

SUMMARY
Proteins can perform completely distinct functions in response to the particular partners to which they bind. Consequently, determination of the mechanism of functional regulation in such systems requires elucidation of the mechanism switching between binding partners. The central protein of the Escherichia coli Biotin Regulatory System, BirA, switches between it’s function as a metabolic enzyme or a transcriptional repressor in response to binding either the Biotin Carboxyl Carrier Protein subunit of acetyl-CoA carboxylase or a second BirA monomer. These two protein:protein interactions are structurally mutually exclusive. Results of previous studies suggest that the system is regulated by kinetic partitioning between the two protein:protein interactions. In this work sedimentation velocity was employed to directly monitor the partitioning. Results of the measurements indicate similar equilibrium parameters governing formation of the two protein:protein interactions. Kinetic analysis of the sedimentation velocity data indicates that holoBirA dimerization is governed by very slow forward and reverse rate constants. The slow kinetics of holoBirA dimerization combined with fluctuations in the intracellular apoBCCP pool are critical determinants in partitioning of BirA between its distinct biological functions.
Keywords: Transcriptional switch, protein:protein interactions, sedimentation velocity, systems biology
INTRODUCTION
Protein function is subject to regulation in response to environmental and metabolic cues. In one common mode of regulation, small molecule metabolites bind to proteins to alter their activities. Alternatively, modulation of protein function can occur via binding of a second protein. An extreme example of this type of regulation is the adoption of completely distinct functions by a protein in response to the specific partner to which it binds. Identification of the protein partners alone is insufficient for understanding the mechanism by which protein function is regulated by a partner. Rather, mechanistic understanding requires determination of the thermodynamic and kinetic rules governing the formation of the alternative protein complexes.
The Escherichia coli Biotin Regulatory System provides a model for investigation of functional switching of a protein via alternative protein partnering (Figure 1). In this system, the protein, BirA, functions as a transcriptional repressor and a metabolic enzyme (1,2). In its metabolic function BirA catalyzes biotin linkage to the biotin-dependent carboxylase, acetyl CoA carboxylase (ACC). This is a two-step reaction in which the enzyme first catalyzes synthesis of bio-5'-AMP from the substrates biotin and ATP and then transfers biotin from the adenylate to the Biotin Carboxyl Carrier Protein (BCCP) subunit of ACC (3). Because ACC catalyzes the first committed step of fatty acid synthesis, post-translational biotin addition is critical for cell viability. As a transcriptional repressor, BirA binds specifically to the 40-base pair biotin operator sequence (bioO) to repress transcription of the biotin biosynthetic operon. Assembly of the repression complex occurs via two coupled reactions, dimerization followed by DNA binding (4,5). The intermediate in biotin transfer, bio-5'-AMP, facilitates repression complex assembly by selectively promoting BirA homo-dimerization (6). Thus in this system the activated form of BirA, holoBirA, is the obligatory species in both metabolism and transcription repression. Furthermore, the functional switch reflects formation two alternative protein:protein interactions; homo-dimerization of holoBirA and hetero-dimerization of holoBirA with apoBCCP.
Figure 1.

The Biotin Regulatory System in Escherichia coli. ApoBirA binds substrates biotin and ATP and catalyzes synthesis of biotinyl-5'-AMP. The resulting holoBirA monomer either homo-dimerizes and binds to bioO or interacts with apoBCCP and catalyzes biotin transfer. BCCP is a subunit of acetyl-CoA carboxylase.
At a structural level the functional switch in BirA is based on formation of mutually exclusive protein:protein interfaces (Figure 2. The structure of BirA dimer bound to the corepressor analog biotinol-5'-AMP (btnOH-AMP), which is a good functional mimic of bio-5'-AMP in promoting homodimerization, has been solved by X-ray crystallography (7,8). Although the holoBirA·apoBCCP heterodimer structure has not been experimentally determined, a model of this structure has been proposed (9). This model was constructed using the experimentally determined structure of a C-terminal apoBCCP fragment, apoBCCP87, which is known to be functionally identical to the intact subunit in the biotin transfer reaction (10). The general features of this model have been confirmed in an experimentally determined structure of the BirA paralog, Pyrococcus horikoshii biotin protein ligase, bound to its homologous acceptor protein (11,12). In both the homo-dimer and hetero-dimer structures the central β-sheet of the holoBirA monomer is extended through a β-sheet interaction with either a second holoBirA monomer or with apoBCCP87. Furthermore, since the same surface of BirA is used for both interactions, formation of one precludes formation of the other.
Figure 2.
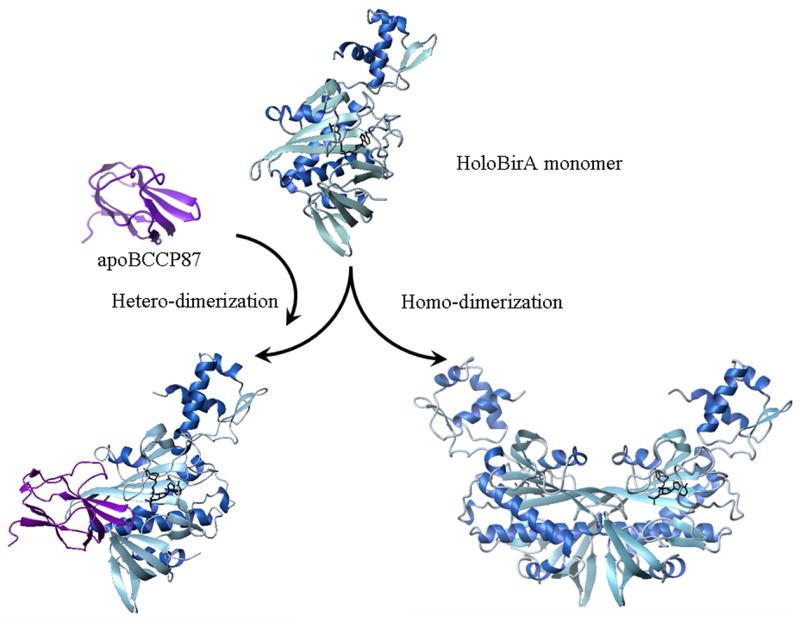
Hetero- and homo-dimerization are mutually exclusive protein-protein interactions. The holoBirA monomer, holoBirA homo-dimer and holoBirA·apoBCCP87 hetero-dimer models were constructed using MolMol (39) with pdb files 2EWN, 1HXD, and 1BDO, respectively.
In vivo studies indicate that the BirA functional switch is controlled by apoBCCP concentration. Li & Cronan have shown that apoBCCP production is controlled by growth rate (13). Furthermore, the intracellular apoBCCP concentration influences transcription repression at the biotin biosynthetic operon control region. Induction of apoBCCP87 synthesis in bacterial cultures in which transcription initiation at the control region is repressed results in derepression (14). A reasonable interpretation of this result is that the increased apoBCCP engages holoBirA, thereby decreasing the availability of holoBirA for homo-dimerization. Consequently, occupancy of bioO by holoBirA is decreased with concomitant derepression.
Although the structural and in vivo data indicate that competitive protein-protein interactions control BirA function, the mechanism of functional switching is not known. In principle, the control can occur at either a kinetic or equilibrium level. The mechanism was previously investigated by measuring the effect of apoBCCP87 on the initial rate of holoBirA binding to bioO (15). The results revealed that although the acceptor protein does not influence either the biphasic kinetic behavior associated with the binding or the rates of the two phases, it does affect the final equilibrium occupancy of bioO by holoBirA. This result is consistent with the idea that apoBCCP87 inhibits bioO binding by depleting the holoBirA pool. Global analysis of inhibition data obtained at a range of apoBCCP87 concentrations yielded an inhibition constant of approximately 2.8 μM, a value similar in magnitude to the equilibrium constant governing holoBirA homo-dimerization of 10 μM. The similar energetics of formation of the two dimers suggest that the functional switch may be subject to kinetic rather than equilibrium control.
Results of combined structural, biological and biochemical studies suggest a model in which partitioning of holoBirA between its metabolic and transcriptional functions is controlled by the relative probability with which it encounters a particular protein partner. In this work this model has been further tested by direct measurements of partitioning of holoBirA between hetero- and homo-dimerization. Partitioning was monitored using a novel approach in which the hetero-dimerization process was measured by quantifying its effect on holoBirA dimerization. The measurements, which were performed using sedimentation velocity, reveal similar equilibrium constants for holoBirA hetero- and homo-dimerization. The sedimentation velocity data were also analyzed to obtain the kinetics of holoBirA dimerization, the results of which indicate unusually slow kinetics. The slow homo-dimerization kinetics combined with the changing intracellular apoBCCP supply are predicted to be critical factors in regulating partitioning of holoBirA between its metabolic and transcriptional functions.
RESULTS
Sedimentation Velocity Measurements of holoBirA Homo-dimerization
The primary technique used in these studies is sedimentation velocity. Previous application of sedimentation equilibrium measurements to characterize the holoBirA monomer-dimer equilibrium revealed that it occurs with an equilibrium dissociation constant of 5–10 μM in Standard Buffer (10 mM Tris-HCl, pH 7.50 ± 0.02, at 20.0 ± 0.1)°C, 200 mM KCl, and 2.5 mM MgCl2) at 20 °C (6,16). In order to apply sedimentation velocity to measure hetero-dimerization of apoBCCP87 with holoBirA it was necessary to first characterize the homo-dimerization reaction using the velocity technique. Velocity measurements were conducted on samples prepared at loading concentrations ranging from 5 to 55μM. Prior to starting centrifugation samples were incubated for at least two hours in order to attain equilibrium. As shown in Figure 3A sedimentation velocity profiles of holoBirA exhibit a single sedimentation boundary. The concentration dependence of the sedimentation coefficient distribution was first obtained by subjecting the data to the c(s) analysis using SEDFIT. Analysis of the samples prepared at the three highest concentrations yields peaks with concentration-independent positions (Figure 3B) of approximately 2.7 S and approximately 3.8 S. The apparent discrepancy between the single sedimentation boundary observed in the raw data and the presence of two peaks in the c(s) analysis is due to the similar magnitudes of the sedimentation coefficients for the species. Finally, the relative populations of the two species change in the concentration-dependent manner anticipated for a protein association equilibrium.
Figure 3.
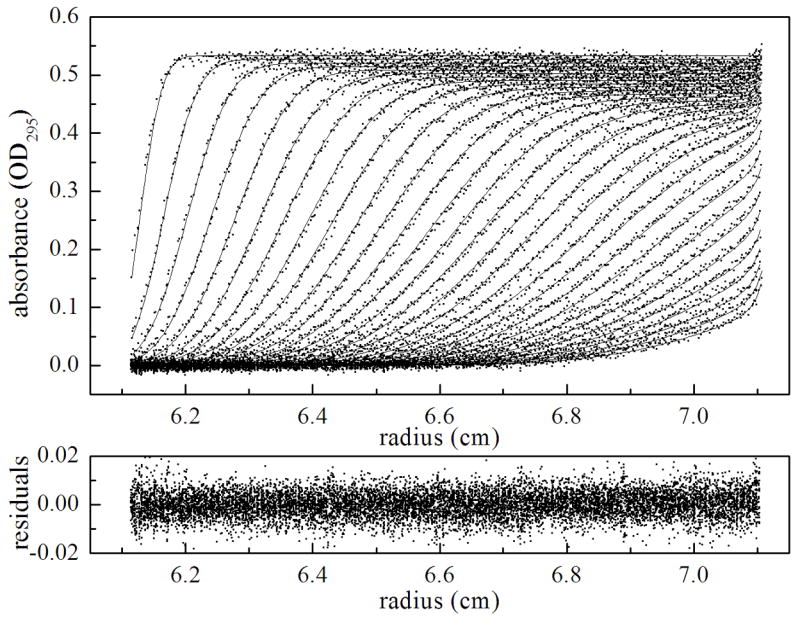
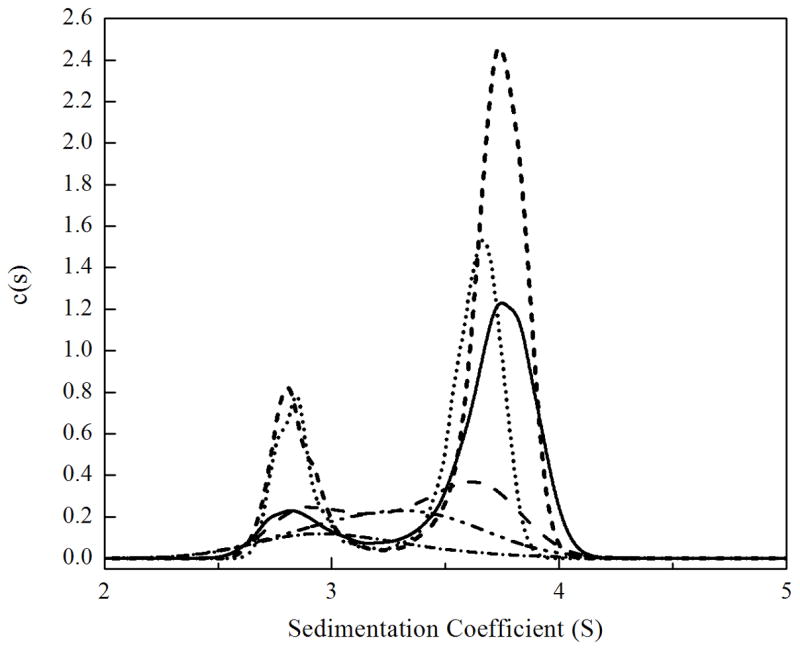
(a). Results of sedimentation velocity measurements of holoBirA dimerization obtained at 20 °C. The measurement was performed with 26 μM holoBirA in standard buffer at a rotor speed of 50,000 rpm. The best-fit curves and residuals were obtained from continuous c(s) analysis with SEDFIT with 68% confidence intervals. (b). Sedimentation coefficient distributions, c(s), obtained at loading concentrations of 5 (-•-), 10 (-• •-), 15 (- -), 20 (---), 40 (…) and 55 (—) μM holoBirA. Scans for the 5, 10, 15, and 20 μM holoBirA samples were obtained at 295 nm and those at and 40 and 55 μM holoBirA were obtained at 300 nm. The smaller extinction coefficient at 300 nm explains why the total area associated with the highest concentration samples is less than that associated with samples prepared at lower concentrations.
In order to determine the equilibrium constant for holoBirA homo-dimerization from the sedimentation velocity data, the isotherm of weight-average sedimentation coefficient sw as a function of protein concentration was analyzed using a monomer-dimer model. Weight-average sedimentation coefficients at the indicated concentrations were resolved by integration of the c(s) profiles. As expected for a self-associating system, the weight-average sedimentation coefficient sw increases with increasing protein concentration (Figure 4). However, the data are limited to the transition region of the isotherm and contain no information about the lower and upper asymptotes. This is due to the limited sensitivity to concentrations relevant to the monomeric endpoint and the very high concentrations required to obtain information at the dimer endpoint. In order to analyze the data using the monomer-dimer model, knowledge of the sedimentation coefficient for monomer (S1) is required. This information was obtained from sedimentation velocity measurements performed on the BirA-R119W mutant that is defective in dimerization (17). Measurements were performed with holoR119W prepared at loading concentrations of 10 and 20 μM, at which the complex is monomeric. The sedimentation coefficient resolved from analysis of these data of 2.72 S (Table 1) was used as the fixed value of S1 in fitting the sw isotherm for wild type holoBirA to a monomer-dimer model. Analysis yielded excellent agreement with the model, a dimer sedimentation coefficient (S2) of 3.9 ± 0.1 S and an equilibrium dissociation constant, KD of 10 ± 1 μM, a value identical to that determined by sedimentation equilibrium measurements (Table 1).
Figure 4.

Dependence of the weight-average sedimentation obtained by integration the c(s), on holoBirA concentration shown with the best-fit isotherm obtained from nonlinear least squares analysis of the data using a monomer-dimer model.
Table 1.
Parameters governing homo- and hetero-dimerization in the Escherichia coli Biotin Regulatory Systema
| Homo-dimerization | Hetero-dimerizationb | |
|---|---|---|
| Kd (μM) | 10 ± 1d | 2.8 ± 0.4 |
| ΔG° (kcal/mole) | −6.7 ± 0.1 | −7.4 ± 0.1 |
| S1, S2c | 2.72±0.03, 3.9±0.1 | NA |
| kon (M−1−s−1) | 20 ± 8 | ND |
| koff (s−1) | 2.7(± 0.5)×10−4 | ND |
All reported values were obtained at 20°C in Standard Buffer.
For hetero-dimerization the parameters are considered apparent equilibrium values. See text for explanation.
The sedimentation coefficient for the monomer was experimentally determined from measurements performed on the mutant R119W and that for the dimer was obtained from nonlinear least squares analysis of the SW isotherm.
Reported errors represent the 68% confidence intervals.
Sedimentation Velocity Measurements of Hetero-dimerization
Hetero-dimerization of apoBCCP87 with holoBirA was determined by measuring perturbation of holoBirA dimerization using sedimentation velocity. While direct measurements are preferred, they are not accessible in this system. First, hetero-dimerization of holoBirA with apoBCCP87 results in chemical transfer of biotin to the acceptor protein with release of holoBCCP87. One approach that was considered for isolating the reversible binding of holoBirA to apoBCCP from the subsequent chemical steps is use of nonhydrolyzable analogs of bio-5'-AMP, including biotinol-5'-AMP and biotinoyl-sulfamoyl adenylate (8). However, trials with these analogs demonstrated that complexes of BirA bound to either do not interact with apoBCCP87. Therefore, the strategy developed to quantify hetero-dimerization was to measure the effect of apoBCCP on holoBirA dimerization as detected in sedimentation velocity measurements. The experimental design is based on the well-established thermodynamic linkage between bio-5'-AMP binding and BirA dimerization. In the conditions used for the measurements reported in this paper apoBirA dimerization is undetectable. By contrast, holoBirA dimerizes with an equilibrium dissociation constant of 10 μM. Thus, combination of apoBCCP87 with an equilibrium mixture of holoBirA monomer and dimer perturbs the self-association equilibrium because interaction of the acceptor protein with holoBirA results in its conversion to the dimerization-incompetent apoBirA. Given that hetero-dimerization is, itself, a concentration dependent process, the magnitude of the perturbation of homo-dimerization reflects the apparent affinity of apoBCCP87 for holoBirA.
Application of sedimentation velocity to measure competing homo- and heterodimerization of holoBirA required careful experimental design. For example, it is important to point out that in the measurements holoBirA was prepared by combining equimolar amounts of apoBirA and bio-5'-AMP. This design ensures that the perturbation of hetero-dimerization reflects a single interaction of a holoBirA monomer with apoBCCP87 and no BirA turnover occurs. A second point about experimental design is that the spectroscopic properties of BCCP87 and bio-5'-AMP are such that use of a wavelength of 295 nm in scans of the sedimentation boundaries avoided any contribution of either of these species to the absorbance.
The c(s) analysis of velocity measurements performed at 20μM holoBirA in the absence and presence of apoBCCP87 are shown in Figure 5. In the absence of acceptor protein the holoBirA population is weighted toward dimer. Inclusion of 5μM apoBCCP87 shifts the population in favor of monomer. In order to obtain the apparent equilibrium constant (KI) governing hetero-dimerization between holoBirA and apoBCCP87 data were subjected to global analysis using the model described in Materials & Methods and constructed in SEDANAL. Eight data sets acquired both at a single holoBirA concentration of 20μM in the presence of apoBCCP87 ranging in concentration from 2 to 20μM and at holoBirA concentrations ranging from 10 to 55μM in the presence of 5μM apoBCCP87 were included in the analysis. In the model indicated in eq 9a and 9b the holoBirA monomer simultaneously participates in both hetero- and homo-dimerization. However, since hetero-dimerization converts holoBirA to a species that is dimerization incompetent, the holoBirA dimer population is controlled by both the equilibrium constant governing homo-dimerization, KD, and the apparent equilibrium dissociation constant, KI, governing hetero-dimerization. In the analysis KD is fixed at the experimentally determined value of 10 μM and KI is a global parameter resolved from the analysis. The value of KI obtained from the analysis is 2.8 ± 0.4 μM, which is similar in magnitude to the equilibrium constant governing homo-dimerization (Table 1).
Figure 5.
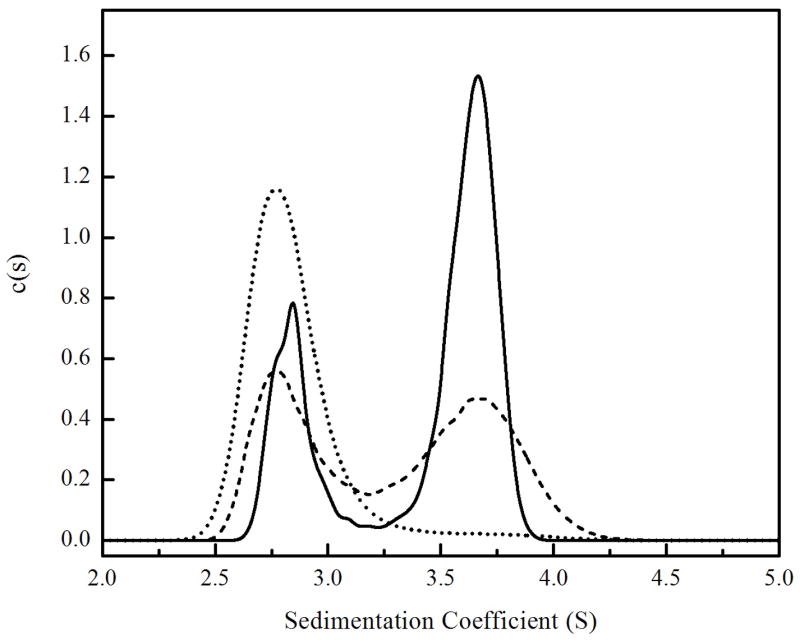
Sedimentation coefficient distributions, c(s), obtained from the sedimentation profiles of 20 μM holoBirA measured in the presence of (—) 0 μM, (---) 5 μM and (....) 20 μM apoBCCP87.
Hetero-dimerization at Physiological Temperature
The similarity of the equilibrium constants governing homo-dimerization and hetero-dimerization of holoBirA at 20°C suggests that the switch from enzyme to transcription repressor is not governed by equilibrium. However, this result does not, necessarily, extend to the physiological temperature of 37°C. Indeed, previous studies of the homo-dimerization reaction indicate that this process becomes significantly more favorable as the temperature is increased from 20 to 37°C (18). Thus, hetero-dimerization was measured at 37°C. Because this temperature is well above the practical range of the commercially available analytical ultracentrifuge, an alternative strategy was used to measure KI at the higher temperature. Measurements of KI at 20°C presented above indicate a value identical to that obtained using an indirect method in which the effect of apoBCCP87 on the occupancy of holoBirA on the biotin operator sequence, bioO, in monitored by DNaseI footprinting (15). This consistency in the KI value not only validates this indirect method for determining the parameter but also provides a means for measuring it at 37°C. In this experiment the fractional saturation of bioO by holoBirA is monitored as a function of apoBCCP87 concentration. Because homo-dimerization obligatorily precedes DNA binding, the perturbation of the homo-dimer population can be observed as a change in the fractional saturation of bioO by holoBirA. As indicated in eq 10 (Materials & Methods) the magnitude of the perturbation depends on the magnitude of KI. In order to establish the reference for the inhibition measurements quantitative DNaseI footprint titrations were first employed to measure assembly of two holoBirA monomers on bioO at 37 °C. These measurements yielded an equilibrium association constant for two-step assembly reaction, Ktotal, of 2.3 × 1017 M−2 and allowed establishment of the conditions for performing the inhibition measurements. In these measurements the holoBirA concentration is set at a value sufficient to just saturate the operator and apoBCCP87 concentration is varied. As indicated in the image obtained from one such experiment, the footprint disappears as apoBCCP87 concentration is increased (Figure 6A). Quantitation of the footprint yielded an inhibition isotherm (Figure 6B) analysis of which using nonlinear least-squares regression with the model shown in eq 10 yields KI. As indicated by the best-fit curve in Figure 6B, the data are well-described by the model and the KI resolved from the analysis is 1.9 ± 0.1 μM, a value similar to that obtained at 20 °C. Previous studies of the temperature dependence of homo-dimerization indicated a KD of 0.12 μM at 37 °C. Thus, the equilibrium constants governing hetero- and homo-dimerization differ by approximately 10-fold at physiological temperature.
Figure 6.


(a). Image of a gel obtained for DNaseI footprint measurements of apoBCCP87-mediated inhibition of holoBirA binding to bioO. The measurement was performed at 37°C with 1×10−8M holoBirA and varying concentrations of apoBCCP87. (b). Inhibition data shown with the best-fit curve obtained from nonlinear least squares analysis using Equation 10.
The Kinetics Governing Homo-dimerization are Slow
The energetic similarity of homo- and hetero-dimerization suggests that the functional switch in BirA is not governed by equilibrium. In order to investigate the kinetics of the system, the results of sedimentation velocity measurements of homo-dimerization were used to estimate the dynamics of the process. This technique can readily reveal whether the kinetics of a macromolecular equilibrium are fast or slow relative to the time scale of sedimentation velocity (19). The kinetics are diagnosed in a series of experiments performed at a range of loading concentrations using c(s) analysis (20). Based on Gilbert-Jenkins theory (21), in fast reactions, c(s) analysis yields peaks at positions that do not necessarily reflect the sedimentation coefficient of either molecular species, but are governed by chemical interconversion. In this case one observes a single peak, the position of which is determined by the relative contributions of the dimer and monomer to the species population. By contrast, in the slow kinetics regime the c(s) analysis yields peaks that reflect the sedimentation coefficients of the contributing species and are concentration independent (19). Inspection of the results of c(s) analysis of sedimentation velocity profiles obtained for holoBirA at a range of loading concentrations in (Figure 3b) suggests that the homo-dimerization kinetics are slow. Concentration-dependent shifts in the peak positions were not observed and, consistent with slow kinetics, at high protein concentrations the peaks corresponding to the monomer and dimer species are baseline separated. Moreover, the approximate values of the sedimentation coefficients estimated from the c(s) analysis of data obtained for the high concentration samples indicates good agreement with the best-fit values obtained from analysis of the sw isotherm (Table 1). Global analysis of the data was conducted using SEDANAL, which is based on finite-element numerical solutions of the Lamm equation, to obtain concentration distribution profiles (22). In the analysis resolution of the kinetic parameters governing the equilibrium is achieved by solving the kinetic differential equations describing the mass-action equilibrium with explicit reaction schemes and stoichiometry (23). The analysis was performed with eight input data sets acquired over a range of holoBirA monomer loading concentrations. Results of the analysis using a monomer-dimer equilibrium model indicate excellent agreement between the data and the model (Figure 7). The rate constant for dimer dissociation, koff, obtained from the analysis is 2.7 (±0.5) × 10−4 s−1. The equilibrium dissociation constant KD of 10 ± 1 μM obtained from the analysis is identical to that resolved from analysis of the sw isotherm (Table 1).
Figure 7.

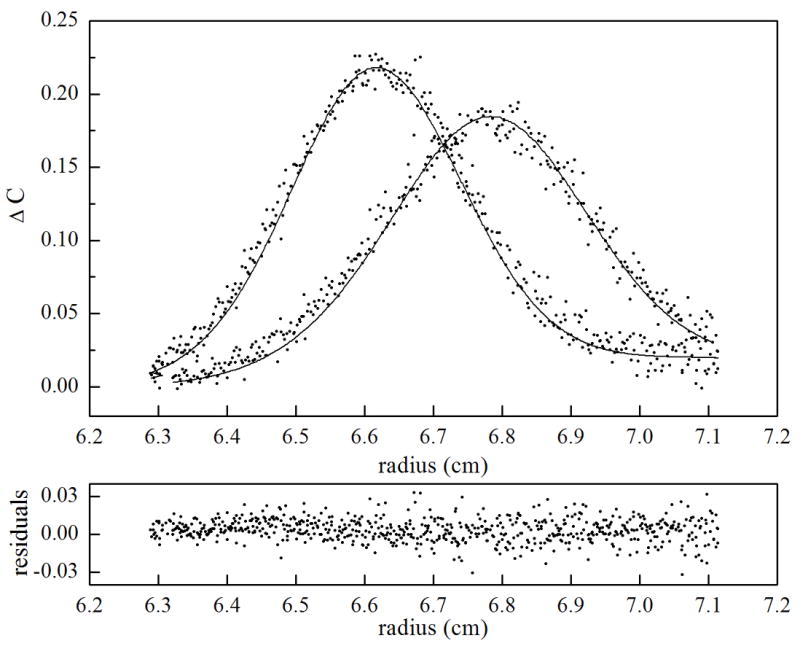
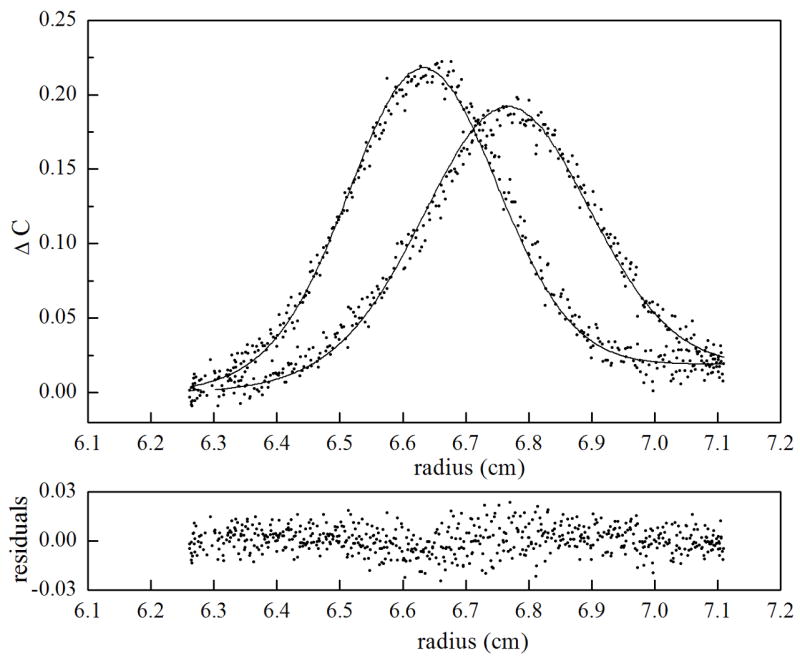
Sedimentation velocity measurements of holoBirA homo-dimerization analyzed using SEDANAL. Time-difference plots are shown for three holoBirA loading concentrations including (a). 5 μM, (b). 20 μM and (c). 55 μM. In the top panel for each plot, raw data are shown as dots and the best-fit curve from global analysis analysis of eight data sets using a monomer-dimer model is shown as continuous lines. The bottom panel for each concentration shows the residuals of the fit.
DISCUSSION
BirA, the central protein of Escherichia coli Biotin Regulatory System funnels biotin into metabolism or regulates biotin biosynthesis by switching between two alternate protein:protein interactions, hetero-dimerization with apoBCCP, and homo-dimerization. The former interaction leads to formation of holoBCCP, an obligate species in catalysis of the first committed step of fatty acid biosynthesis. The latter process precedes DNA binding, which results in transcription repression at the biotin biosynthetic operon. Although structural studies indicate that the two interactions use the same BirA surface, and are, therefore, mutually exclusive, the mechanism by which BirA switches functions is not known. Previous measurements of inhibition of holoBirA binding to bioO by apoBCCP suggest that the two dimerization processes are governed by similar equilibrium constants. Accordingly the functional switch is proposed to be regulated by kinetic partitioning between the two protein-protein interactions. The work in this paper is designed to test this model through direct measurement of partitioning between the two protein:protein interactions.
Sedimentation velocity measurements were applied to determine the relative energetics of hetero- and homo-dimerization. Consistent with results of measurements of the process using sedimentation equilibrium, the velocity technique yielded an equilibrium dissociation constant for homo-dimerization of 10 μM at 20 °C. Direct equilibrium measurements of binding of the holoBirA·apoBCCP binding interaction are complicated by the fact that hetero-dimer formation results in chemistry to produce holoBCCP and apoBirA. However, because apoBirA is a dimerization-incompetent species, addition of apoBCCP to a mixture of holoBirA monomer and dimer perturbs this equilibrium. Consequently, the energetics of hetero-dimerization can be obtained from quantifying the effect of apoBCCP on the homo-dimerization reaction. These measurements are technically facilitated by the spectroscopic characteristic of the system. At the working wavelength, even in a relatively complex mixture of BirA, bio-5'-AMP and BCCP, BirA alone is detected because the absorption signals from all other species are negligible at 295 nm. Because binding of apoBCCP to holoBirA is followed by irreversible chemistry, the inhibition constant, KI, obtained from this analysis is the apparent equilibrium constant governing hetero-dimerization The equilibrium dissociation constant for hetero-dimerization obtained with this method is 2.8 μM (−7.4 kcal/mole), close to the value for homo-dimerization of 10 μM (−6.7 kcal/mole). Thus, partitioning of holoBirA between these two interactions is not controlled by equilibrium. This value of KI is identical to that obtained from previous measurements of apoBCCP-mediated inhibition of bioO binding by holoBirA. Analysis of those data was based on the assumption that the perturbation occurred at the dimerization step and the excellent agreement obtained using the two methods further supports a model in which functional switching of holoBirA is centered on partitioning between hetero- and homo-dimerization.
The relevance of the similar energetics of the two dimerization reactions to physiological conditions was further examined by extending measurements of the apoBCCP interaction with holoBirA to 37 °C. HoloBirA homo-dimerization has been shown to exhibit a steep temperature-dependence (18) and becomes significantly tighter with increasing temperature. Given the limitations of the analytical ultracentrifuge, hetero-dimerization measurements at 37°C were performed by monitoring perturbation of bioO binding by holoBirA. The apparent equilibrium dissociation constant obtained is 1.9 μM (−8.1 kcal/mole), approximately 10-fold greater than that for homo-dimerization of 0.12 μM (−9.8 kcal/mole) at the same temperature. Thus, even at physiological temperature the two alternate protein-protein interactions are energetically similar.
Despite the similarity of the overall energetics of the two dimerization reactions, several lines of evidence suggest that the detailed chemistries of these two processes are not equivalent. First, in addition to bio-5'-AMP, binding of two non-hydrolyzable analogs, biotinol-5'-AMP (btnOH-AMP) and 5'-O-[N-(biotinoyl)-sulfamoyl] adenosine (btnSA), enhance BirA dimerization, the former by −3 kcal/mole and the latter by −1.0 kcal/mole (8). By contrast, the complexes of BirA bound to these analogs do not interact with apoBCCP (Streaker & Beckett, unpublished). Thus the interaction of apoBCCP with BirA is more sensitive to the detailed chemistry of the allosteric effector than is the homo-dimerization process. Second, the homo-dimerization equilibrium is characterized by large temperature dependence, with an enthalpy change of 41 kcal/mol. By contrast, the hetero-dimerization equilibrium constants measured at 20 °C and 37 °C are similar in magnitude. Third, inspection of the homo-dimer and hetero-dimer structures indicates distinct differences in chemistries of their interfaces. Both the holoBirA homo-dimer and the heterodimer with apoBCCP form through β-sheet interactions, and are, therefore superficially similar. However, close inspection of the structures indicates a greater contribution of ionic interactions to the homo-dimer than to the hetero-dimer interface.
The energetic similarity of hetero- and homo-dimerization prompted investigation of the kinetics of homo-dimerization. The sedimentation coefficient distribution, c(s), plots obtained from the scans of the sedimentation velocity measurements performed on holoBirA are consistent with slow kinetics. The hallmark of slow kinetics is full separation of the species boundaries at high protein concentrations. By contrast, rapid kinetics is expected to yield a single peak, representing the average sedimentation coefficient of the rapidly inter-converting mixture of monomer and dimer. Consistent with the baseline separation in the c(s) profiles for holoBirA, the dissociation rate of the dimer, koff, resolved from global analysis of the velocity data is ~3×10−4 s−1. While additional measurements using direct kinetic techniques will be employed to confirm the precise magnitudes of the rates of holoBirA dimerization, the sedimentation velocity results are consistent with slow kinetics for the process. Similarly slow kinetics of protein:protein interactions have been previously reported. TATA box binding protein dimers dissociate slowly (koff =2×10−3 s−1, t1/2 = 6–10 min) and this dissociation is proposed to present a formidable kinetic barrier to the monomer binding to TATA box (24). The slow dissociation rate of the tubulin dimer of 7.8×10−5 s−1 is correlated with a ligand-induced conformational change (25). Sedimentation velocity and equilibrium experiments have demonstrated slow dissociation of the apo IRP1 (iron regulatory protein) dimer, with a koff ~ 5×10−5 s−1 and KD of 10 μM. In the current studies, slow interconversion of holoBirA monomer and dimer indicates a large energetic barrier between these two species. At a chemical level, the slow kinetics of this interaction may be related to the thermodynamic profile of the process, which exhibits a large unfavorable enthalpy change, ΔH°, of +41 kcal/mol and an equally large favorable entropic contribution, −TΔS°, of −48 kcal/mol at 20°C (18). The slow homo-dimerization kinetics are also significant for regulation of holoBirA’s functional switch from metabolic enzyme to transcriptional repressor.
Control of Biological Output in the Biotin Regulatory System
Consideration of the kinetics of hetero- and homo-dimerization of holoBirA in the context of the entire Biotin Regulatory System illuminates the key control points in the system. The extensive kinetic and equilibrium thermodynamic measurements that have been performed on the reactions that contribute to the system are summarized in Figure 8. As indicated in the figure, the activated BirA species, holoBirA, partitions into its two functions in metabolism and transcription regulation. Therefore, the biological output in the system is related to the steady state holoBirA concentration. Although in this system output can be considered in terms of either metabolism or transcription, for the purposes of this discussion the output that is focused upon is the level of transcription initiation at the biotin biosynthetic operon. At a biochemical level the steady state holoBirA concentration reflects three processes. These include its accumulation through synthesis of the adenylate from biotin and ATP, its depletion through interaction with apoBCCP, and its depletion through homodimerization and resulting binding to bioO. Because adenylate synthesis occurs via an obligatorily sequential mechanism, with biotin binding first followed by ATP, the rate of holoBirA accumulation depends directly on biotin concentration. Once the adenylate is synthesized the resulting holoBirA is characterized by a half-life of approximately 30 min at 20 °C and, therefore, minimal spontaneous dissociation of the complex occurs. Partitioning of holoBirA between its metabolic and transcriptional functions depends on the relative rates with which it homo-dimerizes with itself and hetero-dimerizes with apoBCCP. The slow kinetics of holoBirA dimerization reported in this work provides a relatively long window of time in which apoBCCP can hetero-dimerize and partition the system toward metabolism. The hetero-dimerization rate reflects, in turn, the intracellular apoBCCP concentration. In vivo studies indicate that the rate of apoBCCP synthesis is subject to control by growth rate with faster growth rates correlated with higher rates of synthesis. Therefore, at high growth rates the rate of apoBCCP association with holoBirA should be relatively fast. While the rate of hetero-dimerization is not known, measurements of the biotin transfer reaction from holoBirA to apoBCCP indicate a bimolecular rate constant, kcat/KM of 15,000 M−1s−1 and kcat of 5.4 s−1 (10), which are consistent with fast binding kinetics. Thus at high intracellular apoBCCP concentrations the rate of hetero-dimerization exceeds that of homo-dimerization and the system output is transcription initiation at the biotin biosynthetic operon. Importantly, the kinetic demand exerted on holoBirA by apoBCCP not only ensures that biotin is synthesized, but, because of the exquisite control of bio-5'-AMP synthesis by biotin, also drives further replenishment of the adenylate-bound BirA. Since the hetero-dimerization rate depends on apoBCCP supply, once this supply is depleted homo-dimerization should occur with higher probability. The resulting homo-dimer binds very rapidly to bioO and the system output is now switched to transcription repression. In vitro measurements indicate that repression complex dissociation is slow and independent of the apoBCCP concentration (5). Therefore, once the system switches to transcription repression the kinetics of transitioning back to unrepressed transcription are anticipated to be slow. Based on this analysis, three steps in the relatively complex Biotin Regulatory System dictate the transcription output. These steps include bio-5'-AMP synthesis from biotin and ATP, formation of the hetero-dimeric apoBCCP·holoBirA complex and formation of the holoBirA dimer. Ultimately, cellular growth rate, by dictating the apoBCCP concentration, modulates the fluxes through these three steps.
Figure 8.
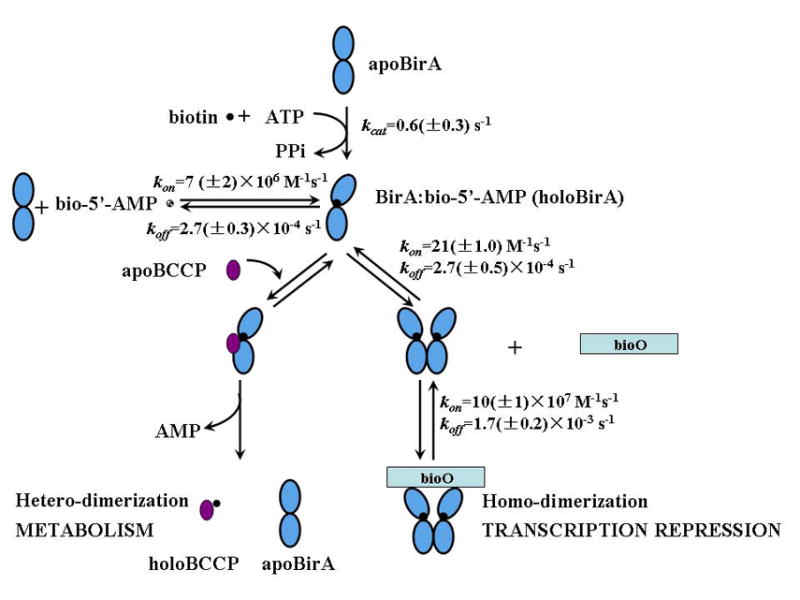
Quantitative features of the Escherichia coli Biotin Regulatory System. The experimentally determined kinetic parameters are shown for four reactions including BirA-catalyzed bio-5'-AMP synthesis (40), holoBirA monomer binding to bio-5'-AMP (41), holoBirA homo-dimerization (this work) and holoBirA homo-dimer binding to bioO (5).
Similar to BirA, the biological functions of many proteins are regulated by the context of interacting partners in which they reside. Recently high throughput proteomics approaches have been developed to define the interaction networks of specific proteins (26,27). This identification of interacting partners provides a starting point for determining the mechanism of regulation of alternative partnering and thus biological output. Several examples of context-dependent regulation of protein function have been identified. For example, the core protein of the archaeal sRNP complex, the Nop56/58 homodimer, functions in sRNP guided methylation (28). Nop56/58 can also dimerize with another core protein, fibrillarin, and the resulting hetero-dimer functions in sRNP assembly. Similar to BirA, the Nop56/58 forms mutually exclusive protein-protein interactions with its two alternative partners. In the yeast GAL system, in the absence of galactose, Gal80 self-associates and binds to Gal4 to inhibit transcription initiation at target genes. Upon galactose accumulation, the Gal80 monomer interacts with Gal3, thus allowing transcription activation by Gal4 (29). In each of these systems determination of how biological output is controlled requires elucidation of the mechanism of regulation of formation of the relevant alternative protein:protein interactions.
MATERIALS AND METHODS
Chemicals and Biochemicals
Biotinyl-5'-AMP was synthesized in this laboratory and purified using a modification of the procedure described by Lane et al (3,30), and BirA was purified as previously described (16). The truncated biotin carboxyl carrier protein (apoBCCP87) was purified as described in Nenortas and Beckett (10) with an additional chromatography step on Hydroxy-apatite ULTROL to remove trace amounts of DNaseI (15). The Klenow fragment of DNA polymerase I and restriction enzymes Hind III and Pst I were obtained from Promega. The [α-32P]dATP and dGTP used in the radiolabeling of DNA were purchased from GE Healthcare Life Sciences. The d-biotin, ATP, and DNaseI used in footprinting experiments were from Sigma-Aldrich. All chemicals used in buffer preparation were at least reagent grade.
Sedimentation Velocity Measurements
Homo-dimerization was measured by sedimentation velocity of holoBirA at six protein concentrations. Hetero-dimerization between holoBirA and apoBCCP87 was measured indirectly through its effect on homo-dimerization. In these experiments, apoBCCP87 was added to the holoBirA at the indicated concentrations. Due to the competition between homo- and hetero-dimerization, the homo-dimerization was perturbed, resulting in a weaker apparent homo-dimerization.
All experiments were carried out using a Beckman Optima XL-I analytical ultracentrifuge equipped with a four-hole An-60Ti rotor. Standard 12 mm double-sectored cells with charcoal-filled Epon centerpieces and saffire windows were used for the experiments. Protein concentration distributions were determined using the absorption optical system of the instrument (Beckman). ApoBirA and apoBCCP87 were fully exchanged into the working buffer [10 mM Tris-HCl, pH 7.50 ± 0.02, at 20 (± 0.1)°C, 200 mM KCl, and 2.5 mM MgCl2] using Biospin6 columns (Biorad). The resulting protein concentrations were determined by UV-Vis spectroscopy using the extinction coefficient at 280 nm for BirA of 47510 M−1cm−1 and 276 nm for BCCP87 of 1450 M−1cm−1 (4,10). The bio-5'-AMP solutions were prepared by diluting the concentrated stock solution in water into the working buffer. HoloBirA was made by combining bio-5'-AMP with apoBirA in a 1:1 ratio, a ratio that is critical to measurement of the hetero-dimerization process. In all measurements bio-5'-AMP and apoBirA concentrations were sufficiently high to ensure saturation of protein with ligand.
Sample volumes used in centrifugation were 390 μL and the reference sector was filled with 400 μL of buffer. Samples were prepared at indicated loading concentrations and equilibrated under vacuum at 20 °C for 2 hours before starting centrifugation. A single rotor speed of 50K RPM was used in all measurements and scans were collected in continuous mode at 295 nm in order to avoid any contribution from bio-5'-AMP or BCCP87.
DNaseI Footprint Titrations
Hetero-dimerization at the physiological temperature of 37 °C was measured by monitoring the inhibitory effect of apoBCCP87 on fractional saturation of bioO by holoBirA (31). In these experiments the fractional saturation of bioO at a single holoBirA concentration, as monitored by DNaseI footprinting, was determined as a function of apoBCCP87 concentration. The holoBirA concentration was 1×10−8 M−1, which is sufficient to saturate of bioO and close to the transition region of the binding isotherm. In the experiments, mixing was performed for only 80 s prior to addition of DNaseI so that holoBirA has sufficient time to bind to bioO but the resulting repressor-DNA complex does not dissociate. A range of apoBCCP87 dilutions were combined in the buffer [10 mM Tris-HCl (pH 7.5 ± 0.02, at 37.0 (± 0.1)°C, 200 mM KCl, 2.5 mM MgCl2, 1 mM CaCl2, 100 μg/mL BSA, 2 μg/mL sonicated calf thymus DNA, and biotin (100 μM) and ATP (1 mM)] with 12,000 cpm labeled bioO DNA (total volume = 45 μL). Reactions were initiated by addition of 5 μL of holoBirA prepared in the same buffer. After an 80 s incubation, 5 μL of 0.244 μg/mL DNaseI were added to each reaction and digestion was allowed to proceed for 20 s. Reactions were quenched by addition of 33 μL of 50 mM Na2EDTA and the DNA was precipitated by the addition of 700 μL of 0.4 M NH4OAc and 50 μg/mL tRNA in absolute ethanol. Following centrifugation, the DNA pellets were washed twice with 500 L of cold 80% (v/v) absolute ethanol in water and lyophilized. The dried pellets were resuspended in 7 μL of gel loading buffer containing 80% deionized formamide, 1 × TBE buffer, 0.02% bromophenol blue, and 0.02% xylene cyanol, heated for 10 min at 90 °C, and the products were separated by electrophoresis on a 10% denaturing acrylamide gel.
Analysis of Sedimentation Velocity Data
Analysis of the Sedimentation Boundaries with SEDFIT
Sedimentation velocity data were analyzed using the c(s) continuous distribution solutions of Lamm equation (32) with the software SEDFIT. The signal a(r,t) from the sedimentation process of an unknown mixture is approximated as a superposition:
| (1) |
of solutions of the Lamm equation
| (2) |
in which c(s) denotes the differential sedimentation coefficient distribution in units of the observed signal, r denotes the distance from the center of rotation, ω the angular velocity, and s and D represent the sedimentation and diffusion coefficients, respectively. In the c(s) analysis, the concentration distribution of a single non-interacting species χ(s,D,r,t) is calculated by the Lamm equation for a large number of sedimentation coefficients ranging from smin to smax (33). The diffusion coefficients D were estimated from a weight-average frictional ratio (f/f0)w (34). The time-invariant signal contributions aTI(r) and the radial-invariant offsets aRI(t) were calculated by algebraic noise decomposition (35). The best-fit distribution, c(s), as well as the weight-average frictional ratio and the meniscus position of the sample were optimized in a nonlinear regression of the experimental data (36).
Sw Isotherm Analysis
In order to determine the energetics of homo-dimerization, peaks of the c(s) distributions were integrated to obtain the weight-average sedimentation coefficients at various loading concentrations. The isotherm of the weight-average sedimentation coefficients takes the general form,
| (3) |
where sw denotes the weight-average s-value, ctot is the total protein concentration, i and si denote each molecular species and its sedimentation coefficient, respectively (37). The isotherm of weight-average sedimentation coefficients was globally fit to a monomer-dimer model written in GraphPad using the following equations,
| (4) |
| (5a) |
| (5b) |
in which c1 and c2 are the concentrations of monomer and dimer respectively, and Keq is the association equilibrium constant governing the dimerization reaction. The sedimentation coefficient used for monomeric holoBirA was determined from sedimentation velocity measurements performed on the holoBirA R119W mutant, which is defective in dimerization (38).
Analysis of Sedimentation Velocity Data using SEDANAL
Sedimentation velocity measurements of homo-dimerization performed at six loading concentrations were globally fit to obtain equilibrium and kinetic parameters governing dimerization using SEDANAL (22). The following equations were used in the analysis,
| (6a) |
| (6b) |
in which A denotes monomer, A2 denotes dimer, Keq is the equilibrium association constant for the reaction, [A] and [A2] are the concentrations for monomer and dimer respectively, kf is the forward rate constant and kr is the reverse rate constant.
Analysis of Sedimentation Velocity for Hetero-dimerization with SEDANAL
In the biotin regulatory system, holoBirA can form homo-dimer, or interact with apoBCCP87 to form hetero-dimer, an enzyme-substrate complex. Once this latter complex forms, biotin is transferred from bio-5'-AMP to apoBCCP87 with release of holoBCCP87 and apoBirA. Therefore, the system includes a reversible homo-dimerization step and hetero-dimerization followed by irreversible chemistry.
| (7a) |
| (7b) |
In this system, the homo-dimerization in equation (7a) is perturbed by the hetero-dimerization (7b). Importantly, once released, apoBirA does not dimerize under the conditions of these measurements. In addition, control experiments indicate that holoBCCP87 does not affect homo-dimerization (data not shown). The following simplified model describes this competition between homo- and hetero-dimerization. This model includes homo-dimer formation of A and displacement interaction between A, B, C and D.
| (8a) |
| (8b) |
In the model, A denotes holoBirA, C denotes apoBirA, and B and D are apoBCCP87 and holoBCCP87, respectively. Information about the interaction between B(apoBCCP87) and A(holoBirA) is obtained by varying both [A] and [B]. Because biotin transfer to apoBCCP87 is irreversible, the model can be further simplified to the following form:
| (9a) |
| (9b) |
where Keq is the association equilibrium constant for homo-dimerization, KI is the equilibrium constant for inhibition by hetero-dimerization. Because hetero-dimerization is coupled to an irreversible chemical step, KI should be considered an apparent equilibrium constant. The model was written in ModelEditor169, a separate program in SEDANAL package (SEDANAL manual). In the analysis, Keq was fixed at the value independently determined by sedimentation velocity measurements of homo-dimerization and KI is a global fitting parameter for all experimental data obtained at a range of apoBCCP87 concentrations.
Quantitation and Analysis of DNaseI Footprint Titrations
Footprinting gels were dried, and images were produced by at least 40 h exposures of the gels to storage phosphor screens, which were directly imaged using the Storm phosphorimaging system (GE Healthcare). The optical densities of bands representing the bioO operator binding site were integrated and isotherms were generated as described by Brenowitz et. al (31). Fractional saturation of operator by repressor was determined as a function of apoBCCP87 concentration and the resulting data were subjected to nonlinear least squares analysis using the following expression (15):
| (10) |
in which ȲBioO is the fractional saturation of bioO with holoBirA, Ktotal is the association equilibrium constant for total assembly which includes two coupled equilibria, homo-dimerization of holoBirA and dimer binding to bioO, and KI is the constant governing apoBCCP-mediated inhibition of bioO binding by holoBirA. In the analysis Ktotal is fixed at the value obtained from analysis of DNaseI footprint titrations of bioO with holoBirA and KI is floated.
Acknowledgments
This work was supported by NIH Grants RO1 GM46511 and S10-RR15899 to DB
The authors thank Drs. Patrick Brown and Jack Correia for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barker DF, Campbell AM. The birA gene of Escherichia coli encodes a biotin holoenzyme synthetase. J Mol Biol. 1981;146:451–467. doi: 10.1016/0022-2836(81)90042-5. [DOI] [PubMed] [Google Scholar]
- 2.Barker DF, Campbell AM. Genetic and biochemical characterization of the birA gene and its product: evidence for a direct role of biotin holoenzyme synthetase in repression of the biotin operon in Escherichia coli. J Mol Biol. 1981;146:469–492. doi: 10.1016/0022-2836(81)90043-7. [DOI] [PubMed] [Google Scholar]
- 3.Lane MD, Rominger KL, Young DL, Lynen F. The Enzymatic Synthesis of Holotranscarboxylase from Apotranscarboxylase and (+)-Biotin. Ii. Investigation of the Reaction Mechanism. J Biol Chem. 1964;239:2865–2871. [PubMed] [Google Scholar]
- 4.Streaker ED, Gupta A, Beckett D. The biotin repressor: thermodynamic coupling of corepressor binding, protein assembly, and sequence-specific DNA binding. Biochemistry. 2002;41:14263–14271. doi: 10.1021/bi0203839. [DOI] [PubMed] [Google Scholar]
- 5.Streaker ED, Beckett D. Coupling of protein assembly and DNA binding: biotin repressor dimerization precedes biotin operator binding. J Mol Biol. 2003;325:937–948. doi: 10.1016/s0022-2836(02)01308-6. [DOI] [PubMed] [Google Scholar]
- 6.Eisenstein E, Beckett D. Dimerization of the Escherichia coli biotin repressor: corepressor function in protein assembly. Biochemistry. 1999;38:13077–13084. doi: 10.1021/bi991241q. [DOI] [PubMed] [Google Scholar]
- 7.Wood ZA, Weaver LH, Brown PH, Beckett D, Matthews BW. Co-repressor induced order and biotin repressor dimerization: a case for divergent followed by convergent evolution. J Mol Biol. 2006;357:509–523. doi: 10.1016/j.jmb.2005.12.066. [DOI] [PubMed] [Google Scholar]
- 8.Brown PH, Cronan JE, Grotli M, Beckett D. The biotin repressor: modulation of allostery by corepressor analogs. J Mol Biol. 2004;337:857–869. doi: 10.1016/j.jmb.2004.01.041. [DOI] [PubMed] [Google Scholar]
- 9.Weaver LH, Kwon K, Beckett D, Matthews BW. Competing protein:protein interactions are proposed to control the biological switch of the E coli biotin repressor. Protein Sci. 2001;10:2618–2622. doi: 10.1110/ps.32701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nenortas E, Beckett D. Purification and characterization of intact and truncated forms of the Escherichia coli biotin carboxyl carrier subunit of acetyl-CoA carboxylase. J Biol Chem. 1996;271:7559–7567. doi: 10.1074/jbc.271.13.7559. [DOI] [PubMed] [Google Scholar]
- 11.Bagautdinov B, Matsuura Y, Bagautdinova S, Kunishima N. Crystallization and preliminary X-ray crystallographic studies of the biotin carboxyl carrier protein and biotin protein ligase complex from Pyrococcus horikoshii OT3. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:334–337. doi: 10.1107/S1744309107011967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bagautdinov B, Matsuura Y, Bagautdinova S, Kunishima N. Protein biotinylation visualized by a complex structure of biotin protein ligase with a substrate. J Biol Chem. 2008 Mar 26; doi: 10.1074/jbc.M709116200. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 13.Li SJ, Cronan JE., Jr Growth rate regulation of Escherichia coli acetyl coenzyme A carboxylase, which catalyzes the first committed step of lipid biosynthesis. J Bacteriol. 1993;175:332–340. doi: 10.1128/jb.175.2.332-340.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cronan JE., Jr Expression of the biotin biosynthetic operon of Escherichia coli is regulated by the rate of protein biotination. J Biol Chem. 1988;263:10332–10336. [PubMed] [Google Scholar]
- 15.Streaker ED, Beckett D. The biotin regulatory system: kinetic control of a transcriptional switch. Biochemistry. 2006;45:6417–6425. doi: 10.1021/bi052599r. [DOI] [PubMed] [Google Scholar]
- 16.Brown PH, Beckett D. Use of binding enthalpy to drive an allosteric transition. Biochemistry. 2005;44:3112–3121. doi: 10.1021/bi047792k. [DOI] [PubMed] [Google Scholar]
- 17.Kwon K, Streaker ED, Ruparelia S, Beckett D. Multiple disordered loops function in corepressor-induced dimerization of the biotin repressor. J Mol Biol. 2000;304:821–833. doi: 10.1006/jmbi.2000.4249. [DOI] [PubMed] [Google Scholar]
- 18.Zhao H, Streaker E, Pan W, Beckett D. Protein-protein interactions dominate the assembly thermodynamics of a transcription repression complex. Biochemistry. 2007;46:13667–13676. doi: 10.1021/bi7013097. [DOI] [PubMed] [Google Scholar]
- 19.Dam J, Schuck P. Sedimentation velocity analysis of heterogeneous protein-protein interactions: sedimentation coefficient distributions c(s) and asymptotic boundary profiles from Gilbert-Jenkins theory. Biophys J. 2005;89:651–666. doi: 10.1529/biophysj.105.059584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dam J, Velikovsky CA, Mariuzza RA, Urbanke C, Schuck P. Sedimentation velocity analysis of heterogeneous protein-protein interactions: Lamm equation modeling and sedimentation coefficient distributions c(s) Biophys J. 2005;89:619–634. doi: 10.1529/biophysj.105.059568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert GA, Jenkins RC. Boundary problems in the sedimentation and electrophoresis of complex systems in rapid reversible equilibrium. Nature. 1956;177:853–854. doi: 10.1038/177853a0. [DOI] [PubMed] [Google Scholar]
- 22.Stafford WF, Sherwood PJ. Analysis of heterologous interacting systems by sedimentation velocity: curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys Chem. 2004;108:231–243. doi: 10.1016/j.bpc.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 23.Stafford WF. Analysis of reversibly interacting macromolecular systems by time derivative sedimentation velocity. Methods Enzymol. 2000;323:302–325. doi: 10.1016/s0076-6879(00)23371-5. [DOI] [PubMed] [Google Scholar]
- 24.Coleman RA, Pugh BF. Slow dimer dissociation of the TATA binding protein dictates the kinetics of DNA binding. Proc Natl Acad Sci U S A. 1997;94:7221–7226. doi: 10.1073/pnas.94.14.7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caplow M, Fee L. Dissociation of the tubulin dimer is extremely slow, thermodynamically very unfavorable, and reversible in the absence of an energy source. Mol Biol Cell. 2002;13:2120–2131. doi: 10.1091/mbc.E01-10-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, Yates JR, 3rd, Lefkowitz RJ. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U S A. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popescu SC, Popescu GV, Bachan S, Zhang Z, Seay M, Gerstein M, Snyder M, Dinesh-Kumar SP. Differential binding of calmodulin-related proteins to their targets revealed through high-density Arabidopsis protein microarrays. Proc Natl Acad Sci U S A. 2007;104:4730–4735. doi: 10.1073/pnas.0611615104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Champion EA, Tran EJ, Brown BA, 2nd, Baserga SJ, Maxwell ES. The coiled-coil domain of the Nop56/58 core protein is dispensable for sRNP assembly but is critical for archaeal box C/D sRNP-guided nucleotide methylation. Rna. 2006;12:1092–1103. doi: 10.1261/rna.2230106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lohr D, Venkov P, Zlatanova J. Transcriptional regulation in the yeast GAL gene family: a complex genetic network. Faseb J. 1995;9:777–787. doi: 10.1096/fasebj.9.9.7601342. [DOI] [PubMed] [Google Scholar]
- 30.Abbott J, Beckett D. Cooperative binding of the Escherichia coli repressor of biotin biosynthesis to the biotin operator sequence. Biochemistry. 1993;32:9649–9656. doi: 10.1021/bi00088a017. [DOI] [PubMed] [Google Scholar]
- 31.Brenowitz M, Senear DF, Shea MA, Ackers GK. Quantitative DNase footprint titration: a method for studying protein-DNA interactions. Methods Enzymol. 1986;130:132–181. doi: 10.1016/0076-6879(86)30011-9. [DOI] [PubMed] [Google Scholar]
- 32.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal Biochem. 2003;320:104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 34.Schuck P, Rossmanith P. Determination of the sedimentation coefficient distribution by least-squares boundary modeling. Biopolymers. 2000;54:328–341. doi: 10.1002/1097-0282(20001015)54:5<328::AID-BIP40>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 35.Dam J, Schuck P. Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods Enzymol. 2004;384:185–212. doi: 10.1016/S0076-6879(04)84012-6. [DOI] [PubMed] [Google Scholar]
- 36.Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J. 2002;82:1096–1111. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuck P. Sedimentation analysis of noninteracting and self-associating solutes using numerical solutions to the Lamm equation. Biophys J. 1998;75:1503–1512. doi: 10.1016/S0006-3495(98)74069-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwon K, Streaker ED, Beckett D. Binding specificity and the ligand dissociation process in the E. coli biotin holoenzyme synthetase. Protein Sci. 2002;11:558–570. doi: 10.1110/ps.33502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 40.Xu Y, Beckett D. Biotinyl-5'-adenylate synthesis catalyzed by Escherichia coli repressor of biotin biosynthesis. Methods Enzymol. 1997;279:405–421. doi: 10.1016/s0076-6879(97)79045-1. [DOI] [PubMed] [Google Scholar]
- 41.Xu Y, Nenortas E, Beckett D. Evidence for distinct ligand-bound conformational states of the multifunctional Escherichia coli repressor of biotin biosynthesis. Biochemistry. 1995;34:16624–16631. doi: 10.1021/bi00051a010. [DOI] [PubMed] [Google Scholar]