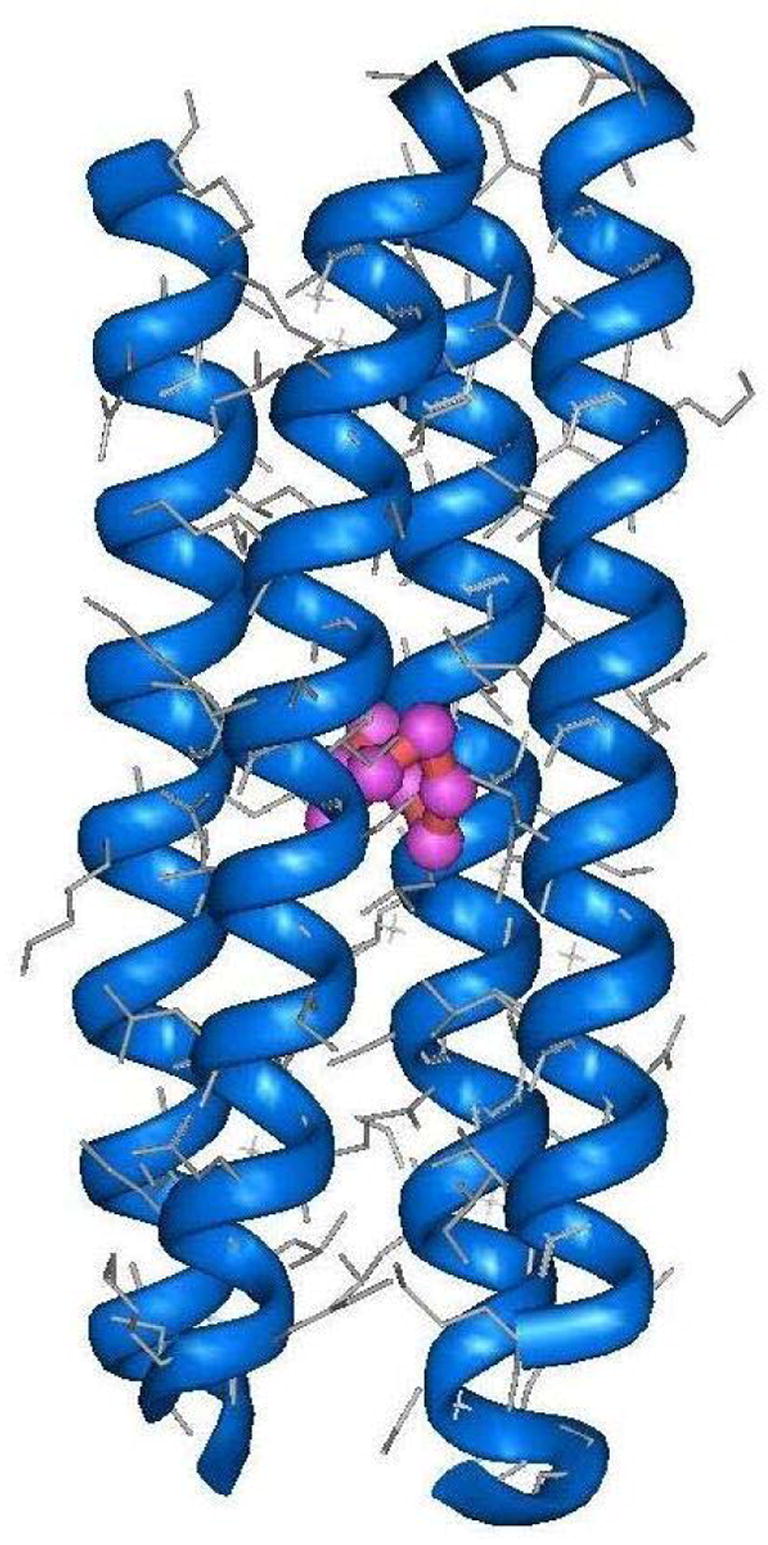
Computer generated model of the tetrameric Cu(I)-C16C19-GGY metalloprotein.
Photoinduced Electron-Transfer Involving the Cu(I)-C16C19-GGY Metalloprotein
The strong room temperature luminescence of Cu(I)-C16C19-GGY suggests that it might function as a photoinduced electron-transfer protein which can be monitored by emission lifetime experiments. As a control, Figure 12 shows that the emission lifetime of this species can be accurately fit to double exponential decay kinetics (eq 1), in which AS, kS and AL, kL are the amplitudes and rate constants of the
Figure 12.

Emission lifetime of 25 μM Cu(I)-C16C19-GGY taken in the a) absence and b) presence of 100 μM [Ru(NH3)5Py]3+ in 0.2 M acetate buffer (pH 5.4). The solid lines are the fits to eq 1 where for trace (a) kS = 9.5 × 105 s-−1, kL = 1.3 × 105 s-−1, and AS/AL = 1 and for trace (b) kS = 1.1 × 106 s−1, kL = 4.2 × 105 s−1, and AS/AL = 1.
| [1] |
shorter and longer lifetime components, respectively. A non-linear least squares fit of the data yield values of kS = 9.5 × 105 s−1 (τS = 1.1 μs), kL = 1.3 × 105 s−1 (τL = 7.7 μs) and AS/AL = 1, where τL and τS are the emission lifetimes of their respective components. These results suggest that the CuI4S4(N/O)4 cofactor of Cu(I)-C16C19-GGY contains two independent lumophores and it is speculated, that the two emissive sites of this protein might be due to the presence of two electronically independent Cu(I)-Cu(I) dimers located within the CuI4S4(N/O)4 cofactor. However, additional explanations for this cannot be ruled out at this time.
Importantly, the data shown in Figure 12 also show that both lifetime components of this protein are quenched in the presence of the electron-acceptor [Ru(NH3)5Py]3+ where Py = pyridine, and it has been shown that this behavior is accompanied by an increased absorption at 400 nm to indicate the formation of the reduced [Ru(NH3)5Py]2+ quencher in a photoinduced electron-transfer event. It is further noted that the relative amplitudes of the fast and slow emission decay components remain approximately equal at all quencher concentrations studied and that plots of the observed emission decay constants kSobs and kLobs are linearly dependent upon the concentration of quencher as pseudo-first order quenching kinetics are observed (Figure 13). Analysis of the data yields values for the bimolecular quenching constants of kSET = (2.46 ± 0.07) × 109 M−1s−1 and kLET = (1.36 ± 0.05) × 109 M−1s−1, respectively. Together, these results show that the luminescent polynuclear copper center in the synthetic metalloprotein Cu(I)-C16C19-GGY can indeed function as a photoinduced electron-transfer protein by undergoing a bimolecular reaction with an exogenous acceptor in free solution.
Figure 13.
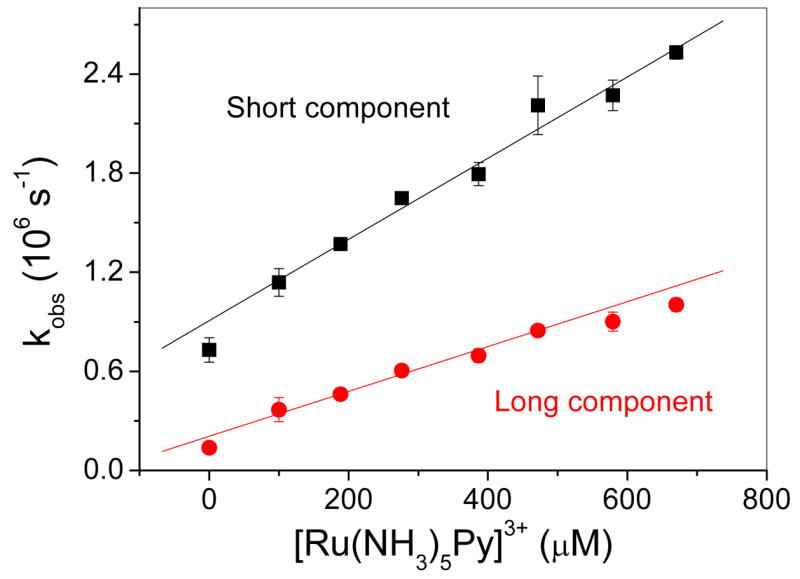
Observed emission decay rate constants for the long and short-lived emission components of Cu(I)-C16C19-GGY (ca. 25 μM) taken as a function of the concentration of added [Ru(NH3)5Py]3+ in 0.2 M acetate buffer (pH 5.4). The error bars are the deviation from the mean of triplicate experiments.
Conclusion
In conclusion, the α-helical coiled-coil motif has served as a robust scaffold for constructing synthetic electron-transfer metalloproteins. Early work from our group appended exogenous, and abiotic, ruthenium-based redox centers to the solvent-exposed surfaces of these proteins for intramolecular electron-transfers studies. Recent work involved the incorporation of a more native-like Cu(I) redox-center into the hydrophobic interior of a synthetic protein. This work showed how the structures of the resulting metalloproteins are controlled by the subtle interplay between the directional bonding properties of their inorganic cofactors and those of their protein environments. Future work from our laboratory will continue to examine how the conformational and chemical (i.e electron-transfer) properties of these model proteins can be controlled by these factors. It is hoped that knowledge gained from this work will contribute to the expanding effort of bioinorganic chemists to prepare new kinds of functionally active synthetic metalloproteins.
Acknowledgments
The authors thank Mikhail Tsurkan, Xianchun Zhu, Fei Xie, Elena Ptchelnikova, and Jiufeng Fan for help in preparing this manuscript. The Ohio Laboratory for Kinetic Spectrometry and Profs. M. A. J. Rodgers and F. Castellano are thanked for use of the laser facilities. This work was sponsored by NIH grant no. GM61171, NSF grant no. CHE-0455441, and ACS-PRF grant no. 34901-AC.
References
- 1.Gilardi G, Fantuzzi A. Trends Biotechnol. 2001;19:468–476. doi: 10.1016/s0167-7799(01)01769-3. [DOI] [PubMed] [Google Scholar]
- 2.Barker PD. Curr Opin Struct Biol. 2003;13:490–499. doi: 10.1016/s0959-440x(03)00108-8. [DOI] [PubMed] [Google Scholar]
- 3.Wittung-Stafshede P. Accounts Chem Res. 2002;35:201–208. doi: 10.1021/ar010106e. [DOI] [PubMed] [Google Scholar]
- 4.Xing G, DeRose VJ. Curr Opin Chem Biol. 2001;5:196–200. doi: 10.1016/s1367-5931(00)00190-3. [DOI] [PubMed] [Google Scholar]
- 5.Lu Y, Berry SM, Pfister TD. Chem Rev. 2001;101:3047–3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- 6.Kennedy ML, Gibney BR. Curr Opin Struct Biol. 2001;11:485–490. doi: 10.1016/s0959-440x(00)00237-2. [DOI] [PubMed] [Google Scholar]
- 7.Baltzer L, Nilsson J. Curr Opin Biotechnol. 2001;12:355–360. doi: 10.1016/s0958-1669(00)00227-5. [DOI] [PubMed] [Google Scholar]
- 8.Bloom JD, Meyer MM, Meinhold P, Otey CR, MacMillan D, Arnold FH. Curr Opin Struct Biol. 2005;15:447–452. doi: 10.1016/j.sbi.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Dwyer MA, Looger LL, Hellinga HW. Science. 2004;304:1967–1971. doi: 10.1126/science.1098432. [DOI] [PubMed] [Google Scholar]
- 10.Calhoun JR, Nastri F, Maglio O, Pavone V, Lombardi A, DeGrado WF. Biopolymers. 2005;80:264–278. doi: 10.1002/bip.20230. [DOI] [PubMed] [Google Scholar]
- 11.Lombardi A, Summa CM, Geremia S, Randaccio L, Pavone V, DeGrado WF. Proc Natl Acad Sci U S A. 2000;97:6298–6305. doi: 10.1073/pnas.97.12.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maglio O, Nastri F, Pavone V, Lombardi A, DeGrado WF. Proc Natl Acad Sci U S A. 2003;100:3772–3777. doi: 10.1073/pnas.0730771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaplan J, DeGrado WF. Proc Natl Acad Sci U S A. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laplaza CE, Holm RH. J Am Chem Soc. 2001;123:10255–10264. doi: 10.1021/ja010851m. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh D, Lee KH, Demeler B, Pecoraro VL. Biochemistry. 2005;44:10732–10740. doi: 10.1021/bi0506674. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh D, Pecoraro VL. Inorg Chem. 2004;43:7902–7915. doi: 10.1021/ic048939z. [DOI] [PubMed] [Google Scholar]
- 17.Lee KH, Matzapetakis M, Mitra S, Neil E, Marsh G, Pecoraro VL. J Am Chem Soc. 2004;126:9178–9179. doi: 10.1021/ja048839s. [DOI] [PubMed] [Google Scholar]
- 18.Farrer BT, Pecoraro VL. Proc Natl Acad Sci U S A. 2003;100:3760–3765. doi: 10.1073/pnas.0336055100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matzapetakis M, Farrer BT, Weng TC, Hemmingsen L, Penner-Hahn JE, Pecoraro VL. J Am Chem Soc. 2002;124:8042–8054. doi: 10.1021/ja017520u. [DOI] [PubMed] [Google Scholar]
- 20.Farrer BT, Pecoraro VL. Current Opinion in Drug Discovery & Development. 2002;5:937–943. [PubMed] [Google Scholar]
- 21.Farrer BT, Harris NP, Balchus KE, Pecoraro VL. Biochemistry. 2001;40:14696–14705. doi: 10.1021/bi015649a. [DOI] [PubMed] [Google Scholar]
- 22.Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL. J Mol Biol. 1998;280:897–912. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- 23.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, Ohalloran TV, PennerHahn JE, DeGrado WF, Pecoraro VL. J Am Chem Soc. 1997;119:6195–6196. [Google Scholar]
- 24.Tanaka T, Mizuno T, Fukui S, Hiroaki H, Oku J, Kanaori K, Tajima K, Shirakawa M. J Am Chem Soc. 2004;126:14023–14028. doi: 10.1021/ja047945r. [DOI] [PubMed] [Google Scholar]
- 25.Kiyokawa T, Kanaori K, Tajima K, Koike M, Mizuno T, Oku JI, Tanaka T. J Pept Res. 2004;63:347–353. doi: 10.1111/j.1399-3011.2004.00109.x. [DOI] [PubMed] [Google Scholar]
- 26.Li XQ, Suzuki K, Kanaori K, Tajima K, Kashiwada A, Hiroaki H, Kohda D, Tanaka T. Protein Sci. 2000;9:1327–1333. doi: 10.1110/ps.9.7.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kharenko OA, Ogawa MY. J Inorg Biochem. 2004;98:1971–1974. doi: 10.1016/j.jinorgbio.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 28.Kharenko OA, Kennedy DC, Demeler B, Maroney MJ, Ogawa MY. J Am Chem Soc. 2005;127:7678–7679. doi: 10.1021/ja042757m. [DOI] [PubMed] [Google Scholar]
- 29.Gibney BR, Huang SS, Skalicky JJ, Fuentes EJ, Wand AJ, Dutton PL. Biochemistry. 2001;40:10550–10561. doi: 10.1021/bi002806h. [DOI] [PubMed] [Google Scholar]
- 30.Welch JT, Kearney WR, Franklin SJ. Proc Natl Acad Sci U S A. 2003;100:3725–3730. doi: 10.1073/pnas.0536562100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kovacic RT, Welch JT, Franklin SJ. J Am Chem Soc. 2003;125:6656–6662. doi: 10.1021/ja0210998. [DOI] [PubMed] [Google Scholar]
- 32.Rossi P, Tecilla P, Baltzer L, Scrimin P. Chemistry-a European Journal. 2004;10:4163–4170. doi: 10.1002/chem.200400160. [DOI] [PubMed] [Google Scholar]
- 33.Discher BA, Noy D, Strzalka J, Ye SX, Moser CC, Lear JD, Blasie JK, Dutton PL. Biochemistry. 2005;44:12329–12343. doi: 10.1021/bi050695m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noy D, Discher BA, Rubtsov IV, Hochstrasser RA, Dutton PL. Biochemistry. 2005;44:12344–12354. doi: 10.1021/bi050696e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye SX, Discher BM, Strzalka J, Xu T, Wu SP, Noy D, Kuzmenko I, Gog T, Therien MJ, Dutton PL, Blasie JK. Nano Letters. 2005;5:1658–1667. doi: 10.1021/nl050542k. [DOI] [PubMed] [Google Scholar]
- 36.Ogawa MY. In: Molecular and Supramolecular Photochemistry. Ramamurthy V, Schanze KS, editors. Vol. 4. Marcel Dekker, Inc.; New York: 1999. pp. 113–150. [Google Scholar]
- 37.Zheng YJ, Case MA, Wishart JF, McLendon GL. J Phys Chem B. 2003;107:7288–7292. [Google Scholar]
- 38.Mutz MW, Case MA, Wishart JF, Ghadiri MR, McLendon GL. J Am Chem Soc. 1999;121:858–859. [Google Scholar]
- 39.Mutz MW, McLendon GL, Wishart JF, Gaillard ER, Corin AF. Proc Natl Acad Sci U S A. 1996;93:9521–9526. doi: 10.1073/pnas.93.18.9521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kennedy ML, Gibney BR. J Am Chem Soc. 2002;124:6826–6827. doi: 10.1021/ja0171613. [DOI] [PubMed] [Google Scholar]
- 41.Cristian L, Piotrowiak P, Farid RS. J Am Chem Soc. 2003;125:11814–11815. doi: 10.1021/ja0292142. [DOI] [PubMed] [Google Scholar]
- 42.Jones G, Vullev V, Braswell EH, Zhu D. J Am Chem Soc. 2000;122:388–389. [Google Scholar]
- 43.Fahnenschmidt M, Bittl R, Schlodder E, Haehnel W, Lubitz W. Physical Chemistry Chemical Physics. 2001;3:4082–4090. [Google Scholar]
- 44.Rau HK, Snigula H, Struck A, Robert B, Scheer H, Haehnel W. Eur J Biochem. 2001;268:3284–3295. doi: 10.1046/j.1432-1327.2001.02231.x. [DOI] [PubMed] [Google Scholar]
- 45.Schnepf R, Haehnel W, Wieghardt K, Hildebrandt P. J Am Chem Soc. 2004;126:14389–14399. doi: 10.1021/ja0484294. [DOI] [PubMed] [Google Scholar]
- 46.Kornilova AY, Wishart JF, Xiao WZ, Lasey RC, Fedorova A, Shin YK, Ogawa MY. J Am Chem Soc. 2000;122:7999–8006. [Google Scholar]
- 47.Tyson DS, Castellano FN. J Phys Chem A. 1999;103:10955–10960. [Google Scholar]
- 48.Fedorova A, Ogawa MY. Bioconjugate Chem. 2002;13:150–154. doi: 10.1021/bc015544k. [DOI] [PubMed] [Google Scholar]
- 49.Fasman GD. Handbook of Biochemistry and Molecular Biology, Proteins, I. 3. CRC Press; 1976. [Google Scholar]
- 50.Mant CT, Chao H, Hodges RS. J Chromatogr A. 1997;791:85–98. doi: 10.1016/s0021-9673(97)00767-x. [DOI] [PubMed] [Google Scholar]
- 51.Kornilova AY, Wishart JF, Ogawa MY. Biochemistry. 2001;40:12186–12192. doi: 10.1021/bi011156u. [DOI] [PubMed] [Google Scholar]
- 52.Lasey RC, Banerji SS, Ogawa MY. Inorg Chim Acta. 2000;300–302:822–828. [Google Scholar]
- 53.Fedorova A, Chaudhari A, Ogawa MY. J Am Chem Soc. 2003;125:357–362. doi: 10.1021/ja026140l. [DOI] [PubMed] [Google Scholar]
- 54.Burkhard P, Stetefeld J, Strelkov SV. Trends Cell Biol. 2001;11:82–88. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- 55.Kohn WD, Hodges RS. Trends Biotechnol. 1998;16:379–389. [Google Scholar]
- 56.Lupas A. Trends Biochem Sci. 1996;21:375–382. [PubMed] [Google Scholar]
- 57.Hodges RS. Biochem Cell Biol. 1996;74:133–154. doi: 10.1139/o96-015. [DOI] [PubMed] [Google Scholar]
- 58.Moser CC, Page CC, Cogdell RJ, Barber J, Wraight CA, Dutton PL. Membrane Proteins. 2003;63:71–109. doi: 10.1016/s0065-3233(03)63004-4. [DOI] [PubMed] [Google Scholar]
- 59.Winkler JR, Di Bilio AJ, Farrow NA, Richards JH, Gray HB. Pure Appl Chem. 1999;71:1753–1764. [Google Scholar]
- 60.Wolfgang J, Risser SM, Priyadarshy S, Beratan DN. J Phys Chem B. 1997;101:2986–2991. [Google Scholar]
- 61.Kurnikov I. Private communication. [Google Scholar]
- 62.Fox MA, Galoppini E. J Am Chem Soc. 1997;119:5277–5285. [Google Scholar]
- 63.Knorr A, Galoppini E, Fox MA. J Phys Org Chem. 1997;10:484–498. [Google Scholar]
- 64.Galoppini E, Fox MA. J Am Chem Soc. 1996;118:2299–2300. [Google Scholar]
- 65.Liu L, Hong J, Ogawa MY. J Am Chem Soc. 2004;126:50–51. doi: 10.1021/ja036579t. [DOI] [PubMed] [Google Scholar]
- 66.Gray HB, Winkler JR. Q Rev Biophys. 2003;36:341–372. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]
- 67.Page CC, Moser CC, Dutton PL. Curr Opin Chem Biol. 2003;7:551–556. doi: 10.1016/j.cbpa.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 68.Archer M, Carvalho AL, Teixeira S, Moura I, Moura JJG, Rusnak F, Romao MJ. Protein Sci. 1999;8:1536–1545. doi: 10.1110/ps.8.7.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Henehan CJ, Pountney DL, Zerbe O, Vasak M. Protein Sci. 1993;2:1756–1764. doi: 10.1002/pro.5560021019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wernimont AK, Huffman DL, Lamb AL, O’Halloran TV, Rosenzweig AC. Nat Struct Biol. 2000;7:766–771. doi: 10.1038/78999. [DOI] [PubMed] [Google Scholar]
- 71.Tanchou V, Gas F, Urvoas A, Cougouluegne F, Ruat S, Averseng O, Quemeneur E. Biochem Biophys Res Commun. 2004;325:388–394. doi: 10.1016/j.bbrc.2004.10.048. [DOI] [PubMed] [Google Scholar]
- 72.Urvoas A, Amekraz B, Moulin C, Le Clainche L, Stocklin R, Moutiez M. Rapid Commun Mass Spectrom. 2003;17:1889–1896. doi: 10.1002/rcm.1130. [DOI] [PubMed] [Google Scholar]
- 73.Urvoas A, Moutiez M, Estienne C, Couprie J, Mintz E, Le Clainche L. Eur J Biochem. 2004;271:993–1003. doi: 10.1111/j.1432-1033.2004.04001.x. [DOI] [PubMed] [Google Scholar]
- 74.Kihlken MA, Leech AP, Le Brun NE. Biochem J. 2002;368:729–739. doi: 10.1042/BJ20021036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kharenko O. PhD Dissertation. Bowling Green State University; 2005. [Google Scholar]
- 76.Gasyna Z, Zelazowski A, Green AR, Ough E, Stillman MJ. Inorganica Chimica Acta-Bioinorganic Chemistry. 1988;153:115–118. [Google Scholar]
- 77.Green AR, Stillman MJ. Inorganica Chimica Acta. 1994;226:275–283. [Google Scholar]
- 78.Heaton DN, George GN, Garrison G, Winge DR. Biochemistry. 2001;40:743–751. doi: 10.1021/bi002315x. [DOI] [PubMed] [Google Scholar]
- 79.Casasfinet JR, Hu S, Hamer D, Karpel RL. Biochemistry. 1992;31:6617–6626. doi: 10.1021/bi00143a036. [DOI] [PubMed] [Google Scholar]
- 80.Cobine PA, George GN, Jones CE, Wickramasinghe WA, Solioz M, Dameron CT. Biochemistry. 2002;41:5822–5829. doi: 10.1021/bi025515c. [DOI] [PubMed] [Google Scholar]
- 81.Stillman MJ. Coord Chem Rev. 1995;144:461–511. [Google Scholar]
- 82.Ford PC, Cariati E, Bourassa J. Chem Rev. 1999;99:3625–3647. doi: 10.1021/cr960109i. [DOI] [PubMed] [Google Scholar]
- 83.Pountney DL, Schauwecker I, Zarn J, Vasak M. Biochemistry. 1994;33:9699–9705. doi: 10.1021/bi00198a040. [DOI] [PubMed] [Google Scholar]
- 84.Bogumil R, Faller P, Pountney DL, Vasak M. Eur J Biochem. 1996;238:698–705. doi: 10.1111/j.1432-1033.1996.0698w.x. [DOI] [PubMed] [Google Scholar]